Back to Journals » Breast Cancer: Targets and Therapy » Volume 17
Prognostic and Immunological Implications of Telomere Maintenance-Related Genes in Breast Cancer: A Mechanistic Exploration
Authors Cao H, Gao F, Wang R, Xie Y, Li B
Received 9 June 2025
Accepted for publication 5 November 2025
Published 12 November 2025 Volume 2025:17 Pages 1063—1082
DOI https://doi.org/10.2147/BCTT.S545745
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Hui Cao,1 Feng Gao,1 Ronghua Wang,1 Yajuan Xie,1 Bin Li2
1Department of Breast Surgery, General Hospital of Tisco (The Sixth Hospital of Shanxi Medical University, The Sixth Clinical Medical College of Shanxi Medical University), Taiyuan, People’s Republic of China; 2Department of General Surgery, General Hospital of Tisco (The Sixth Hospital of Shanxi Medical University, The Sixth Clinical Medical College of Shanxi Medical University), Taiyuan, People’s Republic of China
Correspondence: Bin Li, Email [email protected]
Background: The elongation of telomeres endows cancer cells with the ability of replicative immortality. Nevertheless, the connection between it and breast cancer (BC) has not been clearly defined yet.
Methods: In this study, expression data from multiple BC datasets were analyzed to discover differentially expressed telomere maintenance-related genes (DE-TMRGs). Cox regression analyses were executed to determine prognostic genes, and a risk model was constructed using these genes to predict survival outcomes. Independent prognostic analysis was executed to assess independent prognostic factors. Enrichment analyses were conducted to determine pathways linked to the different risk groups. Immune infiltration levels and immune checkpoint expression were analyzed across risk categories using various algorithms. A regulatory network involving miRNAs and lncRNAs was constructed, and drug sensitivity was assessed. Mendelian randomization (MR) analysis was executed to investigate causal relationships between prognostic genes and BC. Finally, the expression of prognostic genes was further analysed at the single-cell level.
Results: A sum of 81 DE-TMRGs. Through univariate and LASSO regression analyses, five prognostic genes (WT1, TPRXL, RAD54B, JUN, and SEPHS2) were identified to construct a risk model. This model successfully distinguished between high- and low-risk categories with significant differences in survival rates. Immune profiling revealed the high-risk category had increased levels of activated T cells. Eosinophil demonstrated a high correlation with prognostic genes. Furthermore, JUN (OR = 1.1333, P = 0.0085) and RAD54B (OR = 1.0790, P = 0.0015) were identified as causal risk factors for BC. Single-cell RNA sequencing highlighted JUN’s widespread distribution across multiple cell types, suggesting its crucial role in tumor progression.
Conclusion: TM-associated risk model based on WT1, TPRXL, RAD54B, JUN, and SEPHS2 could be used to predict prognosis and treatment of BC.
Keywords: telomere maintenance, breast cancer, risk model, single cell analysis, Mendelian randomization
Introduction
Breast cancer (BC) is among the most prevalent cancers globally, especially affecting women at high rates.1 As of 2023, it is estimated that BC has caused 43,170 deaths among women.2 Currently, standardized multidisciplinary treatment strategies have been established for BC,3 encompassing endocrine therapy, chemotherapy, radiotherapy and targeted therapy. For most early-stage BC patients, these therapeutic approaches significantly improve prognosis. Nonetheless, numerous patients continue to be diagnosed when the disease is in its advanced stages, and sometimes tumors can recur or spread, leading to less than ideal treatment results.4 Therefore, the development of effective prognostic models to guide the selection of appropriate therapeutic strategies and drugs is crucial for improving clinical management and outcomes in BC.
Telomeres are unique formations found at chromosome ends, mainly made up of repetitive DNA sequences and related protein complexes. Their main functions include protecting chromosomes from degradation, rearrangement, and fusion, as well as ensuring the stability of genetic information during cell division.5 Telomerase is a ribonucleoprotein polymerase that maintains telomere length by adding telomere DNA repeat sequences to the 3 ‘-OH terminal of telomeres.6 Cancer cells exhibit telomerase activation, thus the elongation of telomeres provides cancer cells with the ability to achieve “replicative immortality”.7,8 More and more evidence indicates a connection between BC and telomere length.9 Previous studies have shown that high telomerase activity is associated with poor prognosis of BC,10 and telomere length may be a valuable prognostic biomarker for BC.11 Cancer cells establish “replication immortality” by activating the telomere maintenance mechanism (TMM).12 Two major TMMs have been identified, including telomerase-dependent (TEL) and alternative lengthening of telomeres (ALT) mechanisms.12 ALT-TMM is commonly observed in tumors originating from mesenchymal tissues, such as osteosarcoma and liposarcoma, as well as in tumors of the central and peripheral nervous systems or neuroendocrine systems.13 Both telomere maintenance mechanisms have been detected in BC and may be associated with specific histological subtypes or disease stages.14,15 Studies have shown that elevated expression levels of core telomerase components, including HTERT and HTMLR, are closely correlated with reduced overall survival in BC patients.16 Furthermore, other research has highlighted significant associations between changes in telomere length and clinical characteristics of BC, such as tumor stage, recurrence, and metastasis.11 Therefore, telomere maintenance plays a critical role not only in cancer cell growth and survival but also as a potential prognostic biomarker for BC. Treating the great majority of cancer types by targeting telomere maintenance is an exciting prospect.12 More and more evidence indicates that telomere maintenance-related genes (TMRGs) play a significant role in the treatment of cancer.17 For instance, the small molecule inhibitor MST-312 can treat various cancers, including BC, by inducing down-regulation of human telomerase reverse transcriptase (hTERT) expression, reduction of telomerase activity and telomere shortening, thereby causing cell cycle arrest and apoptosis.12 In HER2⁺ BC, the combination of telomerase antagonist Imetelstat and trastuzumab can inhibit the growth of human BC cells.18 These examples prove that TMRGs are not only prognostic biomarkers but also intervenable targets. Therefore, investigating the biological roles and molecular mechanisms of telomere maintenance in BC holds significant clinical value for improving early diagnosis, optimizing therapeutic strategies, and enhancing prognostic evaluation.
