Back to Journals » Journal of Inflammation Research » Volume 16
Programmed Cell Death in Asthma: Apoptosis, Autophagy, Pyroptosis, Ferroptosis, and Necroptosis
Authors Liu L, Zhou L, Wang LL ![]() , Zheng PD, Zhang FQ, Mao ZY, Zhang HJ, Liu HG
, Zheng PD, Zhang FQ, Mao ZY, Zhang HJ, Liu HG
Received 19 April 2023
Accepted for publication 20 June 2023
Published 1 July 2023 Volume 2023:16 Pages 2727—2754
DOI https://doi.org/10.2147/JIR.S417801
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Lu Liu,1,* Ling Zhou,1,* Ling-Ling Wang,1 Peng-Dou Zheng,1 Feng-Qin Zhang,1 Zhen-Yu Mao,1 Huo-Jun Zhang,2 Hui-Guo Liu1
1Department of Respiratory and Critical Care Medicine, Key Laboratory of Pulmonary Diseases of Health Ministry, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, Renmin Hospital of Wuhan University, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hui-Guo Liu; Huo-Jun Zhang, Email [email protected]; [email protected]
Abstract: Bronchial asthma is a complex heterogeneous airway disease, which has emerged as a global health issue. A comprehensive understanding of the different molecular mechanisms of bronchial asthma may be an efficient means to improve its clinical efficacy in the future. Increasing research evidence indicates that some types of programmed cell death (PCD), including apoptosis, autophagy, pyroptosis, ferroptosis, and necroptosis, contributed to asthma pathogenesis, and may become new targets for future asthma treatment. This review briefly discusses the molecular mechanism and signaling pathway of these forms of PCD focuses on summarizing their roles in the pathogenesis and treatment strategies of asthma and offers some efficient means to improve clinical efficacy of therapeutics for asthma in the near future.
Keywords: asthma, bronchial asthma, programmed cell death
Introduction
Asthma is a complex heterogeneous airway disease identified by pulmonary inflammation, airway hyperresponsiveness, mucus hypersecretion, and airway remodeling.1 Asthma has become a global health challenge with more than 300 million patients worldwide. Due to its rising prevalence,2 its sufferers may reach 400 million in 2025.3 Although great improvement has been made in the treatment of asthma in recent decades, there are still 5% to 10% patients are suffering from uncontrollable severe asthma.4 Asthma remains a great cause of heavy disease burden, including reduced quality of life and premature death at all ages.5 Therefore, it is compulsory to further evaluate more effective treatment options for asthma. Fully comprehending the molecular mechanism of asthma may be an effective method to discover new drug targets and enrich asthma treatments.
 |
Figure 1 Signaling pathway of apoptosis. Intrinsic apoptotic pathway: Intracellular damage signals make mitochondrial outer membrane permeabilization (MOMP) release cytochrome C and the second mitochondria-derived activator of caspases (SMAC) into the cytoplasm. Cytochrome C integrates with apoptotic protease activating factor-1 (APAF-1) and pro-caspase - 9 to establish apoptosomes. In the apoptosomes, caspase-9 self-activates and reactivates the apoptotic “executioner” function of caspase-3/7. At the same time, SMAC impedes the function of inhibitors of apoptosis proteins (IAPs) and fosters apoptosis. BH3-only proteins can activate pro-apoptotic proteins and inhibit anti-apoptotic proteins, thereby fostering apoptosis. Extrinsic apoptotic pathway: after the death ligand binds to the death receptor, the death receptor interacts with the adapter protein FAS-associated protein with death domain (FADD) and recruits pro-caspase-8 to create the death-inducing signaling complex (DISC), and caspase-8 is activated, which can directly activate caspase-3/7, and can also cleave Bid into active tBID, thereby inducing MOMP-mediated apoptosis. |
 |
Figure 2 Signaling pathway of Autophagy. Macroautophagy: When a mechanistic target of rapamycin complex 1 (mTORC1) is blocked or 5’-AMP-activated protein kinase (AMPK) is activated, the Unc-51-like kinase (unc-51-like kinase, ULK) complex activates and phosphorylates the PI3K3 complex to start the nucleation phase of the autophagosome. Under the action of ATG4, ATG7, and ATG3, microtubule-linked protein1 light chain 3 (LC3) transforms into mature LC3-II, isolating ubiquitin-like substances into evolving autophagy. The binding of ATG5, ATG12, and ATG16 further stimulate autophagic membrane elongation. Finally, the autophagosome membrane seals to create mature autophagosomes, which merge with a lysosome to form autolysosomes, and the contents of the autophagosome are degraded by lysosomal enzymes. PINK1/Parkin-mediated mitophagy: When mitochondria are damaged, the protein kinase PINK1 is blocked from entering the inner mitochondrial membrane, but accrues in the outer mitochondrial membrane and is activated by autophosphorylation, and then recruits and activates Parkin. Parkin ubiquitinates several mitochondrial membranes substrates on the mitochondrial outer membrane. After these ubiquitinated substances are identified by ubiquitin-binding proteins such as p62, the damaged mitochondria are phagocytized and cleared by autophagosomes. Molecular chaperone-mediated autophagy: Molecular chaperone HSC70 particularly identifies and binds target proteins with KFERQ-like motifs, and transports these target proteins to lysosomes for degradation via the lysosomal membrane protein LAMP-2A. |
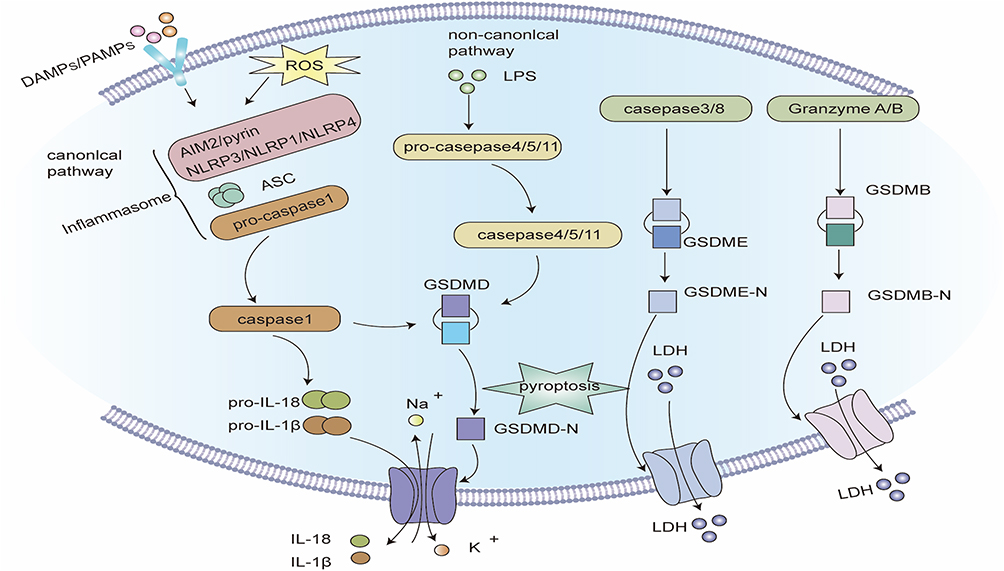 |
Figure 3 Signaling pathway of pyroptosis. Classic pathways of pyroptosis: Pathogen-linked molecular patterns (PAMPs) or damage-linked molecular patterns (DAMPs) activate the corresponding receptor proteins (NLRP3, NLRP1, NLRC4, AIM2 or Pyrin) and recruit ASC and pro-caspase-1 to form inflammasomes, and activate caspase-1. Caspase-1 hydrolyzes GSDMD to create GSDMD-C and GSDMD-N. GSDMD-N multimerizes and forms non-selective membrane pores on the cell membrane, elevating the permeability of the cell membrane, and resulting in cell swelling and rupture. Caspase-1 can also segment pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, and release them into the extracellular space through the above-mentioned membrane pores, inducing a strong inflammatory response. Non-canonical pathway of pyroptosis: Intracellular LPS directly binds and stimulates caspase-4/5/11. Activated caspase-4/5/11 can also cleave GSDMD, but cannot activate pro-IL-1β and pro-IL-18. In some cases, GSDME can also be hydrolyzed to GSDME-N by caspase-3/8, and the oligomerization of GSDME-N can also create plasma membrane pores. Notably, the hydrolysis of GSDMB by granzyme A and granzyme B can also form membrane pores, inducing pyroptosis. |
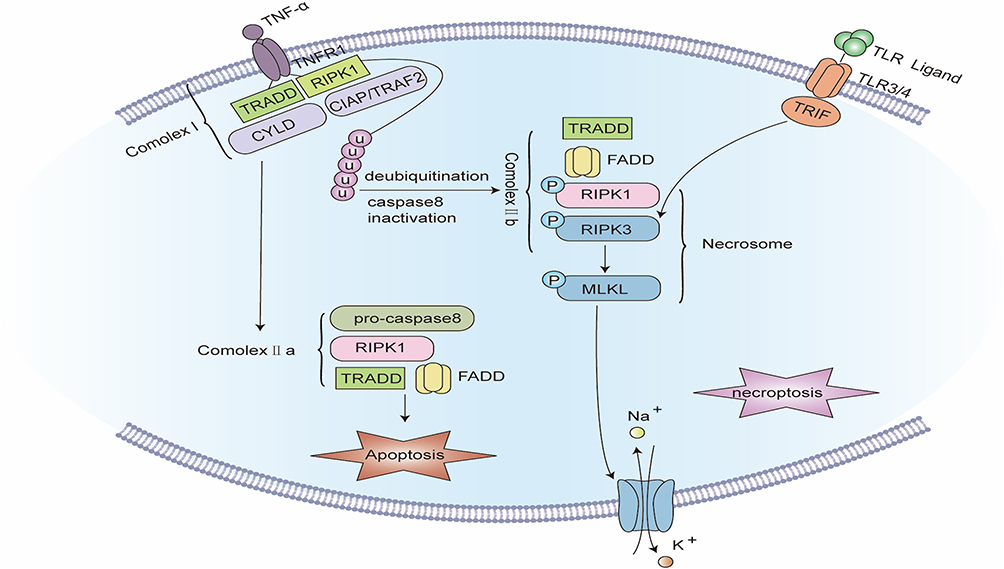 |
Figure 4 Signaling pathway of Ferroptosis. Extracellular Fe3+ is integrated with transferrin receptor 1 (TfR1) and then transported into the endosome, where it is lowered to Fe2+. Fe2+ is transported by divalent metal transporter-1 (divalent metal transporter 1, DMT1) and stored in the labile iron pool (LIP) in the cytoplasm. Nuclear receptor co-activator 4 (NCOA4) can transport ferritin to autophagosomes for lysosomal degradation and release Free iron. When iron metabolism is unbalanced, over-accumulated iron in cells will partake in lipid peroxidation via the Fenton reaction and iron-dependent enzymes such as lipoxygenases (LOXs). Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are major enzymes that control the percentage of polyunsaturated fatty acids (PUFA) in phospholipid membrane, which identifies the difficulty of lipid peroxidation. PUFA in membrane phospholipids is oxidized to phospholipid hydroperoxides (PLOOH) by the Fenton reaction or LOXs enzymatic reaction. GPX4 is a glutathione (GSH)-dependent enzyme that converts GSH into oxidized glutathione (GSSG) while reducing PLOOH to PL-OH, reducing oxidative stress, thereby negatively regulating ferroptosis. System Xc- consists of two subunits: the solute carrier family 3 member 2 (SLC3A2) and the solute carrier family 7 member 11 (SLC7A11). It is the common transport mode of cysteine, the main synthetic raw material of GSH. Inhibition of System Xc- will cause the depletion of GSH and the inactivation of GPX4, thus aggravating ferroptosis. |
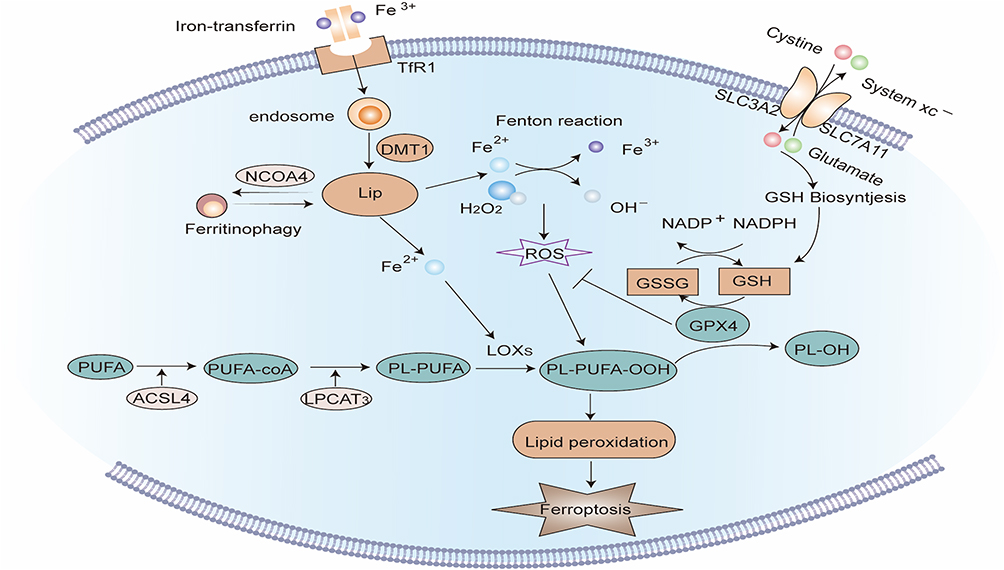 |
Figure 5 Signaling pathway of Necroptosis. The binding of TNF-α to tumor necrosis factor receptor 1 (TNFR1) promotes the assembly of complex I, including TNFR1, TNFR1-associated death domain (TRADD), receptor-interacting serine/threonine protein kinase 1 (RIPK1), TNFR-linked factor 2 (TRAF2), apoptosis protein (cIAP), and deubiquitinating enzymes such as CYLD. Complex I offers a platform for a series of subsequent ubiquitination and de-ubiquitination reactions, in which cIAP keeps RIPK1 ubiquitinated, while CYLD makes RIPK1 de-ubiquitinated. When caspase-8 is blocked, the ubiquitination of RIPK1 causes the formation of complex IIb. In complex IIb, RIPK1 is autophosphorylated, which subsequently recruits and stimulates RIPK3. PIPK3 further recruits and phosphorylates MLKL to create necrosomes. RIPK3 can also be independently stimulated by TIR-domain-containing adapter-inducing interferon-β (TRIF), an adapter protein of Toll-like receptors (TLRs). Eventually, phosphorylated MLKL oligomerizes and translocates to the cell membrane to develop plasma membrane pores, inducing plasma membrane rupture and cell disintegration. |
Cell death is deemed to be the final stage of cell physiology, has physiological and pathological functions, and functions more importantly in the organism onset, the maintenance of physiological homeostasis, and the development of diseases. In adults, 5~70 billion cells will die every day.6 Initially, it was presumed that there were only two different types of cell death: One is necrosis, which is regarded as a passive and accidental cell death with the release of a large number of inflammatory cytoplasmic contents to the extracellular space; The other is apoptosis, also known as programmed cell death (PCD), which is regarded as an active, programmed process of autonomous cellular dismantling without the release of cytoplasmic concentration to the extracellular space.7,8 In recent years, some other types of PCD were found and defined.9 As the study advanced, several studies demonstrated that some forms of PCD, including autophagy, pyroptosis, ferroptosis, and necroptosis, contributed to asthma pathogenesis, and may become new targets for future asthma treatment. In this review, we temporarily described the molecular mechanisms of these PCD forms and summarized and examined their key functions in the pathogenesis and treatment strategies of asthma, to offer some references for future research and PCD application in asthma (Table 1).
 |
Table 1 Comparison of Apoptosis, Autophagy, Pyroptosis, Ferroptosis, and Necroptosis |
Apoptosis
Apoptosis is the most thoroughly studied and described form of programmed cell death. In 1972, Kerr, Wyllie, and Currie introduced the concept of apoptosis and described the morphological characteristics of apoptosis, including cell shrinkage, cell membrane blebbing, nuclear fragmentation, and high chromatin condensation.10 The signal pathway of apoptosis can be classified into intrinsic and extrinsic pathways(Figure 1).