The tumor immune microenvironment (TIME), composed of the interaction between the tissue environment surrounding tumor cells and immune cells, has a significant impact on the prognosis of cancer.17,19 Studies have shown that there are differences in immune cell infiltration among the molecular subtypes derived from TMRGs, suggesting that TMRGs may play a role in mediating different TIME.17 TMRGs play a role in tumor microenvironment, migration and invasion, angiogenesis and fibroblast activation in malignant tumors, which may promote the formation of immunosuppressive environmental homeostasis in the tumor microenvironment.20 Studies have reported that the telomerase catalytic subunit telomerase reverse transcriptase (TERT) promotes the formation of a tumor immunosuppressive microenvironment by activating endogenous retroviruses.21 Research has found that different subtypes of telomere maintenance mechanisms are associated with different patterns of immune cell infiltration in gastric cancer, which indicates a complex interaction between telomere maintenance and the immune microenvironment.20 In conclusion, telomere maintenance and the tumor microenvironment form a complex feedback regulatory network, and this mechanism highlights the significant role of telomere maintenance in shaping the tumor microenvironment.20 Malignant breast cells interact with various cells of the immune system, influencing the proliferation, development, progression of tumors and the efficacy of therapeutic interventions.22 Therefore, systematically clarifying the differences in the immune microenvironment among BC patients classified by different TMRGs subtypes is of great significance for understanding the immune regulatory functions of TMRGs and exploring new immunotherapy targets.
Telomere maintenance is a complex biological process involving the synergistic effects of multiple genes and signaling pathways. We obtained the TMRGs from the TelNet authoritative database.23–25 This collection itself contains genes that are directly involved (such as telomerase components) and indirectly involved (such as enhancers, repressors, transcription factor), as they collectively constitute the molecular basis underpinning the macroscopic phenotype of telomere maintenance. This study utilized clinical information and transcriptomic data of BC from public databases to explore the potential of TMRGs as prognostic biomarkers for BC and to uncover their underlying molecular regulatory mechanisms using bioinformatics approaches. By analyzing BC transcriptomic data, we identified prognostic genes associated with TM and conducted an in-depth analysis of their expression patterns, evaluating their roles in BC prognosis. Clinical samples were employed to further verify the expression levels of prognostic genes. Subsequently, MR analysis was executed to explore the causal relationships between these prognostic genes and BC, shedding light on the potential mechanisms by which TMRGs contribute to BC development and progression. We analyzed the immune microenvironment and the expression levels of immune checkpoints in patients with different subtypes of TMRGs. Additionally, this study examined the expression of these prognostic genes at the single-cell level, providing new perspectives and data support for future clinical research.
Materials and Methods
Data Source
Expression matrix and survival data for BC patients were obtained from UCSC Xena database. The GDC TCGA Breast Cancer (training set) comprised 1118 tumor samples and 113 control samples from BC tissues, with the goal of identifying genes and constructing a risk model for disease prediction. The GSE20685 (validation set) and single-cell dataset (GSE161529) of BC were obtained from the GEO database. The GSE20685 contained 327 BC tissue samples was utilized to validate the risk model. The GSE161529 dataset contained single-cell RNA expression profiles of human breast in normal, pre-cancerous, and tumorigenic states.
The GWAS dataset of BC (ebi-a-GCST90018799) and the eQTL GWAS data were obtained from the IEU OpenGWAS database. Regarding the dataset ebi-a-GCST90018799, it included 257,730 samples, specifically 17,389 BC samples and 240,341 control samples, with a total of 24,133,589 SNP, and the ethnicity was European. Furthermore, 411 TMRGs were obtained from TelNet database.24
Recognition and Enrichment Analysis of Differentially Expressed TMRGs (DE-TMRGs)
In the analysis of the training set, differential expression analysis between tumor versus normal samples was conducted utilizing the “DESeq2” package (v 1.34.0),26 pinpointing DEGs [|log2FC| > 1 and P < 0.05]. The DE-TMRGs were identified through the intersection of DEGs with TMRGs. Subsequently, to explore the potential biological activities and pathways associated with the DE-TMRGs, GO and KEGG pathway analyses were executed utilizing the “ClusterProfiler” package (v 4.2.2)27 (P < 0.05).
Development and Validation of a Risk Model
The identified DE-TMRGs underwent univariate Cox regression analysis to determine genes linked to survival outcomes (P < 0.01). To further streamline the selection of features, a LASSO regression model was implemented utilizing the “glmnet” package (v 4.1–4),28 centered on survival-associated genes. Based on the expression levels of five prognostic genes, the risk score for each BC patient was computed utilizing the following formula: 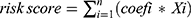 . We determined the optimal cutoff threshold employing the surv_cutpoint function, which classified BC patients into high and low-risk categories across all cohorts. Risk curves were created to demonstrate the distribution of risk scores and the survival status of BC patients. The expression levels of prognostic genes in all cohorts were visualized, and ROC curves for 3, 5, and 7-year survival periods were constructed using the timeROC package (v 0.4),29 computing area under curve (AUC) values to indicate predictive accuracy. Additionally, K-M survival curves were generated to compare survival rates between two-risk categories across all cohorts. Finally, expression heatmaps were produced to display the levels of prognostic genes in the two risk categories.
. We determined the optimal cutoff threshold employing the surv_cutpoint function, which classified BC patients into high and low-risk categories across all cohorts. Risk curves were created to demonstrate the distribution of risk scores and the survival status of BC patients. The expression levels of prognostic genes in all cohorts were visualized, and ROC curves for 3, 5, and 7-year survival periods were constructed using the timeROC package (v 0.4),29 computing area under curve (AUC) values to indicate predictive accuracy. Additionally, K-M survival curves were generated to compare survival rates between two-risk categories across all cohorts. Finally, expression heatmaps were produced to display the levels of prognostic genes in the two risk categories.
Correlation Analysis of Clinical Characteristics and Construction of Nomogram
In the training set, the correlation between risk scores and clinical features was further evaluated by analyzing differences in risk scores across groups with varying clinicopathological characteristics. Furthermore, to assess the efficacy of the risk model and clinical variables as independent prognostic factors, the risk score along with BC patients’ clinical characteristics were progressively included in both univariate (P < 0.01) and multivariate Cox regression analyses. After adjusting for the independent prognostic model, independent prognostic factors for BC patients were recognized. Following this, a nomogram was developed to forecast the survival rates of BC patients using the rms package (v 6.5–1), and calibration curves were drawn to assess the predictive accuracy of the nomogram.
Functional and Annotation Analyses
To conduct a more in-depth and detailed exploration of the signaling pathways associated with the two risk categories in the training cohort, a series of rigorous analysis steps were taken. Firstly, the Spearman correlation analysis between the risk score and all genes was performed utilizing the “psych” package. Subsequently, based on the c2.cp.kegg.v2023.1.Hs.symbols gene set, the GSEA pathway enrichment analysis was executed with the help of the “clusterProfiler” package (v 4.2.2). The threshold conditions for the enrichment analysis were set as follows: |NES| > 1, P < 0.05. Subsequently, using the GSVA package and the limma package, and taking the enriched gene set file (h.all.v2023.1.Hs.symbols.gmt) as the data support, the GSVA enrichment analysis was conducted between the two-risk categories. In this analysis process, the thresholds were set as |t| > 2 and P < 0.05 to draw the GSVA enrichment pathways, thereby presenting the differences in gene enrichment between the two risk categories more intuitively.
Immune Analysis
The tumor microenvironment of the two risk categories were analyzed using the ssGSEA package. This method enabled the identification of 28 distinct immune cell types across all samples in the training cohort, facilitating a thorough comparison of immune infiltration levels between the various risk categories. The association between different immune cell abundance and risk scores per patient was investigated with the support of Spearman correlation analysis. To further enrich our understanding of the tumour microenvironment, we employed the ESTIMATE algorithm (v 1.0.13).30 This yielded quantitative immune, stromal and ESTIMATE scores for each patient sample. The analysis aimed to reveal fundamental differences in the tumour microenvironment between the two risk categories, revealing the intricate interplay between tumour structure and patient prognosis. Ultimately, the research expanded its scope by examining the variations in the expression of 38 immune checkpoints between the two risk categories (P < 0.05).
Constructing Regulatory Networks and Drug Sensitivity Analysis
To further dissect the intricate molecular regulatory mechanisms behind the identified prognostic genes, miRNA (clipExpNum ≥ 5) and lncRNAs (clipExpNum ≥ 30) that might regulate these prognostic genes were retrieved from the starBase database. Using these data, a comprehensive Cytoscape software was used to construct an lncRNA-miRNA-mRNA network, highlighting the complex interactions between these molecular entities. Besides, the study queried 198 chemotherapy agents listed in the GDSC database (http://cancerrxgene.org). The sensitivity of the both risk groups to these chemotherapy drugs was assessed by calculating and comparing the IC50 values using the oncoPredict package (v 0.2)31 (P < 0.0001 and IC50 < 0.01). Moreover, the CTD database (http://ctdbase.org/) was utilized to identify potential chemical compounds associated with these prognostic genes, aiming to gain insights into how these chemicals influence gene expression and functionality.