The intrinsic pathway of apoptosis also referred to as the mitochondrial pathway of apoptosis, is stimulated by the disruption of cellular homeostases such as oxidative stress, endoplasmic reticulum stress, or DNA damage. The intrinsic pathway of apoptosis is a method of cell suicide via mitochondrial outer membrane permeability (MOMP). MOMP causes the release of pro-apoptotic factors, including cytochrome c and the second mitochondria-derived activator of Caspases (SMAC), from the mitochondrial intermembrane space into the cytoplasm. Cytochrome C combines with apoptotic protease activating factor-1 (APAF-1) and pro caspase 9 to form apoptosomes. In the apoptosomes, caspase-9 activates itself and cleaves pro-caspase-3 and pro-caspase-7. The cleavaged caspase-3 and caspase-7 have the function of “executioner” of apoptosis, which ultimately induces apoptosis. Meanwhile, SMAC encourages apoptosis by obstructing the function of the inhibitor of apoptosis proteins (IAPs).11,12
Extrinsic apoptosis is controlled by several B-cell lymphoma 2 (BCL-2) family proteins, which can be categorized into three: (1) pro-apoptotic proteins, including Bcl-2-associated X protein (BAX) and Bcl-2 antagonist/killer 1 (BAK). When receiving apoptosis signals, they will be stimulated and oligomerized at the outer membrane of mitochondria leading to increased permeability of the mitochondrial membrane, which is regarded as a core step of apoptosis;13 (2) anti-apoptosis proteins, including Bcl-2, Bcl-xl, Bcl-w, and Mcl-1, which contributed to the stability of mitochondrial membrane; (3) BH3-only proteins, including BAD, BID, Bim, and Bik, can promote apoptosis by activating pro-apoptotic proteins or neutralizing anti-apoptotic proteins.9,12
The extrinsic pathway of apoptosis also called the death receptor pathway, is activated by the perturbation of the extracellular microenvironment and the signal transduction of the death receptor. Death receptors are some transmembrane proteins that can bind to extracellular specific ligands and transduce apoptotic signals into cells. Death receptors belong to the tumor necrosis factor receptor gene superfamily and have a cysteine-rich extracellular domain and an intracellular domain comprising homologous amino acid residues. This intracellular domain has a proteolytic function and is called the Death Domain (DD), which is a key structure in apoptosis signal transduction. Currently known death receptors include TNFR1, Fas, DR3, DR4, and DR5, of which FAS is the most renowned.14 When FAS and FAS ligands (FasL) are integrated, FAS will be activated by trimerization. FAS interacts with FAS-linked protein with death domain (FADD) through DD, which results in the conformational change of FADD. The conformational altered FADD recruits pro-caspase-8 via its death effector domain to form the Death-inducing signaling complex (DISC), which can initiate pro-caspase-8. Activated caspase-8 can directly activate caspase-3 and caspase-7 to induce apoptosis, or cleaves BID into tBID and triggers MOMP to mediate apoptosis.15,16
Although the provoking signals of the two apoptotic pathways vary, they both undergo a series of caspase activation. The finally activated apoptotic “executioner” caspase-3/7 cleaves the cell substrate and causes apoptosis. Apoptotic cells will discharge some “find me” signals (adenosine triphosphate, uridine triphosphate, etc.), and express some “eat me” molecules (phosphatidylserine, phosphatidylcholine, phosphatidylethanolamine, etc.) on the surface, which gathers phagocytes to engulf them before the loss of cytomembrane integrity.8 Therefore, the cytoplasmic and nuclear contents of apoptotic cells will not be discharged into the extracellular space.
Apoptosis in Asthma
It is well known that multiple cell types, including airway epithelial cells, eosinophils, neutrophils, and immune cells, contribute to chronic airway inflammation, mucus hypersecretion, and airway hyperresponsiveness in asthma. Next, we tried to separately describe the role of apoptosis of different cell types in the pathogenesis of asthma.
Airway epithelial cells comprise the physical barrier between the respiratory system and the external environment, which is at the forefront of defense against harmful inhaled substances and plays a crucial role in inflammatory responses, immune responses, and airway repair.17 The destruction of the airway epithelial barrier is a major issue in the pathogenesis of asthma.18 Injury and abscission of airway epithelium were found in bronchoscopic biopsy tissues of patients with moderate to severe asthma,19,20 which may be a result of excessive apoptosis of airway epithelial cells.21 Prior studies observed that clusters of apoptotic bronchial epithelial cells, called Creola bodies, linger in the induced sputum of asthmatic patients, particularly those with severe asthma.22,23 Apoptotic epithelial cells were discovered in bronchial biopsy specimens of children and adults with asthma, whereas there were no or few in normal controls.24–27 Through immunofluorescence staining, Jiangbo Wang et al discovered that the expression of caspase-3 in bronchial epithelial cells of asthma patients increased, and the increase was most evident in samples from patients with severe asthma.28 Increased apoptosis of airway epithelial cells was also found in asthma mice influenced by OVA,29 HDM,30 and the fungal allergen Alternaria alternata.31 Studies have shown that the increased apoptosis of epithelial cells is a crucial mechanism by which some external factors cause the worsening of asthma. For example, in vitro and in vivo experiments revealed that PM2.5 exposure influenced the severity of experimental asthma by stimulating more epithelial cell apoptosis.32,33 However, the extent of pathological airway epithelial cell apoptosis in asthmatic patients is still unknown, and its importance to the pathogenesis of asthma has not been comprehensively established.
Studies have determined that nearly 50% of patients with slight to moderate asthma have increased eosinophils in the respiratory tract, and the increase of eosinophil count in the peripheral blood is linked to the severity of the disease.34,35 Eosinophils accumulate in the lung tissue of asthma patients and contribute to inflammation and tissue damage by releasing granular cytotoxic substances.36 Unlike healthy individuals, the apoptosis of eosinophils in peripheral blood and lung tissues of asthmatic patients is prolonged,37,38 which was linked to the severity of asthma. Jennifer M Felton et al discovered that, unlike wild-type (WT) mice, mice overexpressing Mcl-1 revealed less eosinophil apoptosis and more severe airway allergic inflammation after OVA sensitization.39 Currently, there is no definitive outcome on the cause of delayed eosinophil apoptosis in asthma, which may be linked to cytokines that improve eosinophil survival rates, such as GM-CSF, IL-5, and IL-3, and may also be linked to thymic stromal lymphopoietin (TSLP) and IL-33 secreted by damaged airway epithelial cells.40 Enhanced adhesion of eosinophils themselves may also be one of the factors.41 Inducing eosinophil in apoptosis has become a treatment strategy for asthma. For example, glucocorticoids can accelerate eosinophil apoptosis, which is deemed to be one of its therapeutic mechanisms for asthma.42 Benralizumab is a humanized IgG1 monoclonal antibody. One the one hand, benralizumab can target the IL-5Rα and IL-5RβC subunits of IL-5R, which results in a sharp decrease in the number of eosinophils in the airway and blood. On the other hand, benralizumab can act on neutrophils, macrophages, and natural killer cells, leading to apoptosis of eosinophils by enhancing the effect of antibody-dependent cell-mediated cytotoxicity.43 In 2017, the FDA approved benralizumab for patients with severe eosinophilic asthma 12 years and older with steroid- resistance.
It was initially presumed that airway inflammation in asthma was primarily eosinophilic inflammation driven by TH2 immune response.44 However, with intensifying studies, it has been discovered that more than 50% of asthmatic patients had no evident eosinophil increase in their airways.45 According to the classification proportion of inflammatory cells in induced sputum, asthma can be classified into eosinophilic asthma, neutrophilic asthma, mixed asthma, and paucigranulocytic asthma.46 Among them, neutrophilic asthma is closely linked to severe asthma and refractory asthma, which has garnered the attention of clinicians and researchers.47 The study revealed that the apoptosis of neutrophils in the airways of patients with severe asthma was prolonged, and culturing blood-derived neutrophils in sputum sols from patients with severe asthma can prolong their apoptosis.48,49 Apoptotic-delayed neutrophils accrue in the airways and contribute to the presence and exacerbation of asthmatic airway inflammation.50
Toluene diisocyanate (TDI) is a commonly used industrial organic compound, which is a prevalent cause of occupational asthma.51 In TDI-induced asthma model mice, the expression defect of TLR4 causes impaired neutrophil apoptosis by up-regulating BCL-2, which exasperates the pathological changes of asthma.52,53 The apoptosis of neutrophils is controlled by multiple cytokines such as IL-6, IL-8, GM-CSF, G-CSF, and MCP-1, but the exact mechanism of delayed apoptosis in asthma remains unclear.54 Inducing neutrophils to apoptosis has become a new strategy for severe asthma treatment. For example, tamoxifen, a non-steroidal estrogen receptor modulator with an immune regulation role, can mitigate the pathological changes of asthma by inducing neutrophil apoptosis in the blood and lung tissue of the horse asthma model.55
The dysfunctional apoptosis of some other forms of cells also causes the pathogenesis of asthma. Research revealed that the number of activated T lymphocytes in the peripheral blood and airways of asthma patients was augmented.56 Unlike healthy controls and COPD patients, mononuclear cells in the airways of asthmatic patients have multiplied Bcl-2 expression, lowered Fas expression, and blocked apoptosis, which may be a critical reason for the accumulation of activated lymphocytes in the airways of asthmatic patients and the long-term persistence of asthmatic airway inflammation.57,58 Unlike TH1 lymphocytes, Th2 lymphocytes are more immune to activation-induced apoptosis, and increasing Th2 apoptosis can help to relieve allergic asthma.56,59
Therapeutic Strategies for Asthma Targeting Apoptosis
Apoptosis has both physiological and pathological effects on asthma. Furthermore, apoptosis of various cell types will indicate different impressions on asthma, which causes a few contradictory treatment effects of drugs regulating cell apoptosis. For example, glucocorticoids, while inducing apoptosis of eosinophils and lymphocytes to suppress inflammation, also augmented the apoptosis of airway epithelial cells, causing harm to the airway barrier.60 Given this reason, although there are several drugs targeting apoptosis, they are less employed in asthma treatment.61
Small molecule inhibitors of Bcl-2, ABT-737, and ABT-199, were found to be more effective in inducing eosinophils and neutrophils to apoptosis than glucocorticoids, may be a promising drug for asthma, particularly severe glucocorticoid-insensitive asthma.62 Bao Ping Tian et al synthesized a nano preparation of ABT-199, Nf-ABT-199, which can explicitly deliver ABT-199 to the mitochondria of airway inflammatory cells, has been confirmed in asthmatic mice that it can specifically and effectively induce eosinophil apoptosis, and considerably reducing airway inflammation.63
Some studies discovered that traditional Chinese medicine ingredients, such as Ephedrae Herba-Armeniacae Semen Amarum,64 Osthole,65 and Apigenin,66 were advantageous to asthma by inhibiting airway epithelial cells from apoptosis. Combined Extracts of Epimedii Folium and Ligustri Lucidi Fructus were determined to induce apoptosis in lung tissue and relieve airway remodeling in asthmatic rats.67
Autophagy
Autophagy, a process of cellular “self-eating”, allows for the degrading of redundant or dysfunctional cellular elements in eukaryotic cells, such as damaged organelles and misfolded proteins, to sustain cellular homeostasis.68 However, excessive or unbridled autophagy has been linked to various forms of caspase-independent cell death coined “autophagic cell death”.69 Here, this section mainly discusses autophagy as the core component of autophagic cell death in asthma.
The morphological feature of autophagy is the formation of a double-membrane autophagosome comprising cytoplasmic components or damaged organelles. Autophagy can be classified into three types: macro-autophagy, micro-autophagy, and chaperone-mediated autophagy, usually, macro-autophagy is called autophagy.70 The entire process of autophagy is tightly regulated by autophagy-related genes (ATG). More than 40 genes encoding Atg proteins have been observed in yeast, most of which are conservative between yeast and mammals, indicating that autophagy is an evolutionarily conserved degradation system(Figure 2).71
The autophagy process comprises several closely related steps, including Induction, assembly, and formation of autophagosome, the fusion of autophagosome and lysosomes, and degradation and recirculation of autophagosomal contents.72 Autophagy can be inducted by nutrition deficiency, organelle damage, protein folding error or aggregation, microbial infection, and other internal and external factors. When the mTORC1 complex is prevented or 5 ‘- AMP-activated protein kinase (AMPK) is activated, the Unc-51-like kinase 1 (ULK1) complex is activated and subcellular localized to the endoplasmic reticulum.73 The ULK1 complex phosphorylates and activates the Beclin-1-PI3KC3 complex, inducing the nucleation phase of autophagy. Then, the Beclin-1-PI3KC3 complex enhanced the activity of phosphatidylinositol 3-phosphate (PI3P) and triggered the formation of the autophagosome membrane. Furthermore, the combination of ATG5, ATG12, and ATG16L results in the extension of the autophagosome membrane. With the aid of ATG5, ATG12, ATG16L, ATG3, and ATG7, microtubule-associated protein 1 light chain 3 (LC3) was modified by cellular lipid phosphatidylethanolamine (PE) and transformed into LC3-II. The recruitment of LC3-II helps to further isolate ubiquitinated substances into advancing autophagosomes. Sealing of the autophagosomal membrane causes the double-membraned mature autophagosome, which is transferred to the perinuclear region and merged with lysosomes to establish autophagic lysosomes. The components of autophagosomes are degraded by lysosomal enzymes, and nutrients are recycled.74,75
Mitochondria are the energy factory of cells and play a significant role in cell physiological activities. It is crucial to clear damaged mitochondria timely and appropriate. The process of overtly degrading damaged mitochondria via autophagy is called mitochondrial autophagy.76 Mitochondrial autophagy can be classified into ubiquitin-mediated mitochondrial autophagy and receptor-mediated mitochondrial autophagy. The PINK1/Parkin pathway of ubiquitin-mediated mitochondrial autophagy is primarily introduced below. PINK1 (PTEN-induced putative kinase 1) is a protein kinase. Normally, PINK1 synthesized in the cytoplasm will enter the inner mitochondrial membrane and be cleaved by presenilins-associated rhomboid-like (PARL), then quickly degraded by the ubiquitin/proteasome system. When mitochondria are destroyed, the continuous depolarization of the inner mitochondrial membrane prevents PINK1 from entering the inner mitochondrial membrane, causing the buildup of PINK1 in the outer mitochondrial membrane. PINK1 is activated by autophosphorylation, and Parkin is moved to the surface of mitochondria. Parkin has E3 ubiquitin ligase activity, ubiquitinates numerous substrates at the outer mitochondrial membrane, and recruits mitochondria to the autophagy pathway after being identified by ubiquitin-binding proteins such as p62 so that ubiquitinated mitochondria are phagocytosed by autophagosomes.77,78
Unlike the preceding two autophagy modes, highly-selective chaperone-mediated autophagy does not depend on double-membrane vesicles. Instead, the molecular chaperone protein HSP70 explicitly recognizes and binds target proteins with KFERQ-like motifs, and induces their transport to lysosomes via the lysosomal membrane protein LAMP-2A.79
Autophagy in Asthma
Studies have demonstrated that the single nucleotide polymorphism of AGT5 is linked to childhood asthma, and the expression level of ATG5 mRNA in children with asthma is greater than that in normal children.80,81 In adults, ATG5 and ATG7 gene polymorphisms are linked to neutrophil counts in the induced sputum of asthmatic patients.82 In patients with refractory asthma, the expression level of ATG5 was positively related to the expression level of Collagen Type V Alpha 1 Chain (COL5A1) in the airway.83 Fan Yang et al determined 66 autophagy-related genes (ARGs) with differential expression levels between mild and severe asthma patients, and classified asthma into high autophagy subtypes and low autophagy subtypes on this premise. There were substantial differences between the two asthma subtypes in the level of symptom control and the reversibility of FEV1. The construction of an autophagy-related risk model may help to forecast the difficulty of asthma clinical control.84 Through electron microscopy, Audrey. H. Poon et al found double-membrane autophagosomes in fibroblasts and epithelial cells of bronchoscopic biopsy tissues from asthma patients, and few or none were found in normal controls.85 Tian Liu et al also got consistent results, and they discovered increased autophagosomes in the lung tissue of OVA asthma mice.86 The above genetic and histological evidence indicates that autophagy contributes to asthma pathogenesis.
Autophagy of airway epithelial cells is linked to airway mucus hypersecretion in asthma. In the airway epithelium of asthmatic patients and asthmatic mice, MTOR activity was decreased and autophagy level was augmented. Specific knockdown of MTOR in mouse bronchial epithelial cells improved allergen-induced airway inflammation and mucus hypersecretion, while knockdown of LC3B had a conflicting effect.87 IL-13 stimulated bronchial epithelial cells in vitro to increase the expression and secretion of intracellular mucin MUC5AC. After using small interfering RNA to reduce the expression of intracellular ATG5 or ATG14, several MUC5AC accumulated in the cells and could not be secreted outside the cells. After lowering the expression of ATG5 or ATG14 with small interfering RNA, numerous MUC5ACs accrued in the cells and could not be secreted. Unlike WT mice, ATG16L1-/- asthmatic mice had more and larger goblet cells in the airways but lowered MUC5AC content in bronchoalveolar lavage fluid (BALF).88 These outcomes indicate that autophagy is important for airway mucus secretion in asthma. However, J M Sweet et al89 found that during peak and resolution phases of mucous metaplasia, autophagy activity rather than secretion is required for the elimination of remaining mucin granules, which suggests that the interplay between autophagy and asthmatic epithelium is complex.
Airway remodeling is one of the major pathological features of asthma, which is linked to epithelial-mesenchymal transition (EMT) and airway fibrosis.90 EMT is a process in which epithelial cells gradually transform into mesenchymal-like cells and lose their epithelial functions and features, manifested by decreased expression of epithelial cell markers such as E-cadherin and increased expression of mesenchymal cell markers such as vimentin.91 Tian Liu et al discovered that inhibition of autophagy with the PI3K inhibitor LY-294002 or AGT5 si-RNA could lower FSTL1-induced EMT in bronchial epithelial cells, and LY-294002 could also lower EMT in the airways of asthmatic mice in vivo.86 TDI-stimulated EMT of bronchial epithelial cells was augmented by the autophagy activator rapamycin and mitigated by the autophagy inhibitor 3-methyladenine (3-MA).92 These outcomes indicate that autophagy contributes to EMT regulation in asthma.
Asthmatic airway fibrosis implies the excessive deposition of extracellular matrix proteins such as interstitial collagen, fibronectin, and proteoglycans in the subepithelial reticular layer, which are mostly controlled by fibroblasts and myofibroblasts.93 TGF-β1, a cytokine significantly increased in asthma, can boost the expression of fibrosis-related proteins by inducing autophagy in bronchial fibroblasts.94 The bioinformatics analysis demonstrated that, unlike healthy controls, mitochondrial autophagy genes were common in fibroblasts from patients with severe asthma, along with increased expression of PINK1, Parkin. Furthermore, the mitochondrial metabolic activity of fibroblasts from patients with severe asthma decreased considerably, implying that the mitochondria of fibroblasts from patients with severe asthma were damaged and autophagy levels increased.95 Rakhee K Ramakrishnan et al indicated that, unlike healthy controls, bronchial fibroblasts from severe asthma patients had more mitophagy, collagen, and fibronectin expression, and extracellular matrix deposition when activated with IL-17 in vitro. Furthermore, the blockade of autophagy with Bafilomycin-A1 attenuated these airway fibrotic changes, indicating that enhanced mitophagy in severe asthma is closely linked to airway fibrosis.96
Group 2 innate lymphoid cells (ILC2) can yield and secrete numerous type 2 cytokines, such as IL-5 and IL-13, which induce eosinophilic infiltration in the asthmatic airways.97 Lauriane Galle-Treger et al activated ILC2s from WT and Atg5 -/- mice with IL-33 in vitro, and the findings revealed that ILC2s from Atg5 -/- mice secreted less IL-5, IL-13, IL- 9, IL-6, and GM-CSF. In vivo, the increase of ILC2s in bone marrow, pulmonary inflammation, and airway hyperresponsiveness of Atg5 -/- asthmatic mice were not as prevalent as those of WT mice.98 In asthma, multiple activated B lymphocytes gather in the lung parenchyma and contribute to the pathogenesis of asthma by producing antigen-specific antibodies and processing and presenting antigens.99 One research demonstrated that the autophagy level of B lymphocytes in the lung tissue of asthmatic mice was increased, and the defective expression of AGT5 in B lymphocytes mitigated airway inflammation in asthmatic mice.100 Unlike healthy controls and patients having slight to moderate asthma, autophagy levels in peripheral blood eosinophils and neutrophils were both augmented in patients with severe asthma.101,102 Autophagy level was also augmented in eosinophils in the airways of asthmatic mice,103 and blocking the autophagy pathway can enhance airway inflammation in asthmatic mice.103,104 These pieces of evidence show that autophagy is closely linked to inflammation in asthma, and controlling autophagy may be a new target for asthma treatment.105
All the preceding studies indicate that enhanced autophagy can worsen asthma, but other studies have discovered that autophagy defect is also not tolerant to asthma. Yuzo Suzuki et al discovered that, unlike WT mice, the proportion of inflammatory cells in the BALF of Atg5 -/- asthmatic mice were substantially increased, and the peribronchial inflammation and tracheobronchial wall thickening were also even worse. The impaired autophagy pathway appeared to aggravate neutrophilic airway inflammation in asthma.106 Further in vitro experiments revealed that the dendritic cells of Atg5 -/- mice secreted more IL-1, IL-23, and IL-17, which led to more severe glucocorticoid-resistant neutrophilic asthma.106 Yuzo Suzuki et al indicated that the level of autophagy in the airway epithelial cells of obese asthmatic mice was not increased like that of non-obese mice, and obesity may hinder the autophagy mechanism of the airway epithelial cells, which caused more adverse airway inflammation and glucocorticoid resistance.107 Surprisingly, one research discovered that P62, a multifunctional adaptor protein that plays a crucial role in autophagy, was increased in the spleen cells of asthmatic mice while decreased in BALF neutrophils, indicating that autophagy is individually regulated in these two systems.108 Recent research found that Apelin-13, an innovative multifunctional protein, increased the pathway of mitophagy and induced mitochondrial homeostasis by activating PINK1/Parkin signaling in bronchial epithelial cells, thereby decreasing the oxidative damage of the airway in asthmatic mice.109 These outcomes reflect the complexity of autophagy in asthma, the impact of autophagy on asthma cannot be generalized, and more studies are required to evaluate the refined regulatory mechanisms.
Therapeutic Strategies for Asthma Targeting Autophagy
The typically used autophagy inhibitors are 3-MA, chloroquine, and bafilomycin A1. 3-methyladenine (3-MA), a selective inhibitor of PI3K, has been confirmed in many in vivo and in vitro studies that it can enhance airway inflammation, AHR, and airway remodeling of asthma by inhibiting autophagy.92,104,110,111 However, 3-MA is not appropriate for clinical treatment due to its toxicity (eg, induction of apoptosis).112 Bafloromycin A1 (BafA1) is a macrolide antibiotic that can hinder the fusion of autophagosomes and lysosomes.113 BafA1 can counteract IL-17-related fibrotic response in bronchial fibroblasts,94 and can also enhance glucocorticoid insensitivity of refractory asthma.114 Chloroquine and its derivative hydroxychloroquine can enhance the pH value of lysosomes and hamper the formation of autophagic lysosomes.115 In asthmatic mice, inhibition of autophagy by chloroquine can relieve airway inflammation, mucus hypersecretion, and airway remodeling.94 Both chloroquine and hydroxychloroquine are well-tolerated drugs, and hydroxychloroquine has obtained satisfactory outcomes in Phase II clinical trials in cancer.116 However, the pharmacological effects of chloroquine are very complex and vague, which restricts its use in asthma treatment. P140 peptide, a therapeutic peptide targeting chaperone-mediated autophagy, which is currently examined in Phase III clinical trial in lupus, can substantially lower the accumulation of eosinophils and lymphocytes in BALF in asthmatic mice, indicating the potential of molecular chaperone-mediated autophagy modulator in asthma.108 Several other autophagy inhibitors such as ULK-1 inhibitors SBI-0206965 and MRT68921 are under innovation in cancer and may be employed in asthma treatment in the future.117 Future studies should also evaluate the site-specificity of autophagy modulators, such as developing novel inhalers or nanoparticle-based cell-targeting approaches to prevent potential side effects from action-site nonspecificity.118
Rapamycin (RAPA), also called “sirolimus”, is a macrolide drug that initiates autophagy by inhibiting the activity of mTOR protein. It has been authorized by the US Food and Drug Administration (FDA) for the prevention of immune rejection after organ transplantation and the treatment of sporadic lymphangioleiomyomatosis.119 The function of RAPA in asthma is contentious. Fredriksson et al discovered that the simultaneous administration of RAPA during the development of the HDM asthma model can well inhibit airway inflammation, airway hyperresponsiveness, and goblet cell proliferation. However, when the HDM asthma model is effectively established, PAPA actually magnified these pathological reactions.120 Further studies discovered that RAPA can mitigate airway inflammation in asthmatic mice by limiting the ability of ILC2s to yield IL-5, regulating the balance of Th1/Th2 cytokines, and inhibiting eosinophil differentiation.121–123
Statins, such as simvastatin, can also relieve asthma by regulating autophagy. In asthmatic mice, simvastatin lowered airway remodeling by increasing autophagy levels in bronchial smooth muscle cells, lowering IL-4, IL-5, and IL-13 secretion, and reversing extracellular matrix deposition.124 Statins enhanced the anti-inflammatory effect of inhaled glucocorticoids by modulating autophagy and may become an innovative method for the treatment of glucocorticoid-resistant asthma.125 Research revealed that yeast fermentation prebiotics126 and vitamin D127 can relieve asthma by regulating autophagy.
Some traditional Chinese medicine ingredients, including Chrysophanol,128 Luteolin,129 Rhynchophylline,130 Vitexin,131 Cycloastragenol,132 Astragalin,133 Paeoniflorin,134 Sesamin,135 and some Chinese medicine mixtures, including Wuhu decoction,136 Qingfei oral liquid,137 Pingchuanning decoction,138 have been shown to relieve airway inflammation, AHR, and airway remodeling in asthmatic mice by regulating autophagy. Furthermore, acupuncture, as a widely used TCM treatment, was determined to treat asthma by inhibiting ATG5-mediated autophagy to regulate endoplasmic reticulum stress and CD4+ T lymphocyte differentiation.139
Pyroptosis
Pyroptosis is an inflammatory PCD described by Cookson and Brennan 2001.140 Pyroptosis is the formation of pores in the cell membrane, and the cells continue to swell until rupture, resulting in the release of inflammatory factors into the extracellular space, which causes a strong inflammatory response.141 The signaling pathways of pyroptosis mostly include the classical pathway and the non-classical pathway (Figure 3).
The classical pyrolytic pathway is mediated by the assembly of inflammasomes and depends on Caspase-1. Most inflammasomes comprise three components: (1) Receptor proteins, such as NLRP1, NLRP3, NLRC4, AIM2, and pyrin, of which NLRP3 is the most researched. (2) The adaptor protein ASC, contains CARD. CARD is essential for the recruitment of pro-caspase-1. Some pattern recognition receptors, such as PRR, also have CARD and can directly recruit pro-caspase-1.142 (3) The effector protein pro-caspase-1.143 After the inflammasome is formed, pro-caspase-1 is hydrolyzed to mature cleaved caspase-1. On the one hand, caspase-1 can cleave GSDMD to create GSDMD-C and GSDMD-N. Multiple GSDMD-Ns oligomerize and transfer to the cell membrane, creating non-selective pores with an inner diameter of about 10–14 nm, leading to cell swelling and rupture. On the other hand, caspase-1 also cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, which are released to the extracellular space via the membrane pores mentioned above, which induces a strong inflammatory response.12,143,144
The non-classical pyroptotic pathway does not rely on caspase-1 but on caspase-4/5/11. Caspase-4/5 (the mouse orthologous caspase-11) can be directly stimulated by LPS in the cytoplasm. Activated caspase-4/5/11 can cleave GSDMD, but not pro-IL-1β and pro-IL-18. The membrane pores comprising GSDMD-Ns cause the efflux of intracellular potassium ions, which in turn activates the NLRP3 inflammasome and the classical pathway of pyroptosis, and ultimately results in the cleavage and release of pro-IL-1β and pro-IL-18.34,143
Furthermore, GSDME may also be cleaved to GSDME-N by caspase-3/8 when cells are stimulated by a viral infection, chemotherapeutic drugs, and tumor necrosis factor (TNF). Notably, GSDME-Ns can also assemble into cytoplasmic membrane pores and induce pyroptosis.145,146 In recent years, studies have discovered that granzyme A and granzyme B can also hydrolyze GSDMB to create cytoplasmic membrane pores, thus proposing a granzyme-mediated pathway of pyroptosis.147,148
Pyroptosis in Asthma
Developing researches indicate that pyroptosis is linked to the pathogenesis of asthma. Queiroz, G.A. et al discovered that SNPs in NLRP3 and caspase-1 were linked to susceptibility to asthma, and the higher their risk alleles, the higher the risk of asthma.149 Unlike the healthy control, the levels of NLRP3 and caspase-1 in BALF supernatant of asthma patients were greatly increased,150 and the expressions of NLRP3 and IL-18 in airway epithelial cells of lung biopsy tissues were also greatly increased.151 There was also a substantial variation between the levels of pyroptosis in airway epithelial cells between mild-moderate and severe asthma patients.152 In the lung tissue of asthmatic mice caused by OVA alone or OVA in combination with adjuvant aluminum hydroxide, the expression levels of NLRP3, caspase-1, and IL-1β were greatly increased. Furthermore, NLRP3-/-, ASC-/-, and IL-1R-/- mice had more slight pathological changes in AHR, airway mucus secretion, and airway inflammation than WT mice.153,154 Consistent findings were also found in the asthma murine models induced by HDM, Der f1, and TDI.155–157
GSDMB is highly demonstrated in well-differentiated airway epithelial cells, and studies discovered that exon-coding variants of the GSDMB gene were linked to a lowered risk of asthma. For instance, a splice variant in GSDMB (rs11078928) skipped a key exon from the transcript, inhibiting the pyroptotic activity of GSDMB, thereby reducing the disease risk of asthma.158 Xingnan Li et al indicated that SNPs of GSDMB were linked to asthma severity and exacerbation frequency.159 Other researchers discovered that GSDMB expression was up-regulated in the airway epithelial cells of asthma patients,160 and asthmatic mice overexpressing GSDMB showed more severe airway hyperresponsiveness and airway remodeling.159 These outcomes indicate that GSDMB-mediated pyroptosis is one of the molecular mechanisms influencing asthma susceptibility and severity.