Single-Cell Data Analysis
Using the expression matrix from the single-cell dataset GSE161529 sample as a basis, the data was first filtered using the Seurat package (v 4.3.0).32 Cells containing fewer than 200 genes and genes covered by fewer than 3 cells were filtered out. Also exclude cells with over 20% mitochondrial genes, remove cells with ≤200 and ≥6000 genes in the cell, and remove genes with counts ≤200 and ≥40000. After log-normalisation of the retained data, 2000 highly variable genes with high intercellular coefficients of variation were extracted for subsequent analysis using the vst method. PCA was then performed on the high-variance genes. Based on the PCA displacement test and inflection plots, the PC values were selected to cluster cells when they levelled off. Following the PCA dimensionality reduction, we conducted an unsupervised clustering analysis of the cells using the FindNeighbors and FindClusters functions from the Seurat package. The resulting cell clusters were then annotated based on marker genes for each cell type found in the CellMarker database, with the identified cell types presented in a UMAP clustering visualization. Finally, we further analysed the distribution of prognostic genes across cell types.
Pseudo-Temporal Analyses and Cellular Communication Analysis
Pseudo-temporal analyses of key cells were performed using the Monocle software package (v 2.26.0)33 and pseudo-temporal trajectory maps of the cells were constructed, which were sorted according to the temporal ordering of each key cell. These sequencing results show the dynamics of the cells in time, allowing further insights into the function and developmental processes of each cell type or state. In addition, the expression of prognostic genes in the key cells was visualised and analysed at different times. In this study, the CellChat package (v 1.6.1)34 was used for intercellular communication and interaction analysis to infer communication probabilities at the signalling pathway level by calculating the communication correlations of all ligand-receptor interactions associated with each signalling pathway and constructing ligand-receptor networks. To accurately determine the node sizes and edge weights of the networks across various subgroups, we computed the maximum number of interactions and the interaction weights for each cell in the Control and BC groups.
MR Analysis
This study adhered to the Mendelian reporting guidelines for randomized studies (STROBE-MR).35 To further elucidate the causal association between prognostic genes and BC, we performed MR analysis, considering the prognostic genes as the exposure variable and BC as the outcome. MR studies are based on three essential assumptions: (1) there is a strong and significant correlation between the instrumental variables (IVs) and the exposure; (2) the IVs are not associated with any confounding factors; and (3) the IVs affect the outcomes exclusively through their effect on the exposure, without any other paths. At the outset, we utilized extract_instruments function from “TwoSampleMR” package (v 0.5.8)36 to extract exposure factors and filter out IVs exhibiting a significant correlation with exposure factors (P < 5*10−5). Subsequently, to remove IVs with LD from our analysis, we specified r2 = 0.01 and kb = 100. We also calculated the F-statistic and deemed IVs to be suitably robust when F > 10.
After the screening of IVs, we conducted MR analysis to investigate causal link between prognostic genes and BC. Primarily, harmonise_data function was utilized to harmonize effect equivalents with effect sizes, ensuring consistency across datasets. Subsequently, MR analysis was performed utilizing the mr function alongside five algorithms: MR-Egger, Weighted median, Inverse variance weighted (IVW), Simple mode, and Weighted mode. To assess the robustness of the results, a sensitivity analysis was conducted.
RT-qPCR
This study collected 5 pairs of BC patients’ tumor and adjacent normal tissue samples from General Hospital of Taiyuan Iron and Steel (Group) Co., Ltd. All participants executed informed consent forms. The study was approved by the Medical Ethics Committee of General Hospital of Taiyuan Iron and Steel (Group) Co., Ltd. (Ethics ID: k202502). The FastPure Complex Tissue/Cell Total RNA Isolation Kit (RC113-01, Vazyme, China) was used to extract total RNA from tissue samples, and the Nano-500 micro-spectrophotometer was employed to evaluate RNA purity. Following the manufacturer’s guidelines, RNA was reverse-transcribed into cDNA utilizing the ABScript III RT Master Mix (RK20429, ABclonal, China). RT-qPCR was then conducted utilizing the Genious 2X SYBR Green Fast RT-qPCR Mix (RK21205, ABclonal, China) to ascertain the expression levels of biomarkers. Relative gene expression normalized to GAPDH was calculated utilizing the 2−ΔΔCt approach. Primer sequences are enumerated in Table 1.
 |
Table 1 Sequences of Primers for Prognostic Genes |
Western Blotting (WB)
Proteins were extracted from 3 BC and 3 control tissue samples using RIPA buffer (P0013B, Beyotime Biotechnology, China) and 100×PSMF protease inhibitor (CR2404006, Servicebio, China). Protein samples in equal quantities were applied to SDS-PAGE and then transferred onto a PVDF membrane (0000279048, Millipore, USA). The membrane was treated with 5% skim milk for blocking, followed by incubation with the primary antibody at 4°C overnight. Following this, the secondary antibody was utilized, and protein bands were detected utilizing a chemiluminescence reagent. Protein bands were quantified using ImageJ software.