Among the multiple phenotypes of asthma, pyroptosis appears to be more markedly linked to severe, corticosteroid-resistant, neutrophilic asthma. Unlike patients with mild to moderate asthma, the levels of NLRP3 activators such as C5a in the induced sputum of patients with serious neutrophilic asthma were augmented,161 and the expression of NLRP3, caspase-1, caspase-4, and caspase-5 in induced sputum cells were increased.162 Furthermore, there was a connection between the level of pyroptosis and airway inflammation, asthma symptom control, and lung function in neutrophilic asthma.163 Some studies demonstrated that there were numerous activated neutrophils in the airways of patients with severe asthma, and the extracellular traps of neutrophils and their products can stimulate the pyroptosis pathway, which may be a crucial reason why these patients are not sensitive to glucocorticoid treatment and their symptoms are hard to control.164,165
It was discovered that, unlike the control group, the protein expressions of NLRP3, caspase-1, IL-1β, and IL-18 in the airway tissue of the neutrophilic asthma group were markedly increased. Furthermore, highly-selective NLRP3 inhibitor MCC950 and specific caspase-1 inhibitor Ac-YVAD-cmk substantially lowered AHR and airway inflammation in asthmatic mice.150,166 Hern-Tze Tina Tan et al built three different phenotypes (eosinophilic, mixed, and neutrophilic) in asthma mouse models through several sensitization and challenge methods. The whole-transcriptome sequencing of the lung determined that Pyroptosis-related genes such as Nlrp3, Nlrc4, Casp-1, and IL-1β were common in neutrophilic asthma.167 Furthermore, Richard Y. Kim et al showed that in the process of eosinophilic asthma turning into glucocorticoid-resistant neutrophilic asthma induced by chlamydia and Haemophilus infection, the pyroptotic pathway in mouse lung tissue was markedly activated, and targeted blockade of pyroptotic signaling with MCC950, z-VAD-fmk, Ac-YVAD-cho can hinder this transition process.163 These outcomes indicate that pyroptosis may advance the refractory, glucocorticoid-resistant, neutrophilic asthma and may be a possible therapeutic target for these asthma subtypes. These outcomes indicate that pyroptosis may advance the refractory, glucocorticoid-resistant, neutrophilic asthma and may be a possible therapeutic target for these asthma subtypes.
Obese-asthma is a unique asthma phenotype that responds minimally to traditional treatment and is predisposed to severe refractory asthma.168 Unlike non-obese patients, obese asthmatic patients had elevated levels of NLRP3 and IL-1β in induced sputum and were positively linked to body mass index.169,170 In animal experiments, obesity brought on by a high-fat diet aggravates lung lesions in asthmatic mice, and prevention of NLRP3 or blockade of IL-1β decreased airway allergic inflammation and glucocorticoids resistance in obese mice,170,171 which indicates that pyroptosis may be a potential mechanism of obesity-induced asthma exasperation. Glucagon-like peptide 1 receptor agonists can lower airway inflammation in obese asthmatic mice by preventing NLRP3 inflammasome activation and have been recommended as a novel drug for obese-asthma.172
Virus infection is a significant cause of asthma worsening, and cell death contributes to virus infection causing asthma exacerbation. One study revealed that virus infection of bronchial epithelial cells in vitro induced NLRP3 and NLRC5 inflammasome activation.173 Furthermore, bronchial epithelial cells from asthmatic patients showed higher levels of pyroptosis following influenza virus infection than healthy controls.174 Viral infection also increases macrophage pyroptosis in the lung tissue of asthmatic patients, thus worsening asthma.175 Unlike WT asthmatic mice, NLRP3-/- asthmatic mice did not have asthma exacerbation after human rhinovirus infection, indicating that inhibition of pyroptosis can hinder virus-induced asthma exacerbations.176 Pyroptosis is also linked to severe illness after SARS-CoV-2 infection in patients with severe asthma. Jeong, J.S.et al discovered that pyroptosis was also linked to critical illness after SARS-CoV-2 infection in patients with severe asthma. Unlike mild asthmatic mice, the protein expressions of NLRP3, ASC, caspase-1, and IL-1β in the lungs of severe asthmatic mice caused by Aspergillus fumigatus were substantially increased after SARS CoV-2 infection. It is presumed that pre-existing severe asthma may elevate pyroptosis induced by SARS-CoV-2 infection, and targeting pyroptosis may be a promising technique for the early treatment of COVID-19 in severe asthma patients.177
Silica nanoparticles, an extensively used chemical material and a critical element of air pollution in Asia can damage the respiratory system of healthy people and also exasperate the clinical symptoms of patients with respiratory diseases.178 SiONPs nasal drip dose-dependently augmented the activation of NLRP3 in the lung tissue of asthmatic mice, followed by the worsening of asthma pathological changes,179 implying that pyroptosis also contributed to the aggravation of asthma caused by SiONPs. Studies demonstrated that the polymorphism of human NLRP3 was linked to the susceptibility to aspirin-related asthma.180 Prostaglandin E2 (PGE2) is an endogenous inhibitor of caspase-11, which can efficiently inhibit pyroptosis in the lung tissue of asthmatic mice. Zbigniew Zasłona et al discovered that indomethacin lowered PGE2 production and increased pyroptosis and inflammation in the lung tissue of asthmatic mice, which appears to explain nonsteroidal antiinflammatory drugs-induced asthma exacerbations, but it should be further validated.181
The preceding studies all indicate that pyroptosis is not conducive to the onset of asthma, but some studies have acquired inconsistent outcomes. In the analysis of Irving C. Allen et al, there was no considerable difference in airway inflammation, airway mucus secretion, and AHR between WT and Nlrp3-/- asthmatic mice. The authors recommended that some modifications in the gut microbiome of Nlrp3-/- mice affected the immune response to external allergens, thus masking the effect of pyroptosis on asthma.182,183 Fahima Madouri et al discovered that activation of NLRP3 and caspase-1 attenuated HDM-induced allergic airway inflammation. The authors presumed that this was when the caspase-1 expression was deficient, macrophages generated and released large amounts of IL-33, thereby indirectly worsening allergic asthma.184 That is to say, impaired pyroptosis mechanism is not the direct cause of asthma exacerbation. However, the function of pyroptosis in asthma remains unknown.
Therapeutic Strategies for Asthma Targeting Pyroptosis
There are several small molecule inhibitors targeting NLRP3 for the treatment of inflammatory diseases. So far, MCC950, OLT1177, and RRx-001 have been researched in asthma.185 MCC950 can directly and specifically bind to the NACHT domain of NLRP3, inhibiting the assembly of inflammasomes.186 In asthmatic mice, MCC950 reduced airway inflammation and mucus hypersecretion and lowered serum IgE and cytokine levels.187,188 Comparable results have been found in mouse models of bacterial infection mimicking neutrophilic asthma,163 fungal asthma,187 and TDI occupational asthma.189 However, the clinical trial of MCC950 has been discontinued because MCC950 had inflammatory hepatotoxicity in the phase II clinical trial in rheumatoid arthritis.190
OLT1177 can efficiently reduce allergic airway inflammation in experimental asthma mice.191 OLT1177 has demonstrated a favorable safety profile in preclinical toxicology studies and is currently in Phase IIa clinical trials for the treatment of gout.192 RRx-001 binds covalently to cysteine 409 of NLRP3 via its bromoacetyl group, inhibiting the NLRP3-NEK7 interaction, which is crucial for the assembly and activation of the NLRP3 inflammasome.193 Currently, RRx-001 is in phase III clinical trials for lung cancer and has a good curative effect in asthmatic mice, and is believed to be an innovative drug for asthma treatment.155 Some novel NLRP3 inhibitors such as dapansutra and IZD-174 have also entered clinical trials, but have not been researched in asthma.194
VX-765 is an effective and nontoxic small-molecule inhibitor of caspase-1 that has been demonstrated to be safe in humans.195 VX-765 attenuated airway inflammation and airway hyperresponsiveness in asthmatic mice,166 and also enhanced the response to glucocorticoid therapy in refractory neutrophilic asthma.163 Ac-YVAD-CHO is also a potent and specific caspase-1 inhibitor, which can efficiently reduce AHR, airway inflammation, and airway remodeling in TDI asthmatic mice.189 Other inhibitors of caspase-1, such as CrmA, z-VAD-fmk, Ac-FLTD-CMK, etc., have not been studied in asthma. GSDMD is the executor of cell death, some recognized GSDMD inhibitors, such as necrotizing sulfonamide, dimethyl fumarate, disulfiram, and Ac-FLTD-CMK, have revealed therapeutic effects in some inflammatory diseases, but there is no substantial study in asthma.196
Considering the excessive release of IL-1β in pyroptosis, blocking IL-1β may be an innovative strategy for the treatment of asthma. Canakinumab, a humanized anti-IL-1β monoclonal antibody, was found to safely and effectively attenuate the response to inhaled allergens in asthmatic patients in a randomized controlled clinical trial.197 However, because the role of IL-1β is complex, in addition to being a product of pyroptosis, it is also a very important inflammatory factor, which plays many other functions such as anti-infection and induction of immune differentiation in asthma.
So far, canakinumab is no longer being studied as an asthma treatment because of its limited efficacy in asthmatic patients.198
Some traditional Chinese medicine ingredients and formulations, including Oridonin,199 parthenolide,200 curcumin,201 Schisandrin B,202 Abscisic acid,203 Yanghe Pingchuan Granules,204 Yupingfeng San,205 and Suhuang antitussive capsule206 were also shown to have a protective effect on asthma by controlling the pyroptosis pathway.
Ferroptosis
Ferroptosis, first suggested by Dr. Brent R. Stockwell in 2012, is an iron-dependent PCD brought on by lipid peroxidation. Unlike other forms of PCD, there is no cell swelling, plasma membrane rupture, chromatin condensation, and nuclear fragmentation during ferroptosis, and its major manifestations are increased mitochondrial density, outer membrane rupture, and reduced or even disappearance of mitochondrial cristae (Figure 4).207
The abnormal accumulation of iron ions in the cytoplasm is considered the trigger of ferroptosis. Extracellular Fe3+ integrates with the transferrin receptor (TfR) on the cell membrane and enters the endosomes of the cell. Then Fe3+ is reduced to Fe2+ by ferredoxin. Under the mediation of divalent metal transporter 1 (DMT1), Fe2+ is released from endosomes into the cytoplasmic labile iron pool (LIP). Nuclear receptor co-activator 4 (NCOA4) is a unique transport receptor for ferritinophagy, which can transport ferritin to autophagosomes and deliver it to lysosomes for degradation to release free iron.208,209 The dysfunction of these iron-related proteins will cause an imbalance of intracellular iron concentration. When Fe2+ extremely accumulates in LIP, it will contribute to the process of lipid peroxidation via the Fenton reaction and iron-dependent oxidase such as lipoxygenase (LOX).210
Lipid peroxidation is a central step in ferroptosis. Because polyunsaturated fatty acids comprise more double bonds, they are more easily oxidized, so the percentage of polyunsaturated fatty acids in the plasma membrane determines the difficulty of lipid peroxidation. Acyl-CoA synthase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are core enzymes that control the synthesis of polyunsaturated fatty acids in phospholipid membranes.211 There are two pathways for cellular lipid peroxidation, namely non-enzymatic lipid peroxidation and enzymatic lipid peroxidation. In non-enzymatic lipid peroxidation, polyunsaturated fatty acids are oxidized to lipid hydroperoxides by hydroxyl radicals formed by the Fenton reaction. In enzymatic lipid peroxidation, LOX directly catalyzes the oxidation of free polyunsaturated fatty acids to lipid hydroperoxides. The lipid hydroperoxides produced by both pathways can yield lipid peroxyl radicals under the catalysis of iron ions, and then conduct the chain reaction of lipid peroxidation.212 Currently, the exact mechanism by which lipid peroxidation results in ferroptosis remains unclear. It may be linked to the modifications in cell membrane fluidity and permeability due to diminished polyunsaturated fatty acids, structural lipid gap formation, and loss of membrane integrity.213
Glutathione (glutathione, GSH) and glutathione peroxidase (Glutathione Peroxidase 4, GPX4) are major regulators of ferroptosis. GPX4 is a GSH-dependent enzyme. While converting GSH to oxidized glutathione (GSSG), GSH lowers lipid hydroperoxides to the corresponding lipid alcohols or free hydrogen peroxide to water, thereby negatively controlling ferroptosis.214–216 Cysteine is a crucial raw material for GSH synthesis and is typically transported into cells through System Xc- on the cell membrane.217 System Xc - comprises two subunits, namely, the solute carrier family 3 Member 2 (SLC3A2) and the solute carrier family 7 Member 11 (SLC7A11). When System Xc- is damaged, the synthesis of GSH is hindered, and the intracellular antioxidant capacity is decreased, which will worsen lipid peroxidation and ferroptosis.215,218
Ferroptosis in Asthma
Currently, little is known regarding the role of ferroptosis in asthma. Studies have demonstrated that there is abnormal iron metabolism in the airway cells of asthmatic patients. Unlike healthy controls, free iron was mitigated in BALF supernatants of asthmatic patients, conversely, the iron load was augmented in BALF cells. The expression of DMT1 and Transferrin Receptor 1 (TFR1) in the lung tissue of asthma patients was also augmented. Furthermore, these iron metabolism abnormalities are linked to the lung function decline and airway inflammation of asthma patients. Consistent results were found in asthmatic mice, indicating that abnormal iron metabolism in lung tissue contributes to the pathogenesis of asthma.219 Ferritin phagocytosis is a selective autophagy that depends on nuclear receptor coactivator 4 (NCOA4) to transport ferritin to autophagosomes for degradation, promoting the generation of unstable iron ions.220 In a murine asthmatic model, HDM-induced ferritin phagocytosis in lung tissue generated large amounts of free iron, which caused ferroptosis in airway epithelial cells, and contributed to the airway inflammation in asthma.221
The phosphatidylethanolamine-binding protein 1 (PEBP1)/15-lipoxygenase (15-LO) complex is a major regulator of lipid peroxidation. Sally E. Wenzel et al demonstrated higher co-localized levels of PEBP1 and 15-LO in the airway epithelial cells of asthmatics than in healthy controls.222 Unlike healthy controls, GSSG levels were increased and GSH: GSSG lowered in the BALF supernatants of patients with severe asthma, implying that the redox balance in the lung tissue of patients with asthma was modified, and this change was linked to 12/15-lipoxygenase and airway inflammation. IL-13 stimulated bronchial epithelial cells in vitro to activate 15 lipoxygenase-1 to produce hydroperoxyphospholipids, reducing intracellular GSH and increasing extracellular GSSG, and increasing the susceptibility of bronchial epithelial cells to iron death. Based on this, prevention of SLC7A11 further decreased GSH and promoted ferroptosis, meanwhile, increasing TH2 inflammatory factors expression.223 In vitro stimulation of bronchial epithelial cells by IL-6 disrupted intracellular iron homeostasis, causing lipid peroxidation and ferroptosis. Furthermore, pretreatment with the ferroptosis inhibitor Ferrostatin-1 (Fer-1) and the antioxidant N-Acetyl-L-cysteine (NAC) relieved these changes.224 In animal experiments, unlike the control group, the expressions of ACSL4 and 15-LO1 in the lung tissue of asthmatic mice were up-regulated, the activity of GPX4 was lowered, and the morphological changes of mitochondria were found under the electron microscope, implying that the ferroptosis pathway was activated in experimental asthma. Inhibition of ferroptosis with deferoxamine mesylate (DFO), Fer-1, and 3-methyladenine (3-MA) attenuated iron accumulation and lipid peroxidation in lung tissue of asthmatic mice, improving airway inflammation in asthmatic mice.225,226 Furthermore, 12/15-LO-deficient asthmatic mice had reduced serum IgE antibody levels and less airway inflammation than WT mice.227
Ferroptosis is also critical in neutrophilic asthma. Simultaneous stimulation of bronchial epithelial cells by LPS and IL-13 reduced the expression of GPX4 and SLC7A11 in vitro, and the reduction of intracellular mitochondrial cristae, outer membrane rupture, mitochondrial vacuolar degeneration, and swelling were found under the electron microscope, implying that bronchial epithelial cells had undergone ferroptosis. In the lung tissue of OVA/LPS-induced neutrophilic asthmatic mice, the expressions of SLC7A11 and GPX4 were lowered, and the ferroptosis inhibitor Liproxstatin-1 relieved the pathological changes of asthma.228 These outcomes indicate that ferroptosis contributes to the pathogenesis of asthma, and inhibition of ferroptosis may be an innovative strategy for asthma treatment.
Therapeutic Strategies for Asthma Targeting Ferroptosis
Currently prevalently used ferroptosis inhibitors are deferoxamine (DFO), Liproxstatin-1, Ferrostatin-1, 3-methyladenine (3-MA), and N-acetyl -L-cysteine (N-Acetyl-L-cysteine, NAC). The mechanisms by which they inhibit ferroptosis vary: DFO is an iron chelator that can bind to excess iron ions and mitigate the accumulation of iron ions in cells;229 Ferrostatin-1 is a synthetic antioxidant that can inhibit lipid peroxidation through a reducing mechanism;230 Liproxstatin-1 inhibits lipid peroxidation by lowering the accumulation of ROS;231 NAC is a sulfhydryl-containing antioxidant that helps increase intracellular cysteine levels; Although 3-methyladenine is an autophagy inhibitor, it can also prevent ferroptosis by regulating system Xc- and activating GPX4.232 Till this moment, the therapeutic effects of DFO, Fer-1, Liproxstatin-1, and 3-MA on asthma by inhibiting ferroptosis have been confirmed in in vitro cell experiments and asthma mouse models,222,224,226,228 NAC has only been researched in vitro.224
Surprisingly, the ferroptosis inducers FINs also improved allergic airway inflammation. One study demonstrated FIN-induced ferroptosis in eosinophils in vitro. In vivo, FIN alone or in combination with dexamethasone-induced eosinophil ferroptosis in the airways of asthmatic mice, thus reducing allergic airway inflammation.233
A recent analysis indicated that quercetin, a conventional Chinese medicine, could mitigate ferroptosis-associated neutrophil airway inflammation in asthmatic mice.234 Furthermore, the mechanism of acupuncture to relieve asthma may also involve the regulation of ferroptosis. The expression of SLC3A2 is elevated in the lung tissue of asthmatic mice, and acupuncture can down-regulate its expression, indicating that acupuncture may regulate SLC3A2-related ferroptosis.235 Weifeng Tang et al also discovered that acupuncture can reduce lipid peroxidation and ferroptosis in the lung tissue of asthmatic mice, causing reduced airway inflammation in asthma.236 However, the theory that acupuncture has a regulating effect on ferroptosis still requires more rigorous experiments to be validated. Mesenchymal stem cell treatment is an innovative approach to asthma that has been demonstrated to be effective in several studies.237 Studies have shown that increasing intracellular GSH levels can improve the function of mesenchymal stem cells and lead to better therapeutic effects.238 Ruth Lee Kim et al discovered that Liproxstatin-1-pretreated mesenchymal stem cells had a stronger ability to inhibit inflammation. In IL-13-induced chronic asthmatic mice, Liproxstatin-1-treated human umbilical cord blood mesenchymal stem cells also revealed better asthma therapeutic effects. It is indicated that inhibiting ferroptosis may be a modification method for mesenchymal cell therapy.239
So far, the analysis of ferroptosis in asthma is still far from inadequate. Although the effect of ferroptosis inhibitors in asthma has been confirmed in animal experiments, there is no major research on clinical translation. In the future, researchers should evaluate the mechanism of ferroptosis in asthma comprehensively and search for ferroptosis-targeting drugs that can be employed clinically.240
Necroptosis
Necroptosis, suggested by Degterev et al in 2005,241 is identified as a caspase-independent PCD with morphological features comparable to necrosis but a regulated death process. The triggers of necroptosis are comparable to those of apoptosis, while the morphological changes are cell swelling, plasma membrane rupture, chromosome condensation, and release of damage-associated molecular patterns (DAMPs), inflammatory cytokines, and chemotaxis Factor.11,242,243 Necroptosis is stimulated when specific death receptors such as TNFR1, FAS, PRRs, and TLR3 identify extracellular and extracellular danger signals, and caspase-8, which mediates apoptosis, is inhibited (Figure 5).244
In response to TNFα binding, the intracellular segment of TNFR1 recruits various proteins to create a signaling complex called complex I. In complex I, a cellular inhibitor of apoptosis proteins (cIAPs) keep Receptor Interacting Serine/Threonine Kinase 1 (RIPK1) ubiquitinated,245 while CYLD deubiquitinates RIPK1 and converts complex I to complex II. In complex II, FADD recruits procaspase-8, while RIPK1 recruits RIPK3. When RIPK3 is absent or at a low level, caspase-8 is activated and causes apoptosis. In RIPK3-rich cells with caspase-8 inhibited, RIPK1 recruits RIPK3 and mixed lineage kinase domain-like protein (MLKL) to create necrosomes. The kinase activity of RIPK3 is a required condition for necroptosis, aside from being activated by RIPK1 phosphorylation, it can also be directly activated by TIR-domain-containing adapter-inducing interferon-β (TRIF), an adapter protein of Toll-like receptors (TLRs). Activated RIPK3 phosphorylates MLKL, which oligomerizes and moves to the cell membrane to form plasma membrane pores, which encourage the inflow of extracellular calcium ions and the outflow of intracellular potassium ions, increase the permeability of the cell membrane to water, and eventually results in cell swelling and disintegration. Cellular elements such as DAMP, HMGB1, mitochondrial DNA, uric acid, histones, and ATP are also simultaneously released into the extracellular space via the plasma membrane pores, inducing an inflammatory response.243,246,247
Necroptosis in Asthma
Airway eosinophil infiltration is a hallmark of TH2 asthma, and particulate matter released after the lysis of adherent eosinophils promotes airway epithelial damage, which is closely linked to asthma severity.248 Radonjic-Hoesl et al discovered that MLKL was phosphorylated in adherent eosinophils under inflammatory conditions in vivo and in vitro, and pharmacological inhibition of RIPK3 and MLKL blocked eosinophil lysis,249 which indicated that eosinophil lysis in asthmatic airways may be RIPK3-MLKL-dependent necroptosis. In recent research, immunofluorescence staining demonstrated that the expression of p-MLKL in lung biopsies from asthmatic patients was considerably higher than that in healthy individuals, and comparable results were found in asthmatic mice. It is presumed that necroptosis contributes to the disease process of asthma. Furthermore, p-MLKL mainly emerged in eosinophils rather than neutrophils.250 Unlike neutrophils, eosinophils had lower levels of caspase-8 and higher levels of RIPK1, which could be a reason why eosinophils are more vulnerable to necroptosis. However, research revealed that neutrophils in the airways of neutrophilic asthma mice also had necroptosis and contributed to the formation of neutrophil extracellular traps, exasperating airway inflammation in asthmatic mice.251
IL-33 is a pro-inflammatory cytokine that can trigger TH2 immune responses by activating immune cells and contributes to the formation of eosinophilic airway inflammation in asthma.252–254 Due to the absence of secretory signal sequences, the mechanism of IL-33 release is a mystery. Inbar Shlomovitz et al discovered that the MLKL inhibitor GW806742X can block the secretion of IL-33 by bronchial epithelial cells in vitro and in vivo, and lower airway eosinophil inflammation in asthmatic mice, suggesting that necrotic apoptosis may be a mechanism for the release of IL-33 from epithelial cells.255
In FADD -/- mice, inhibition of RIPK1 significantly enhanced HDM-induced allergic airway inflammation. However, in WT mice with normal expression of FADD, inhibition of necrotic apoptosis cannot promote airway inflammation in asthmatic mice,256 which indicates that necroptosis is a backup mechanism for apoptosis, which functions more when apoptosis is inhibited. This situation is better explained by viral infection. In the process of trying to evade host defenses and multiply in cells, viruses have aided some mechanisms to interfere with apoptosis, such as expressing some caspase inhibitors.257 At this time, necroptosis becomes a critical antiviral defense mechanism.258 Replication of Rhinovirus in infected cells activates the synthesis and release of interferon β (IFN-β), thereby causing peripheral cells to go into an antiviral state.259 Bao, Cet al discovered that bronchial epithelial cells from asthmatic patients could not produce IFN-β upon rhinovirus infection and dsRNA stimulation, and could not fully utilize their antiviral function.260 When using dsRNA to imitate viral infection, the expression of RIPK3 and p-MLKL was elevated in the lung tissue of WT asthmatic mice, and even more in IFN-β-/- mice. It is presumed that necroptosis contributes to the antiviral response in asthmatic states, however, the release of inflammatory factors during necroptosis will worsen asthma.261
Bronchiolitis from Respiratory syncytial virus (RSV) infection in infants is an identified risk factor for asthma.262 A study discovered that RSV infection-induced bronchial epithelial cell death was linked to increased expression of RIPK1 and MLKL, but not with caspase-3 activity. Inhibition of RIPK1 or MLKL reduced HMGB1 release from bronchial epithelial cells upon RSV infection. In animal experiments, RIPK1 and MLKL expression was elevated in the airway epithelium of mice after pneumovirus infection, and this was linked to airway epithelial shedding, HMGB1 release, and airway inflammation. Genetic or pharmacological inhibition of RIPK1 or MLKL considerably attenuated these pathological changes and inhibited subsequent airway remodeling and TH2 immune responses. It is believed that inhibition of necroptosis may be a viable method to limit the severity of viral bronchiolitis and prevent its advancement to asthma.263
PM2.5 exposure exasperates asthma and augments the risk of asthma exacerbations.264,265 Yunxia Zhao et al discovered that PM 2.5 exposure elevated the expression of RIPK3 and MLKL in mouse lung tissue, and blocking necroptosis lowered PM2.5-induced airway inflammation and airway hyperresponsiveness.266 TNF-α is a proinflammatory cytokine closely linked to neutrophilic asthma and could be beneficial in refractory asthma.267 TNF-α activates necroptosis of bronchial epithelial cells in vitro while up-regulating the expression of Mucin 1 (MUC1). Decreasing the expression of MUC1 significantly increased TNF-α-induced necroptosis and glucocorticoid-resistance of bronchial epithelial cells, and these changes could be partially transposed by Nec-1. It is presumed that necroptosis mediates the protective effect of MUC1 and the therapeutic effect of glucocorticoids on asthma.268,269
Therapeutic Strategies for Asthma Targeting Necroptosis
Necrostatins are a sequence of small-molecule inhibitors of necroptosis, among which Necrostatin-1 (Nec-1) is the most utilized. Nec-1 can efficiently and explicitly inhibit the kinase activity of RIPK1 and block the signal transduction of RIPK1-RIPK3-MLKL.270,271 Nec-1 has been determined to relieve asthma severity in vitro experiments and mouse asthma models,251,268 but there is no significant clinical trial currently. To date, there is minimal clinical evidence to correlate necroptosis with asthma, and the function of necroptosis in asthma has not been comprehensively studied, which limits the possibility of necroptosis as a therapeutic target for asthma.
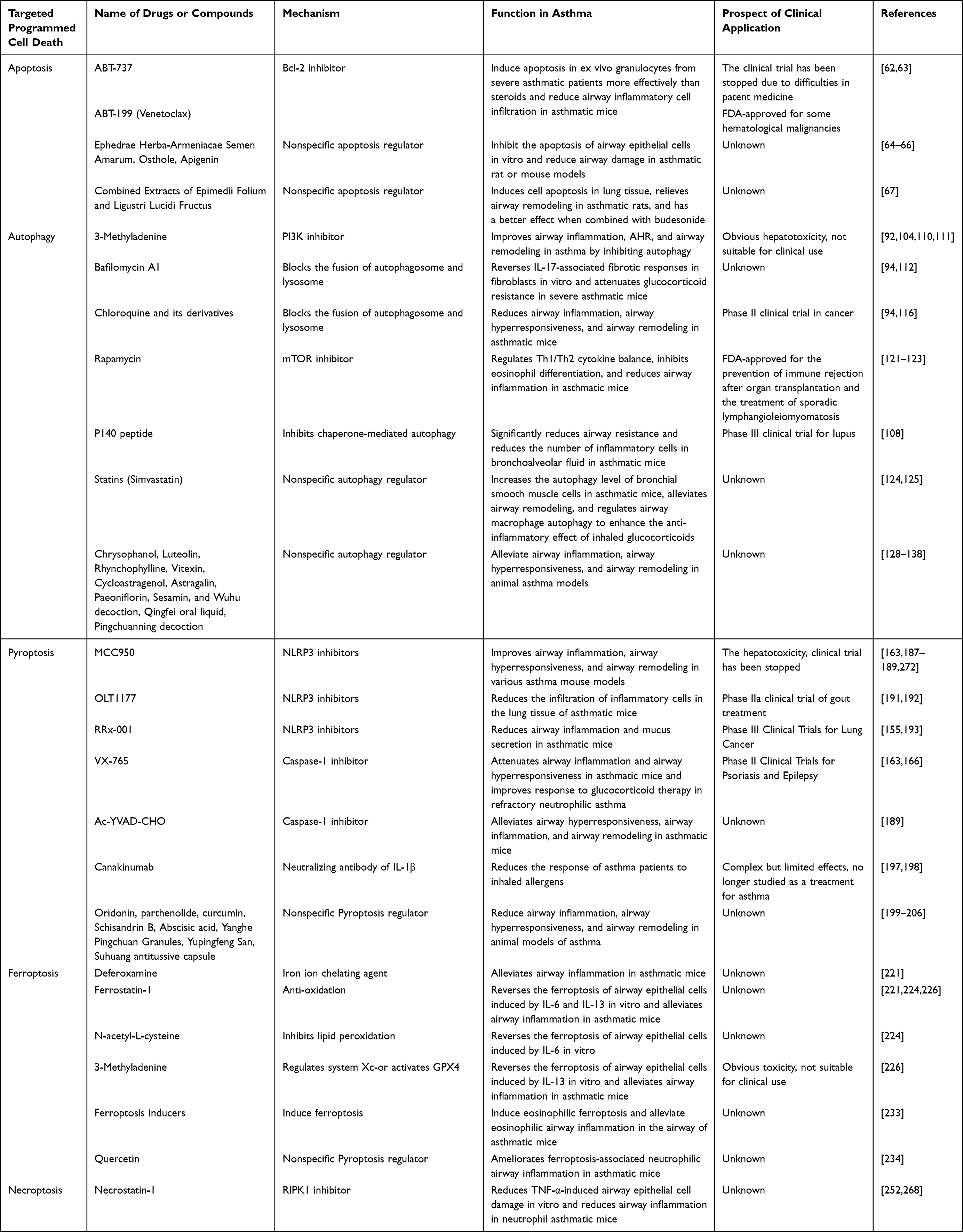 |
Table 2 Therapeutic Strategies for Asthma Targeting Programmed Cell Death |
Conclusion
In this research, we summarize the existing PCD-related studies in asthma and concluded thus: (1) Several types of PCD, including apoptosis, autophagy, pyroptosis, ferroptosis and necroptosis, have been determined to contribute to the pathogenesis of asthma in clinical samples, in vitro cell experiments and animal models of asthma. Furthermore, the functions of various forms of PCD in asthma seem to have different focuses. For instance, autophagy appears to have a considerable influence on airway remodeling in asthma, pyroptosis is more closely linked to refractory asthma such as neutrophilic asthma and obesity asthma, and necroptosis is mostly involved in the antiviral response in asthma. However, these findings cannot be determined as artifacts caused by research bias. The real function and exact molecular mechanism of these PCD forms in asthma are still unknown; (2) Multiple studies have found some connections between these PCD forms and clinical indicators of asthmatic patients, so the identification of PCD levels in asthmatic patients allows for asthma diagnosis and prognostic assessment. However, the current research in this aspect is still inconclusive; (3) Drugs targeting PCD have been studied in asthma, but there is insufficient evidence to indicate that intervention in any PCD can be safe and effective in the treatment of asthma, and most of the PCD-targeted drugs have no indications of clinical translation. Some traditional Chinese medicines have been discovered to have a protective effect on asthma by regulating certain PCD pathways, but their particular targets and mechanisms are unclear, which restricts the use of these drugs in clinical asthma treatment (Table 2).