Statistical Analysis
All analyses were conducted utilizing R software (v 4.2.2). The R packages for visualizing DEGs include the “ggplot2” package (v 3.4.1)37 and “pheatmap” package (v 0.7.7).38 Group differences were assessed utilizing the Wilcoxon test. P < 0.05 was deemed statistically significant.
Results
Identification and Analysis of DE-TMRGs
An analysis of gene expression levels in tumor samples versus control samples from the GDC TCGA Breast Cancer dataset revealed a total of 6289 DEGs. Of these, 3640 genes exhibited increased expression, while 2649 genes showed decreased expression (Figures 1A and Figure S1). Subsequently, the intersection of the above-mentioned DEGs and 411 TMRGs was taken, resulting in 81 overlapping genes, which were named DE-TMRGs (Figure 1B). Then, GO and KEGG enrichment analyses were executed to further analyze the molecular functions and action mechanisms of these DE-TMRGs. In the GO analysis, it was found that from the perspective of BP, 623 entries were significantly enriched, such as DNA recombination, DNA-templated DNA replication, and recombinational repair; from the aspect of CC, 38 entries were significantly enriched, covering chromosomal region, chromosome, telomeric region, and transcription regulator complex; from the perspective of MF, 69 entries were significantly enriched, including catalytic activity and DNA helicase activity (Figure 1C). After that, 25 KEGG pathways were found to be significantly enriched, specifically including the Fanconi anemia pathway, DNA replication, and Cell cycle, etc (Figure 1D).
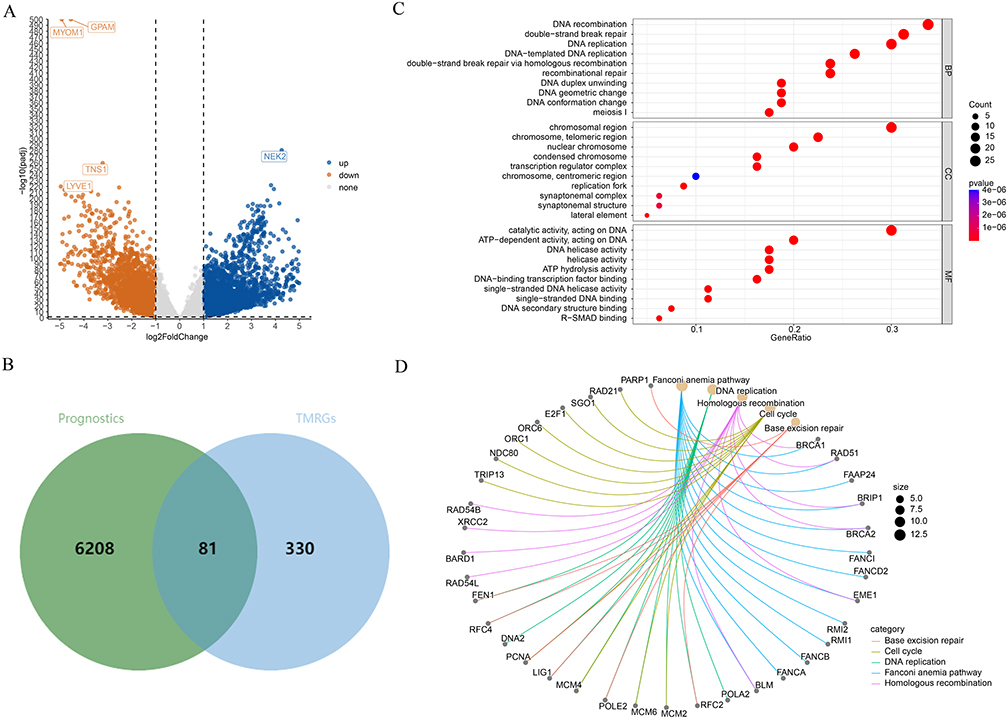 |
Figure 1 Identification and functional enrichment analysis of DE-TMRG. (A) Volcano plots of differently expressed genes (DEGs) in BC and control. (B) Venn diagram showing the intersection of DEGs with telomere maintenance related genes (TMRGs) to identify DE-TMRG. (C and D) Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses highlight the biological functions and signaling pathways enriched among these DE-TMRG. |
Creation of a Risk Model Based on Five Prognostic Genes
After the identification of 81 DE-TMRGs, these genes were incorporated into a univariate Cox regression analysis. Through this analysis, six genes that showed significant correlations with survival were screened out (Figure 2A). Among them, five genes, namely WT1, TPRXL, RAD54B, JUN, and SEPHS2, passed the PH assumption test, and were therefore further incorporated into the LASSO analysis. Ultimately, five prognostic genes were determined, namely, WT1, TPRXL, RAD54B, JUN, and SEPHS2 (Figure 2B). The risk scores for each BC patient were determined using the expression levels of these five prognostic genes. The optimal cut-off threshold (median) of the training set was 2.303, and that of the validation set was 7.626. Therefore, we classified patients above the critical value as the high-risk group and those below the critical value as the low-risk group (Figure S1). The K-M survival analysis indicated a significant difference in survival rates between two risk categories. (Figure 2C). Specifically, the mortality rate of patients in the high-risk category was higher. The AUC values of the ROC curve at 3 years, 5 years, and 7 years were 0.60, 0.62, and 0.65, respectively (Figure 2D). These results displayed that the prognostic risk model constructed with these five prognostic genes can effectively predict the mortality status of BC patients, and the performance of the prognostic risk model is relatively good. This result was further verified in the validation set (Figure 2E and F).