Following these conclusions, we make some suggestions for future research: (1) The study of PCD in asthma should be more in-depth and specific. For example, the study of apoptosis in asthma has been specific to various cells, and different types of cells have different effects on asthma, which may be advantageous or detrimental. Most studies of other types of PCD in asthma have fixated on an ambiguous overall effect, and whether these PCD forms also have two-sidedness in asthma needs additional investigation. (2) Some studies have revealed that there are some correlations between various forms of PCD, but there are few studies on this aspect in asthma, and then the cross-pathway between these PCDs in asthma should be thoroughly studied, to determine which type of PCD functions more in different aspects of asthma. Furthermore, the correlation between PCD and other asthma-related mechanisms should also be investigated, and attention should be given to the upstream and downstream regulatory molecules of PCD in asthma, which is of great importance in perfecting the pathogenesis of asthma. (3) In future research, it is important to assess the link between various PCD forms and clinical indicators of asthma patients, to offer a clinical basis for PCD-targeted asthma treatment. (4) Concerning the treatment research of PCD-target drugs in asthma, it is crucial, however, to validate the possibility of existing drugs in asthma, and also, it is necessary to develop safer and more effective new drugs.
Acknowledgments
This work was supported by the National Natural Science Foundation of China, 82270104.This study was supported by “the Fundamental Research Funds for the Central Universities” (2042023kf0044).
Author Contributions
Huo-Jun Zhang and Hui-Guo Liu contributed to the conception of the study. Lu Liu drafted the manuscript under the help of Ling Zhou. Lingling Wang and Peng-Dou Zheng, Feng-Qin Zhang and Zhen-Yu Mao took part in drafting, revising or critically reviewing the article. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Agache I, Palmer E, Sanver D, Kirtland M, Shamji MH. Molecular allergology approach to allergic asthma. Mol Aspects Med. 2022;85:101027.
2. Arteaga-Badillo DA, Portillo-Reyes J, Vargas-Mendoza N, et al. Asthma: new integrative treatment strategies for the next decades. Medicina. 2020;56:438.
3. Nunes C, Pereira AM, Morais-Almeida M. Asthma costs and social impact. Asthma Res Pract. 2017;3:1.
4. Guntur VP, Manka LA, Moore CM, et al. Refractory neutrophilic asthma and ciliary genes. J Allergy Clin Immunol. 2022;149:1970–1980.
5. Tong S, Yin Y, Bao Y. Climatotherapy for asthma: research progress and prospect. Environ Res. 2022;214:113988.
6. Liu J, Hong M, Li Y, Chen D, Wu Y, Hu Y. Programmed cell death tunes tumor immunity. Front Immunol. 2022;13:847345.
7. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147.
8. Shi Z, Yuan S, Shi L, et al. Programmed cell death in spinal cord injury pathogenesis and therapy. Cell Prolif. 2021;54:e12992.
9. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
10. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257.
11. Sauler M, Bazan IS, Lee PJ. Cell death in the lung: the apoptosis-necroptosis axis. Annu Rev Physiol. 2019;81:375–402.
12. Ketelut-Carneiro N, Fitzgerald KA. Apoptosis, pyroptosis, and necroptosis-oh my! The many ways a cell can die. J Mol Biol. 2022;434:167378.
13. Peña-Blanco A, García-Sáez AJ. Bax, Bak and beyond - mitochondrial performance in apoptosis. Febs j. 2018;285:416–431.
14. Ashkenazi A, Salvesen G. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol. 2014;30:337–356.
15. Kashyap D, Garg VK, Goel N. Intrinsic and extrinsic pathways of apoptosis: role in cancer development and prognosis. Adv Protein Chem Struct Biol. 2021;125:73–120.
16. Santagostino SF, Assenmacher CA, Tarrant JC, Adedeji AO, Radaelli E. Mechanisms of regulated cell death: current perspectives. Vet Pathol. 2021;58:596–623.
17. Barros B, Almeida BR, Barros DTL, Toledo MS, Suzuki E. Respiratory epithelial cells: more than just a physical barrier to fungal infections. J Fungi. 2022;8:438.
18. Lloyd CM, Saglani S. Asthma and allergy: the emerging epithelium. Nat Med. 2010;16:273–274.
19. Martínez-Girón R, van Woerden HC. Disruption of airway epithelium in asthma pathogenesis: are protozoa responsible? Proc Am Thorac Soc. 2010;7:161.
20. Chanez P. Severe asthma is an epithelial disease. Eur Respir J. 2005;25:945–946.
21. Truong-Tran AQ, Grosser D, Ruffin RE, Murgia C, Zalewski PD. Apoptosis in the normal and inflamed airway epithelium: role of zinc in epithelial protection and procaspase-3 regulation. Biochem Pharmacol. 2003;66:1459–1468.
22. Yamada Y, Yoshihara S, Arisaka O. Creola bodies in wheezing infants predict the development of asthma. Pediatr Allergy Immunol. 2004;15:159–162.
23. Naylor B. The shedding of the mucosa of the bronchial tree in asthma. Thorax. 1962;17:69–72.
24. White SR. Apoptosis and the airway epithelium. J Allergy (Cairo). 2011;2011:948406.
25. Zhou C, Yin G, Liu J, Liu X, Zhao S. Epithelial apoptosis and loss in airways of children with asthma. J Asthma. 2011;48:358–365.
26. Juncadella IJ, Kadl A, Sharma AK, et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547–551.
27. Barbato A, Turato G, Baraldo S, et al. Epithelial damage and angiogenesis in the airways of children with asthma. Am J Respir Crit Care Med. 2006;174:975–981.
28. Wang J, Sun H, Liu Y. The proliferative and anti-apoptosis functions of KGF/KGFR contributes to bronchial epithelial repair in asthma. Pulm Pharmacol Ther. 2020;63:101931.
29. Wan J, Cao Y, Abdelaziz MH, et al. Downregulated Rac1 promotes apoptosis and inhibits the clearance of apoptotic cells in airway epithelial cells, which may be associated with airway hyper-responsiveness in asthma. Scand J Immunol. 2019;89:e12752.
30. Yuan X, Wang E, Xiao X, et al. The role of IL-25 in the reduction of oxidative stress and the apoptosis of airway epithelial cells with specific immunotherapy in an asthma mouse model. Am J Transl Res. 2017;9:4137–4148.
31. James BN, Oyeniran C, Sturgill JL, et al. Ceramide in apoptosis and oxidative stress in allergic inflammation and asthma. J Allergy Clin Immunol. 2021;147:1936–1948.e1939.
32. He X, Zhang L, Hu L, et al. PM2.5 Aggravated OVA-Induced Epithelial Tight Junction Disruption Through Fas Associated via Death Domain-Dependent Apoptosis in Asthmatic Mice. J Asthma Allergy. 2021;14:1411–1423.
33. Zhao C, Wang Y, Su Z, et al. Respiratory exposure to PM2.5 soluble extract disrupts mucosal barrier function and promotes the development of experimental asthma. Sci Total Environ. 2020;730:139145.
34. Aglietti RA, Estevez A, Gupta A, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113:7858–7863.
35. Duncan CJ, Lawrie A, Blaylock MG, Douglas JG, Walsh GM. Reduced eosinophil apoptosis in induced sputum correlates with asthma severity. Eur Respir J. 2003;22:484–490.
36. Jackson DJ, Korn S, Mathur SK, et al. Safety of eosinophil-depleting therapy for severe, eosinophilic asthma: focus on benralizumab. Drug Saf. 2020;43:409–425.
37. Kankaanranta H, Lindsay MA, Giembycz MA, Zhang X, Moilanen E, Barnes PJ. Delayed eosinophil apoptosis in asthma. J Allergy Clin Immunol. 2000;106:77–83.
38. Tai PC, Sun L, Spry CJ. Effects of IL-5, granulocyte/macrophage colony-stimulating factor (GM-CSF) and IL-3 on the survival of human blood eosinophils in vitro. Clin Exp Immunol. 1991;85:312–316.
39. Felton JM, Dorward DA, Cartwright JA, et al. Mcl-1 protects eosinophils from apoptosis and exacerbates allergic airway inflammation. Thorax. 2020;75:600–605.
40. Ilmarinen P, Kankaanranta H. Eosinophil apoptosis as a therapeutic target in allergic asthma. Basic Clin Pharmacol Toxicol. 2014;114:109–117.
41. Januskevicius A, Janulaityte I, Kalinauskaite-Zukauske V, Gosens R, Malakauskas K. The enhanced adhesion of eosinophils is associated with their prolonged viability and pro-proliferative effect in asthma. J Clin Med. 2019;8:127.
42. Druilhe A, Létuvé S, Pretolani M. Glucocorticoid-induced apoptosis in human eosinophils: mechanisms of action. Apoptosis. 2003;8:481–495.
43. Zhu M, Yang J, Chen Y. Efficacy and safety of treatment with benralizumab for eosinophilic asthma. Int Immunopharmacol. 2022;111:109131.
44. Peters MC, Wenzel SE. Intersection of biology and therapeutics: type 2 targeted therapeutics for adult asthma. Lancet. 2020;395:371–383.
45. Zhang J, Zhu Z, Zuo X, et al. The role of NTHi colonization and infection in the pathogenesis of neutrophilic asthma. Respir Res. 2020;21:170.
46. Tanaka A, Sato H, Akimoto K, Matsunaga T, Sagara H. Spontaneous sputum discriminates inflammatory phenotypes in patients with asthma. Ann Allergy Asthma Immunol. 2021;126:54–60.e51.
47. Chen F, Yu M, Zhong Y, Hua W, Huang H. The role of neutrophils in asthma. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2021;50:123–130.
48. Uddin M, Nong G, Ward J, et al. Prosurvival activity for airway neutrophils in severe asthma. Thorax. 2010;65:684–689.
49. Parfrey H, Farahi N, Porter L, Chilvers ER. Live and let die: is neutrophil apoptosis defective in severe asthma? Thorax. 2010;65:665–667.
50. Ciepiela O, Ostafin M, Demkow U. Neutrophils in asthma--a review. Respir Physiol Neurobiol. 2015;209:13–16.
51. Sharifi L, Karimi A, Shokouhi Shoormasti R, et al. Asthma Symptoms and Specific IgE Levels among Toluene Diisocyanate (TDI) Exposed Workers in Tehran, Iran. Iran J Public Health. 2013;42:397–401.
52. Choi Y, Sim S, Park HS. Is TLR4 critical for neutrophil apoptosis in occupational asthma? Allergy Asthma Immunol Res. 2020;12:560–562.
53. Chen S, Deng Y, He Q, et al. Toll-like Receptor 4 Deficiency Aggravates Airway Hyperresponsiveness and Inflammation by Impairing Neutrophil Apoptosis in a Toluene Diisocyanate-Induced Murine Asthma Model. Allergy Asthma Immunol Res. 2020;12:608–625.
54. Kim DH, Gu A, Lee JS, et al. Suppressive effects of S100A8 and S100A9 on neutrophil apoptosis by cytokine release of human bronchial epithelial cells in asthma. Int J Med Sci. 2020;17:498–509.
55. Olave C, Alvarez P, Uberti B, et al. Tamoxifen induces apoptosis and inhibits respiratory burst in equine neutrophils independently of estrogen receptors. J Vet Pharmacol Ther. 2019;42:248–254.
56. Tsai YG, Chien JW, Chen WL, Shieh JJ, Lin CY. Induced apoptosis of TH2 lymphocytes in asthmatic children treated with Dermatophagoides pteronyssinus immunotherapy. Pediatr Allergy Immunol. 2005;16:602–608.
57. Hamzaoui A, Hamzaoui K, Salah H, Chabbou A. Lymphocytes apoptosis in patients with acute exacerbation of asthma. Mediators Inflamm. 1999;8:237–243.
58. Lamb JP, James A, Carroll N, Siena L, Elliot J, Vignola AM. Reduced apoptosis of memory T-cells in the inner airway wall of mild and severe asthma. Eur Respir J. 2005;26:265–270.
59. Mo Y, Ye L, Cai H, et al. SERPINB10 contributes to asthma by inhibiting the apoptosis of allergenic Th2 cells. Respir Res. 2021;22:178.
60. Dorscheid DR, Wojcik KR, Sun S, Marroquin B, White SR. Apoptosis of airway epithelial cells induced by corticosteroids. Am J Respir Crit Care Med. 2001;164:1939–1947.
61. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417.
62. Tian BP, Xia LX, Bao ZQ, et al. Bcl-2 inhibitors reduce steroid-insensitive airway inflammation. J Allergy Clin Immunol. 2017;140:418–430.
63. Tian BP, Li F, Li R, et al. Nanoformulated ABT-199 to effectively target Bcl-2 at mitochondrial membrane alleviates airway inflammation by inducing apoptosis. Biomaterials. 2019;192:429–439.
64. Ma JX, Xiao X, Zhou KF, et al. Herb pair of Ephedrae Herba-Armeniacae Semen Amarum alleviates airway injury in asthmatic rats. J Ethnopharmacol. 2021;269:113745.
65. Tang J, Liu J, Zhang X. The Role of Osthole on TGF-β-Induced Lung Epithelium Apoptosis Injury and Epithelial-Mesenchymal Transition-Mediated Airway Remodeling in Pediatric Asthma. J Healthc Eng. 2022;2022:7099097.
66. Yu H, Huang X, Zhu HH, et al. Apigenin ameliorates non-eosinophilic inflammation, dysregulated immune homeostasis and mitochondria-mediated airway epithelial cell apoptosis in chronic obese asthma via the ROS-ASK1-MAPK pathway. Phytomedicine. 2023;111:154646.
67. Ma Z, Tang X, Gao Y, Wang H, Yu P, Liu R. Combined Extracts of Epimedii Folium and Ligustri Lucidi Fructus with Budesonide Attenuate Airway Remodeling in the Asthmatic Rats by Regulating Apoptosis and Autophagy. Evid Based Complement Alternat Med. 2020;2020:2319409.
68. Kobayashi S. Choose Delicately and Reuse Adequately: the Newly Revealed Process of Autophagy. Biol Pharm Bull. 2015;38:1098–1103.
69. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010.
70. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19:12.
71. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
72. Ornatowski W, Lu Q, Yegambaram M, et al. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020;36:101679.
73. Hara T, Takamura A, Kishi C, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510.
74. Racanelli AC, Choi AMK, Choi ME. Autophagy in chronic lung disease. Prog Mol Biol Transl Sci. 2020;172:135–156.
75. Sachdeva K, Do DC, Zhang Y, Hu X, Chen J, Gao P. Environmental Exposures and Asthma Development: autophagy, Mitophagy, and Cellular Senescence. Front Immunol. 2019;10:2787.
76. Springer MZ, Macleod KF. In Brief: mitophagy: mechanisms and role in human disease. J Pathol. 2016;240:253–255.
77. Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40:151–166.
78. Li A, Gao M, Liu B, et al. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022;13:444.
79. Catarino S, Pereira P, Girão H. Molecular control of chaperone-mediated autophagy. Essays Biochem. 2017;61:663–674.
80. Martin LJ, Gupta J, Jyothula SS, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One. 2012;7:e33454.
81. Ahmad ES, Diab SM, Behiry EG, Bassyoni S, Ishak SR, Ramadan A. Autophagy-related 5 gene mRNA expression and ATG5 rs510432 polymorphism in children with bronchial asthma. Pediatr Pulmonol. 2022;57:2659.
82. Pham DL, Kim SH, Losol P, et al. Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma. Korean J Intern Med. 2016;31:375–385.
83. Poon AH, Choy DF, Chouiali F, et al. Increased autophagy-related 5 gene expression is associated with collagen expression in the airways of refractory asthmatics. Front Immunol. 2017;8:355.
84. Yang F, Kong J, Zong Y, et al. Autophagy-related genes are involved in the progression and prognosis of asthma and regulate the immune microenvironment. Front Immunol. 2022;13:897835.
85. Poon AH, Chouiali F, Tse SM, et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol. 2012;129:569–571.
86. Liu T, Liu Y, Miller M, et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am J Physiol Lung Cell Mol Physiol. 2017;313:L27–l40.
87. Li W, Wu Y, Zhao Y, et al. MTOR suppresses autophagy-mediated production of IL25 in allergic airway inflammation. Thorax. 2020;75:1047–1057.
88. Dickinson JD, Alevy Y, Malvin NP, et al. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. 2016;12:397–409.
89. Sweeter JM, Kudrna K, Hunt K, et al. Autophagy of mucin granules contributes to resolution of airway mucous metaplasia. Sci Rep. 2021;11:13037.
90. Winkler T, Frey U. Airway remodeling: shifting the trigger point for exacerbations in asthma. J Allergy Clin Immunol. 2021;148:710–712.
91. Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. 2014;69:760–765.
92. Jiao B, Chen Y, Yang Y, et al. Toluene diisocyanate-induced inflammation and airway remodeling involves autophagy in human bronchial epithelial cells. Toxicol In Vitro. 2021;70:105040.
93. Joseph C, Tatler AL. Pathobiology of airway remodeling in asthma: the emerging role of integrins. J Asthma Allergy. 2022;15:595–610.
94. McAlinden KD, Deshpande DA, Ghavami S, et al. Autophagy activation in asthma airways remodeling. Am J Respir Cell Mol Biol. 2019;60:541–553.
95. Ramakrishnan RK, Bajbouj K, Hachim MY, et al. Enhanced mitophagy in bronchial fibroblasts from severe asthmatic patients. PLoS One. 2020;15:e0242695.
96. Ramakrishnan RK, Bajbouj K, Al Heialy S, et al. IL-17 Induced Autophagy Regulates Mitochondrial Dysfunction and Fibrosis in Severe Asthmatic Bronchial Fibroblasts. Front Immunol. 2020;11:1002.
97. Hurrell BP, Shafiei Jahani P, Akbari O. Social Networking of Group Two Innate Lymphoid Cells in Allergy and Asthma. Front Immunol. 2018;9:2694.
98. Galle-Treger L, Hurrell BP, Lewis G, et al. Autophagy is critical for group 2 innate lymphoid cell metabolic homeostasis and effector function. J Allergy Clin Immunol. 2020;145:502–517.e505.
99. Matsushita K, Yoshimoto T. B cell-intrinsic MyD88 signaling is essential for IgE responses in lungs exposed to pollen allergens. J Immunol. 2014;193:5791–5800.
100. Xia F, Deng C, Jiang Y, et al. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy. 2018;14:450–464.
101. Ban GY, Pham DL, Trinh TH, et al. Autophagy mechanisms in sputum and peripheral blood cells of patients with severe asthma: a new therapeutic target. Clin Exp Allergy. 2016;46:48–59.
102. Pham DL, Ban GY, Kim SH, et al. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin Exp Allergy. 2017;47:57–70.
103. Liu JN, Suh DH, Trinh HK, Chwae YJ, Park HS, Shin YS. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med. 2016;48:e243.
104. Silveira JS, Antunes GL, Kaiber DB, et al. Autophagy induces eosinophil extracellular traps formation and allergic airway inflammation in a murine asthma model. J Cell Physiol. 2020;235:267–280.
105. Barnes PJ, Baker J, Donnelly LE. Autophagy in asthma and chronic obstructive pulmonary disease. Clin Sci (Lond). 2022;136:733–746.
106. Suzuki Y, Maazi H, Sankaranarayanan I, et al. Lack of autophagy induces steroid-resistant airway inflammation. J Allergy Clin Immunol. 2016;137:1382–1389.e.
107. Suzuki Y, Aono Y, Akiyama N, et al. Involvement of autophagy in exacerbation of eosinophilic airway inflammation in a murine model of obese asthma. Autophagy. 2022;18:2216–2228.
108. Daubeuf F, Schall N, Petit-Demoulière N, Frossard N, Muller S. An autophagy modulator peptide prevents lung function decrease and corrects established inflammation in murine models of airway allergy. Cells. 2021;10:2659.
109. Liu M, Zhang Y, Dong L, Guo Z. Apelin-13 facilitates mitochondria homeostasis via mitophagy to prevent against airway oxidative injury in asthma. Mol Immunol. 2023;153:1–9.
110. Zhang Y, Do DC, Hu X, et al. CaMKII oxidation regulates cockroach allergen-induced mitophagy in asthma. J Allergy Clin Immunol. 2021;147:1464–1477.e1411.
111. Yu Y, Men S, Zhang Y. miR-20a-5p ameliorates ovalbumin (OVA)-induced mouse model of allergic asthma through targeting ATG7-regulated cell death, fibrosis and inflammation. Int Immunopharmacol. 2021;95:107342.
112. Chicote J, Yuste VJ, Boix J, Ribas J. Cell Death Triggered by the Autophagy Inhibitory Drug 3-Methyladenine in Growing Conditions Proceeds With DNA Damage. Front Pharmacol. 2020;11:580343.
113. Redmann M, Benavides GA, Berryhill TF, et al. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017;11:73–81.
114. McAlinden KD, Kota A, Haghi M, Ghavami S, Sharma P. Pharmacologic Inhibition of Vacuolar H(+)ATPase Attenuates Features of Severe Asthma in Mice. Am J Respir Cell Mol Biol. 2020;62:117–120.
115. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455.
116. Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9:1167–1181.
117. Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511.
118. Theofani E, Xanthou G. Autophagy: a Friend or Foe in Allergic Asthma? Int J Mol Sci. 2021;22.
119. Craparo EF, Drago SE, Quaglia F, Ungaro F, Cavallaro G. Development of a novel rapamycin loaded nano- into micro-formulation for treatment of lung inflammation. Drug Deliv Transl Res. 2022;12:1859–1872.
120. Fredriksson K, Fielhaber JA, Lam JK, et al. Paradoxical effects of rapamycin on experimental house dust mite-induced asthma. PLoS One. 2012;7:e33984.
121. Hua W, Liu H, Xia LX, et al. Rapamycin inhibition of eosinophil differentiation attenuates allergic airway inflammation in mice. Respirology. 2015;20:1055–1065.
122. Boberg E, Weidner J, Malmhäll C, Calvén J, Corciulo C, Rådinger M. Rapamycin Dampens Inflammatory Properties of Bone Marrow ILC2s in IL-33-Induced Eosinophilic Airway Inflammation. Front Immunol. 2022;13:915906.
123. Zhang Y, Jing Y, Qiao J, et al. Activation of the mTOR signaling pathway is required for asthma onset. Sci Rep. 2017;7:4532.
124. Gu W, Cui R, Ding T, et al. Simvastatin alleviates airway inflammation and remodelling through up-regulation of autophagy in mouse models of asthma. Respirology. 2017;22:533–541.
125. Maneechotesuwan K, Kasetsinsombat K, Wongkajornsilp A, Barnes PJ. Role of autophagy in regulating interleukin-10 and the responses to corticosteroids and statins in asthma. Clin Exp Allergy. 2021;51:1553–1565.
126. Gong S, Ji X, Su J, et al. Yeast Fermentate Prebiotic Ameliorates Allergic Asthma, Associating with Inhibiting Inflammation and Reducing Oxidative Stress Level through Suppressing Autophagy. Mediators Inflamm. 2021;2021:4080935.
127. Huang C, Peng M, Tong J, et al. Vitamin D ameliorates asthma-induced lung injury by regulating HIF-1α/Notch1 signaling during autophagy. Food Sci Nutr. 2022;10:2773–2785.
128. Song G, Zhang Y, Yu S, et al. Chrysophanol attenuates airway inflammation and remodeling through nuclear factor-kappa B signaling pathway in asthma. Phytother Res. 2019;33:2702–2713.
129. Wang S, Wuniqiemu T, Tang W, et al. Luteolin inhibits autophagy in allergic asthma by activating PI3K/Akt/mTOR signaling and inhibiting Beclin-1-PI3KC3 complex. Int Immunopharmacol. 2021;94:107460.
130. Li H, Bi Q, Cui H, Lv C, Wang M. Suppression of autophagy through JAK2/STAT3 contributes to the therapeutic action of rhynchophylline on asthma. BMC Complement Med Ther. 2021;21:21.
131. Tirpude NV, Sharma A, Kumari M, Bhardwaj N. Vitexin restores lung homeostasis by targeting vicious loop between inflammatory aggravation and autophagy mediated via multiple redox cascade and myeloid cells alteration in experimental allergic asthma. Phytomedicine. 2022;96:153902.
132. Zhu X, Cao Y, Su M, et al. Cycloastragenol alleviates airway inflammation in asthmatic mice by inhibiting autophagy. Mol Med Rep. 2021;24:1.
133. Cho IH, Choi YJ, Gong JH, Shin D, Kang MK, Kang YH. Astragalin inhibits autophagy-associated airway epithelial fibrosis. Respir Res. 2015;16:51.
134. Han X, Hu S, Yang Q, Sang X, Tang D, Cao G. Paeoniflorin ameliorates airway inflammation and immune response in ovalbumin induced asthmatic mice: from oxidative stress to autophagy. Phytomedicine. 2022;96:153835.
135. Bai Q, Wang Z, Piao Y, et al. Sesamin alleviates asthma airway inflammation by regulating mitophagy and mitochondrial apoptosis. J Agric Food Chem. 2022;70:4921–4933.
136. Chen X, Luo Y, Wang M, et al. Wuhu Decoction Regulates Dendritic Cell Autophagy in the Treatment of Respiratory Syncytial Virus (RSV)-Induced Mouse Asthma by AMPK/ULK1 Signaling Pathway. Med Sci Monit. 2019;25:5389–5400.
137. Yu L, Wang J, Zou Y, Zeng H, Cheng W, Jing X. Qingfei oral liquid inhibited autophagy to alleviate inflammation via mTOR signaling pathway in RSV-infected asthmatic mice. Biomed Pharmacother. 2021;138:111449.
138. Wang X, Gao Y, Yang Q, Fang X, Li Z. Pingchuanning decoction attenuates airway inflammation by suppressing autophagy via phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway in rat models of asthma. J Cell Biochem. 2019;120:3833–3844.
139. Zhao H, Dong F, Li Y, et al. Inhibiting ATG5 mediated autophagy to regulate endoplasmic reticulum stress and CD4(+) T lymphocyte differentiation: mechanisms of acupuncture’s effects on asthma. Biomed Pharmacother. 2021;142:112045.
140. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–114.
141. Yang J, Hu S, Bian Y, et al. Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol. 2021;9:789948.
142. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420.
143. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6:128.
144. Hsu SK, Li CY, Lin IL, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics. 2021;11:8813–8835.
145. Wang Y, Yin B, Li D, Wang G, Han X, Sun X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun. 2018;495:1418–1425.
146. Hou J, Zhao R, Xia W, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–1275.
147. Zhou Z, He H, Wang K, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:eaaz7548.
148. Liu Y, Fang Y, Chen X, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020;5:435.
149. Queiroz GA, da Silva RR, Pires AO, et al. New variants in NLRP3 inflammasome genes increase risk for asthma and Blomia tropicalis-induced allergy in a Brazilian population. Cytokine X. 2020;2:100032.
150. Kim SR, Kim DI, Kim SH, et al. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014;5:e1498.
151. Sebag SC, Koval OM, Paschke JD, et al. Mitochondrial CaMKII inhibition in airway epithelium protects against allergic asthma. JCI Insight. 2017;2:e88297.
152. Yang F, Wang T, Yan P, et al. Identification of pyroptosis-related subtypes and establishment of prognostic model and immune characteristics in asthma. Front Immunol. 2022;13:937832.
153. Ritter M, Straubinger K, Schmidt S, et al. Functional relevance of NLRP3 inflammasome-mediated interleukin (IL)-1β during acute allergic airway inflammation. Clin Exp Immunol. 2014;178:212–223.
154. Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126.
155. Ma M, Li G, Qi M, Jiang W, Zhou R. Inhibition of the Inflammasome Activity of NLRP3 Attenuates HDM-Induced Allergic Asthma. Front Immunol. 2021;12:718779.
156. Tsai YM, Chiang KH, Hung JY, et al. Der f1 induces pyroptosis in human bronchial epithelia via the NLRP3 inflammasome. Int J Mol Med. 2018;41:757–764.
157. Zhuang J, Cui H, Zhuang L, et al. Bronchial epithelial pyroptosis promotes airway inflammation in a murine model of toluene diisocyanate-induced asthma. Biomed Pharmacother. 2020;125:109925.
158. Panganiban RA, Sun M, Dahlin A, et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J Allergy Clin Immunol. 2018;142:1469–1478.e1462.
159. Li X, Christenson SA, Modena B, et al. Genetic analyses identify GSDMB associated with asthma severity, exacerbations, and antiviral pathways. J Allergy Clin Immunol. 2021;147:894–909.
160. Das S, Miller M, Beppu AK, et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc Natl Acad Sci U S A. 2016;113:13132–13137.
161. Rossios C, Pavlidis S, Hoda U, et al. Sputum transcriptomics reveal upregulation of IL-1 receptor family members in patients with severe asthma. J Allergy Clin Immunol. 2018;141:560–570.
162. Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J. 2014;43:1067–1076.
163. Kim RY, Pinkerton JW, Essilfie AT, et al. Role for NLRP3 Inflammasome-mediated, IL-1β-Dependent Responses in Severe, Steroid-Resistant Asthma. Am J Respir Crit Care Med. 2017;196:283–297.
164. Awad F, Assrawi E, Jumeau C, et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS One. 2017;12:e0175336.
165. Lachowicz-Scroggins ME, Dunican EM, Charbit AR, et al. Extracellular DNA, neutrophil extracellular traps, and inflammasome activation in severe asthma. Am J Respir Crit Care Med. 2019;199:1076–1085.
166. Chen L, Hou W, Liu F, et al. Blockade of NLRP3/Caspase-1/IL-1β Regulated Th17/Treg Immune Imbalance and Attenuated the Neutrophilic Airway Inflammation in an Ovalbumin-Induced Murine Model of Asthma. J Immunol Res. 2022;2022:9444227.
167. Tan HT, Hagner S, Ruchti F, et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy. 2019;74:294–307.
168. Miethe S, Karsonova A, Karaulov A, Renz H. Obesity and asthma. J Allergy Clin Immunol. 2020;146:685–693.
169. Wood LG, Li Q, Scott HA, et al. Saturated fatty acids, obesity, and the nucleotide oligomerization domain-like receptor protein 3 (NLRP3) inflammasome in asthmatic patients. J Allergy Clin Immunol. 2019;143:305–315.
170. Pinkerton JW, Kim RY, Brown AC, et al. Relationship between type 2 cytokine and inflammasome responses in obesity-associated asthma. J Allergy Clin Immunol. 2022;149:1270–1280.
171. Kim HY, Lee HJ, Chang YJ, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20:54–61.
172. Hur J, Kang JY, Kim YK, Lee SY, Lee HY. Glucagon-like peptide 1 receptor (GLP-1R) agonist relieved asthmatic airway inflammation via suppression of NLRP3 inflammasome activation in obese asthma mice model. Pulm Pharmacol Ther. 2021;67:102003.
173. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol. 2013;49:923–934.
174. Bauer RN, Brighton LE, Mueller L, et al. Influenza enhances caspase-1 in bronchial epithelial cells from asthmatic volunteers and is associated with pathogenesis. J Allergy Clin Immunol. 2012;130:958–967.e914.