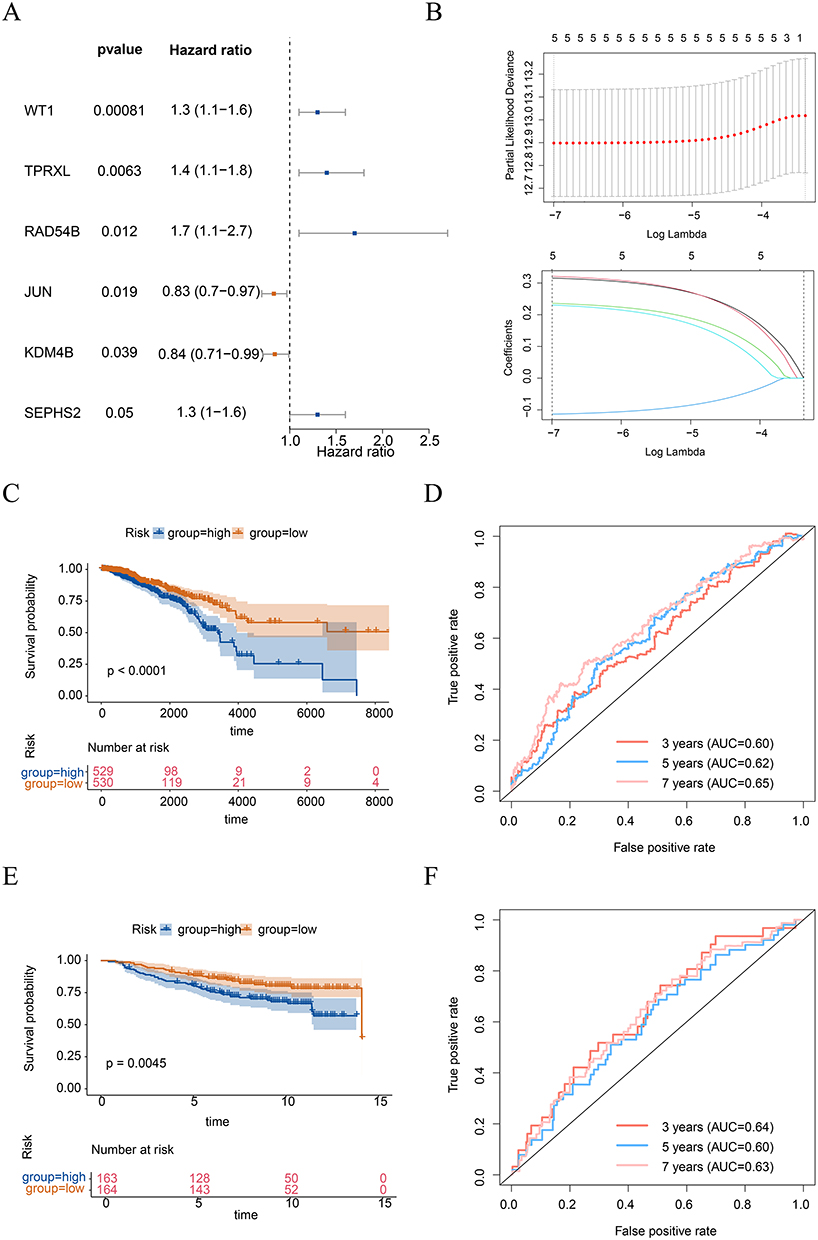 |
Figure 2 Identification of prognostic genes and construction risk model. (A) Univariate cox regression analysis to screen for survival-related genes. (B) Least Absolute Shrinkage and Selection Operator (LASSO) regression analysis applied to survival-related genes identified. Above: The x-axis represented the log (Lambda), the y-axis denoted the cross-validation error. Deviance signified the proportion of residuals explained by the model. Below: The x-axis was the log (Lambda), and the y-axis represented the coefficients of the genes. The curves depicted the different genes, including WT1, TPRXL, RAD54B, JUN, and SEPHS2. Kaplan-Meier (K-M) survival curves depicting the differences in overall survival (OS) between high-risk and low-risk groups in training (C) and validation (E) cohorts. Receiver operating characteristic (ROC) curves of the risk model, with the calculation of area under the curve (AUC) for 3, 5, and 7 years in training (D) and validation (F) cohorts. |
Association Between Clinical Characteristics and Risk Scores
The following analysis focused on examining the correlation between clinical characteristics and risk scores. In BC patients, significant differences in risk scores were observed across various T stages. (Figure 3A). To assess whether the risk model and clinical characteristics of BC patients could independently predict prognosis, we conducted a series of analyses. Initially, we performed univariate Cox regression analysis incorporating various clinical factors and risk groups. The results indicated that all included factors were significantly correlated with prognosis (P < 0.01) (Figure 3B). We then employed the stepwise selection method to develop a multivariate Cox regression model. This analysis revealed that stage, age, and risk score were significant independent prognostic factors (Figure 3C). Based on these results, we created a nomogram to predict the 3-year, 5-year, and 7-year survival rates of BC patients (Figure 3D). To assess the accuracy of this nomogram, we utilized a calibration curve, which showed a slope close to 1, indicating strong predictive capability (Figure 3E).
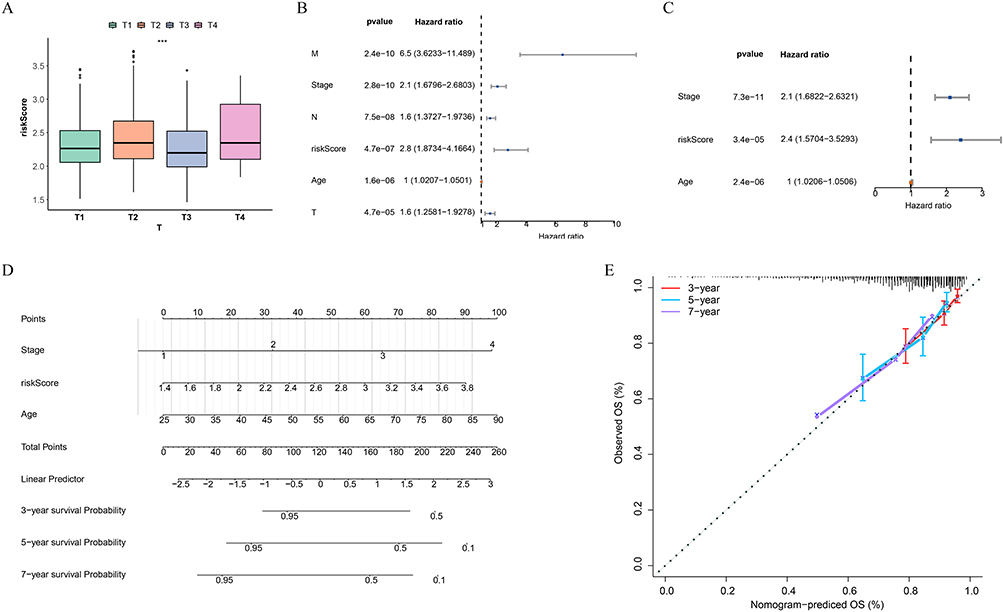 |
Figure 3 Relationship between risk scores and clinical characteristics and construction of nomogram. (A) Differences in risk scores between different subgroups of T staging. ***P < 0.001. (B and C) Univariate and Multivariate Cox regression analysis to identify independent prognostic factors. (D) Construction of a nomogram based on the identified independent prognostic factors and risk scores. (E) Calibration curve for the nomogram, assessing the accuracy of the model predictions. |
Functional Relevance of Distinct Risk Groups in BC
Subsequently, the enrichment analysis was conducted to further explore the pathways involved in the two risk groups. The GSEA showed that the cell cycle, oocyte meiosis, DNA replication, and proteasome pathways were activated in the high-risk category, while the dilated cardiomyopathy pathway was inhibited in the high-risk category (Figure 4A). The GSVA further emphasized the pathways with differences between the two risk categories, including MTORC1 signaling, E2F targets, myogenesis, etc (Figure 4B).