175. Gordon EM, Yao X, Xu H, et al. Apolipoprotein E is a concentration-dependent pulmonary danger signal that activates the NLRP3 inflammasome and IL-1β secretion by bronchoalveolar fluid macrophages from asthmatic subjects. J Allergy Clin Immunol. 2019;144:426–441.e423.
176. Han M, Bentley JK, Rajput C, et al. Inflammasome activation is required for human rhinovirus-induced airway inflammation in naive and allergen-sensitized mice. Mucosal Immunol. 2019;12:958–968.
177. Jeong JS, Choi JY, Kim JS, et al. SARS-CoV-2 infection in severe asthma is associated with worsening of COVID-19 through respiratory NLRP3 inflammasome activation. Allergy. 2022.
178. Kashima S, Yorifuji T, Suzuki E. Asian dust and daily emergency ambulance calls among elderly people in Japan: an analysis of its double role as a direct cause and as an effect modifier. J Occup Environ Med. 2014;56:1277–1283.
179. Ko JW, Shin NR, Je-Oh L, et al. Silica dioxide nanoparticles aggravate airway inflammation in an asthmatic mouse model via NLRP3 inflammasome activation. Regul Toxicol Pharmacol. 2020;112:104618.
180. Hitomi Y, Ebisawa M, Tomikawa M, et al. Associations of functional NLRP3 polymorphisms with susceptibility to food-induced anaphylaxis and aspirin-induced asthma. J Allergy Clin Immunol. 2009;124:779–785.e776.
181. Zasłona Z, Flis E, Wilk MM, et al. Caspase-11 promotes allergic airway inflammation. Nat Commun. 2020;11:1055.
182. Allen IC, Jania CM, Wilson JE, et al. Analysis of NLRP3 in the development of allergic airway disease in mice. J Immunol. 2012;188:2884–2893.
183. Huang C, Wang J, Zheng X, et al. Commensal bacteria aggravate allergic asthma via NLRP3/IL-1β signaling in post-weaning mice. J Autoimmun. 2018;93:104–113.
184. Madouri F, Guillou N, Fauconnier L, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol. 2015;7:351–365.
185. Williams EJ, Negewo NA, Baines KJ. Role of the NLRP3 inflammasome in asthma: relationship with neutrophilic inflammation, obesity, and therapeutic options. J Allergy Clin Immunol. 2021;147:2060–2062.
186. Bakhshi S, Shamsi S. MCC950 in the treatment of NLRP3-mediated inflammatory diseases: latest evidence and therapeutic outcomes. Int Immunopharmacol. 2022;106:108595.
187. Wang L, Zha B, Shen Q, et al. Sevoflurane Inhibits the Th2 Response and NLRP3 Expression in Murine Allergic Airway Inflammation. J Immunol Res. 2018;2018:9021037.
188. Primiano MJ, Lefker BA, Bowman MR, et al. Efficacy and Pharmacology of the NLRP3 Inflammasome Inhibitor CP-456773 (CRID3) in Murine Models of Dermal and Pulmonary Inflammation. J Immunol. 2016;197:2421–2433.
189. Chen S, Yao L, Huang P, et al. Blockade of the NLRP3/caspase-1 axis ameliorates airway neutrophilic inflammation in a toluene diisocyanate-induced murine asthma model. Toxicol Sci. 2019;170:462–475.
190. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606.
191. Lunding LP, Skouras DB, Vock C, Dinarello CA, Wegmann M. The NLRP3 inflammasome inhibitor, OLT1177(®), ameliorates experimental allergic asthma in mice. Allergy. 2022;77:1035–1038.
192. Klück V, Jansen T, Janssen M, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020;2:e270–e280.
193. Chen Y, He H, Lin B, et al. RRx-001 ameliorates inflammatory diseases by acting as a potent covalent NLRP3 inhibitor. Cell Mol Immunol. 2021;18:1425–1436.
194. Corcoran SE, Halai R, Cooper MA. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol Rev. 2021;73:968–1000.
195. Wen S, Deng F, Li L, Xu L, Li X, Fan Q. VX-765 ameliorates renal injury and fibrosis in diabetes by regulating caspase-1-mediated pyroptosis and inflammation. J Diabetes Investig. 2022;13:22–33.
196. Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43:653–668.
197. Pascoe S, Kanniess F, Bonner J, Lloyd P, Lowe P, Beier J. A monoclonal antibody to IL-1β attenuates the late asthmatic response to antigen challenge in patients with mild asthma. Annu Congr Eur Resp Soc. 2006;752.
198. Theofani E, Semitekolou M, Morianos I, Samitas K, Xanthou G. Targeting NLRP3 inflammasome activation in severe asthma. J Clin Med. 2019;8:1615.
199. Wang J, Li F, Ding J, et al. Investigation of the anti‑asthmatic activity of Oridonin on a mouse model of asthma. Mol Med Rep. 2016;14:2000–2006.
200. Arıkan-Ayyıldız Z, Karaman M, Özbal S, et al. Efficacy of parthenolide on lung histopathology in a murine model of asthma. Allergol Immunopathol. 2017;45:63–68.
201. Jaiswal A, Dash D, Singh R. Intranasal curcumin and dexamethasone combination ameliorates inflammasome (NLRP3) activation in lipopolysaccharide exposed asthma exacerbations. Toxicol Appl Pharmacol. 2022;436:115861.
202. Chen X, Xiao Z, Jiang Z, Jiang Y, Li W, Wang M. Schisandrin B Attenuates Airway Inflammation and Airway Remodeling in Asthma by Inhibiting NLRP3 Inflammasome Activation and Reducing Pyroptosis. Inflammation. 2021;44:2217–2231.
203. Zhao CC, Xu J, Xie QM, Zhang HY, Fei GH, Wu HM. Abscisic acid suppresses the activation of NLRP3 inflammasome and oxidative stress in murine allergic airway inflammation. Phytother Res. 2021;35:3298–3309.
204. Pan L, Chen Y, Jiang Y, Sun Y, Han Y, Wang Y. Yanghe Pingchuan Granules Alleviate Airway Inflammation in Bronchial Asthma and Inhibit Pyroptosis by Blocking the TLR4/NF-κB/NRLP3 Signaling Pathway. Mediators Inflamm. 2022;2022:6561048.
205. Liu X, Shen J, Fan D, et al. Yupingfeng San Inhibits NLRP3 Inflammasome to Attenuate the Inflammatory Response in Asthma Mice. Front Pharmacol. 2017;8:944.
206. Qin W, Wu X, Jia Y, et al. Suhuang antitussive capsule inhibits NLRP3 inflammasome activation and ameliorates pulmonary dysfunction via suppression of endoplasmic reticulum stress in cough variant asthma. Biomed Pharmacother. 2019;118:109188.
207. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072.
208. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85.
209. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
210. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379.
211. Shui S, Zhao Z, Wang H, Conrad M, Liu G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol. 2021;45:102056.
212. Zhang C, Liu N. Ferroptosis, necroptosis, and pyroptosis in the occurrence and development of ovarian cancer. Front Immunol. 2022;13:920059.
213. Lu S, Wang XZ, He C, et al. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron. Acta Pharmacol Sin. 2021;42:1690–1702.
214. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–4975.
215. Xu W, Deng H, Hu S, et al. Role of Ferroptosis in Lung Diseases. J Inflamm Res. 2021;14:2079–2090.
216. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331.
217. McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids. 2012;42:199–205.
218. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
219. Ali MK, Kim RY, Brown AC, et al. Crucial role for lung iron level and regulation in the pathogenesis and severity of asthma. Eur Respir J. 2020;55:5443.
220. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109.
221. Zeng Z, Huang H, Zhang J, et al. HDM induce airway epithelial cell ferroptosis and promote inflammation by activating ferritinophagy in asthma. FASEB j. 2022;36:e22359.
222. Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–641.e626.
223. Nagasaki T, Schuyler AJ, Zhao J, et al. 15LO1 dictates glutathione redox changes in asthmatic airway epithelium to worsen type 2 inflammation. J Clin Invest. 2022;132.
224. Han F, Li S, Yang Y, Bai Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered. 2021;12:5279–5288.
225. Tang W, Dong M, Teng F, et al. Environmental allergens house dust mite-induced asthma is associated with ferroptosis in the lungs. Exp Ther Med. 2021;22:1483.
226. Yang N, Shang Y. Ferrostatin-1 and 3-Methyladenine Ameliorate Ferroptosis in OVA-Induced Asthma Model and in IL-13-Challenged BEAS-2B Cells. Oxid Med Cell Longev. 2022;2022:9657933.
227. Hajek AR, Lindley AR, Favoreto S, Carter R, Schleimer RP, Kuperman DA. 12/15-Lipoxygenase deficiency protects mice from allergic airways inflammation and increases secretory IgA levels. J Allergy Clin Immunol. 2008;122:633–639.e633.
228. Bao C, Liu C, Liu Q, et al. Liproxstatin-1 alleviates LPS/IL-13-induced bronchial epithelial cell injury and neutrophilic asthma in mice by inhibiting ferroptosis. Int Immunopharmacol. 2022;109:108770.
229. Farr AC, Xiong MP. Challenges and Opportunities of Deferoxamine Delivery for Treatment of Alzheimer’s Disease, Parkinson’s Disease, and Intracerebral Hemorrhage. Mol Pharm. 2021;18:593–609.
230. Miotto G, Rossetto M, Di Paolo ML, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328.
231. Zilka O, Shah R, Li B, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232–243.
232. Li Y, Yang Y, Yang Y. Multifaceted Roles of Ferroptosis in Lung Diseases. Front Mol Biosci. 2022;9:919187.
233. Wu Y, Chen H, Xuan N, et al. Induction of ferroptosis-like cell death of eosinophils exerts synergistic effects with glucocorticoids in allergic airway inflammation. Thorax. 2020;75:918–927.
234. Wang Y, Wan R, Peng W, Zhao X, Bai W, Hu C. Quercetin alleviates ferroptosis accompanied by reducing M1 macrophage polarization during neutrophilic airway inflammation. Eur J Pharmacol. 2023;938:175407.
235. Tang W, Dong M, Teng F, et al. TMT-based quantitative proteomics reveals suppression of SLC3A2 and ATP1A3 expression contributes to the inhibitory role of acupuncture on airway inflammation in an OVA-induced mouse asthma model. Biomed Pharmacother. 2021;134:111001.
236. Tang W, Qin J, Zhou Y, et al. Regulation of ferroptosis and ACSL4-15LO1 pathway contributed to the anti-asthma effect of acupuncture. Int Immunopharmacol. 2023;115:109670.
237. Mathias LJ, Khong SM, Spyroglou L, et al. Alveolar macrophages are critical for the inhibition of allergic asthma by mesenchymal stromal cells. J Immunol. 2013;191:5914–5924.
238. Lim J, Heo J, Ju H, et al. Glutathione dynamics determine the therapeutic efficacy of mesenchymal stem cells for graft-versus-host disease via CREB1-NRF2 pathway. Sci Adv. 2020;6:eaba1334.
239. Kim RL, Bang JY, Kim J, et al. Mesenchymal stem cells exert their anti-asthmatic effects through macrophage modulation in a murine chronic asthma model. Sci Rep. 2022;12:9811.
240. Lv X, Dong M, Tang W, et al. Ferroptosis, novel therapeutics in asthma. Biomed Pharmacother. 2022;153:113516.
241. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119.
242. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320.
243. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and Relevance to Disease. Annu Rev Pathol. 2017;12:103–130.
244. Kaiser WJ, Upton JW, Long AB, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372.
245. Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700.
246. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. 2022;7:196.
247. Ning J, Qiao L. The role of necroptosis in common respiratory diseases in children. Front Pediatr. 2022;10:945175.
248. Muniz-Junqueira MI, Barbosa-Marques SM, Junqueira LF. Morphological changes in eosinophils are reliable markers of the severity of an acute asthma exacerbation in children. Allergy. 2013;68:911–920.
249. Radonjic-Hoesli S, Wang X, de Graauw E, et al. Adhesion-induced eosinophil cytolysis requires the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase-like (MLKL) signaling pathway, which is counter-regulated by autophagy. J Allergy Clin Immunol. 2017;140:1632–1642.
250. He A, Chen J, Guan J, et al. Selective eosinophil necroptosis contributes to airway inflammation and remodeling in asthma. Allergy. 2022;77:3456–3459.
251. Han XA, Jie HY, Wang JH, et al. Necrostatin-1 Ameliorates Neutrophilic Inflammation in Asthma by Suppressing MLKL Phosphorylation to Inhibiting NETs Release. Front Immunol. 2020;11:666.
252. Hamzaoui A, Berraies A, Kaabachi W, Haifa M, Ammar J, Kamel H. Induced sputum levels of IL-33 and soluble ST2 in young asthmatic children. J Asthma. 2013;50:803–809.
253. Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490.
254. Kurowska-Stolarska M, Stolarski B, Kewin P, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–6477.
255. Shlomovitz I, Erlich Z, Speir M, et al. Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. Febs j. 2019;286:507–522.
256. Oikonomou N, Schuijs MJ, Chatzigiagkos A, et al. Airway epithelial cell necroptosis contributes to asthma exacerbation in a mouse model of house dust mite-induced allergic inflammation. Mucosal Immunol. 2021;14:1160–1171.
257. Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell. 2014;54:273–280.
258. Zhang T, Yin C, Boyd DF, et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell. 2020;180:1115–1129.e1113.
259. Gaajetaan GR, Geelen TH, Vernooy JH, et al. Interferon-β induces a long-lasting antiviral state in human respiratory epithelial cells. J Infect. 2013;66:163–169.
260. Wark PA, Johnston SL, Bucchieri F, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947.
261. Cerps SC, Menzel M, Mahmutovic Persson I, Bjermer L, Akbarshahi H, Uller L. Interferon-β deficiency at asthma exacerbation promotes MLKL mediated necroptosis. Sci Rep. 2018;8:4248.
262. Feldman AS, He Y, Moore ML, Hershenson MB, Hartert TV. Toward primary prevention of asthma. Reviewing the evidence for early-life respiratory viral infections as modifiable risk factors to prevent childhood asthma. Am J Respir Crit Care Med. 2015;191:34–44.
263. Simpson J, Loh Z, Ullah MA, et al. Respiratory Syncytial Virus Infection Promotes Necroptosis and HMGB1 Release by Airway Epithelial Cells. Am J Respir Crit Care Med. 2020;201:1358–1371.
264. Liu K, Hua S, Song L. PM2.5 exposure and asthma development: the key role of oxidative stress. Oxid Med Cell Longev. 2022;2022:3618806.
265. Luo J, Liu H, Hua S, Song L. The Correlation of PM2.5 Exposure with Acute Attack and Steroid Sensitivity in Asthma. Biomed Res Int. 2022;2022:2756147.
266. Zhao Y, Zhang H, Yang X, Zhang Y, Feng S, Yan X. Fine particulate matter (PM(2.5)) enhances airway hyperresponsiveness (AHR) by inducing necroptosis in BALB/c mice. Environ Toxicol Pharmacol. 2019;68:155–163.
267. Niessen NM, Gibson PG, Baines KJ, et al. Sputum TNF markers are increased in neutrophilic and severe asthma and are reduced by azithromycin treatment. Allergy. 2021;76:2090–2101.
268. Zhang H, Ji J, Liu Q, Xu S. MUC1 downregulation promotes TNF-α-induced necroptosis in human bronchial epithelial cells via regulation of the RIPK1/RIPK3 pathway. J Cell Physiol. 2019;234:15080–15088.
269. Zhang H, Liu Q, Kong L, Xu S. Mucin 1 downregulation impairs the anti-necroptotic effects of glucocorticoids in human bronchial epithelial cells. Life Sci. 2019;221:168–177.
270. Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321.
271. Takahashi N, Duprez L, Grootjans S, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437.
272. Jeong JS, Lee KB, Kim SR, et al. Airway epithelial phosphoinositide 3-kinase-δ contributes to the modulation of fungi-induced innate immune response. Thorax. 2018;73:758–768.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.