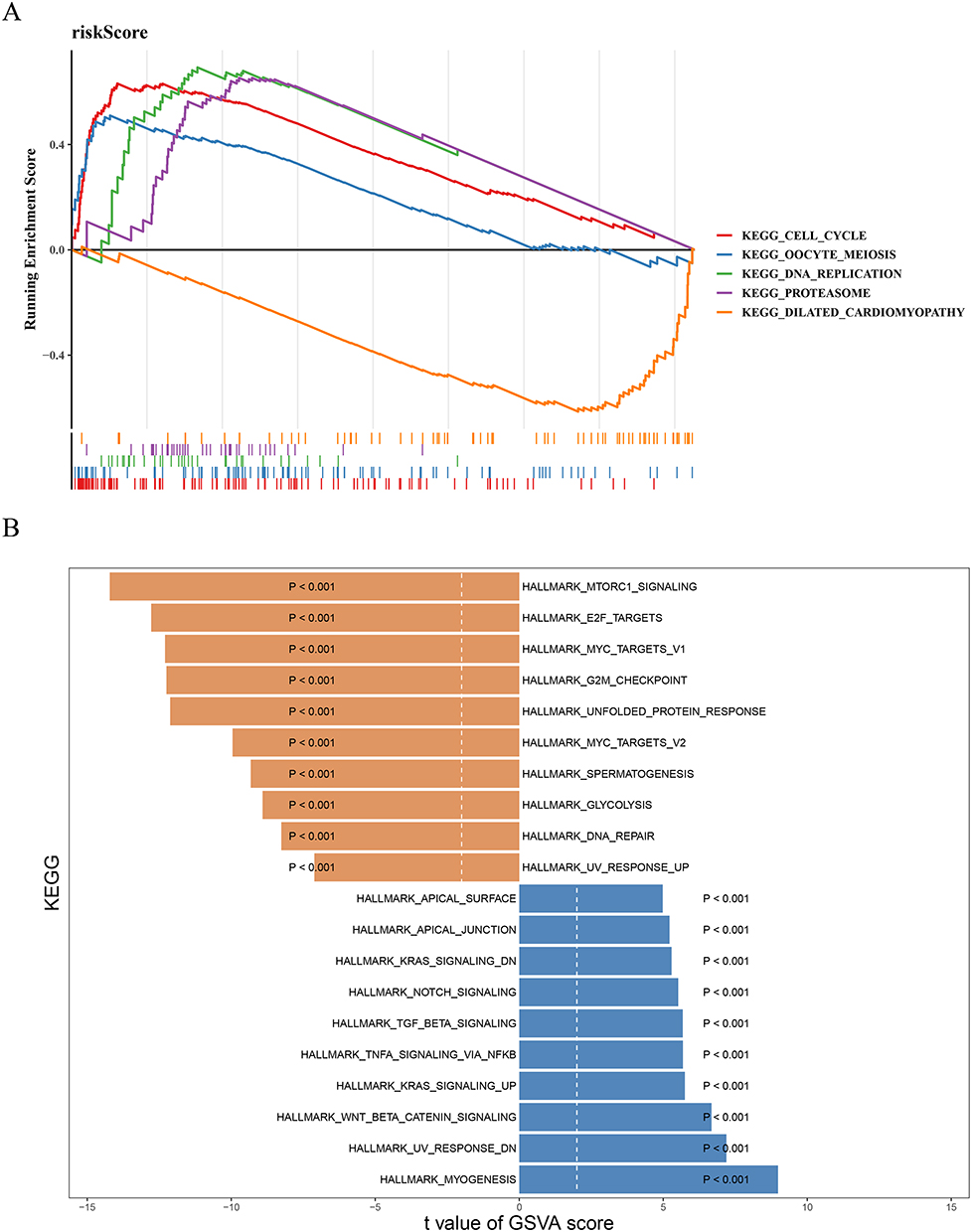 |
Figure 4 Enrichment analysis. (A) Gene set enrichment analysis (GSEA) of high and low risk groups. (B) GSVA reveals pathway differences between high and low risk groups. |
Multifaceted Analysis of the Immune Microenvironment Variations Among Patients with Differing Risk Levels
We then analyzed the differences in the immune microenvironment across patients with varying risk levels. The bar chart illustrated the abundance of 28 types of immune cells in the two risk categories (Figure 5A). The box plot further presented the significant differences in 14 types of immune cells between the two groups (P < 0.05) (Figure 5B). In terms of immune cell infiltration levels, the high-risk category exhibited significantly higher levels of Central memory CD8 T cells, Activated CD4 T cells, and Gamma delta T cells. Conversely, the low-risk category showed heightened infiltration of Central memory CD4 T cells, Activated B cells, and Effector memory CD4 T cells. Additionally, correlation study indicated that JUN had the highest positive correlation with Eosinophils (cor = 0.41, P < 0.05), while RAD54B displayed the highest negative correlation with Eosinophils (cor = -0.39, P < 0.05) (Figure 5C).
 |
Figure 5 Immune analysis. (A) Histogram depicting the abundance of 22 types of immune cells in high and low risk groups. (B) Box plot visualizing the differences in immune cell infiltration abundance between high and low risk groups for 22 immune cell types. *P < 0.05, **P < 0.01, ****P < 0.0001, ns P > 0.05. (C) Heatmap of the correlation between prognostic genes and differential immune cells. (D) Box line plot of Stromalscore and Estimate Score for high and low risk groups. **P < 0.01, ****P < 0.0001. (E) Box plot of differences in immune checkpoints between high and low risk groups. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns P > 0.05. |
The ESTIMATE analysis revealed that the Stroma Score and Estimate Score of patients in the low-risk category were higher (P < 0.05) (Figure 5D). This implied that in the low-risk category, the stromal environment around the tumor tissue might be relatively more conducive to the body’s immune defense against the tumor. Furthermore, it was found that the expression levels of 19 immune checkpoint molecules differed significantly between the two risk categories (P < 0.05), and most of these molecules had higher expression levels in the high-risk category (Figure 5E). This might be because the immune system of high-risk patients was relatively weak in attacking tumors, so more immune checkpoint molecules were needed to regulate the immune reaction to maintain the relative stability of the immune system.
Regulatory Network Construction and Drug Sensitivity Analysis
After conducting the prediction analysis, it was found that JUN could predict 51 miRNAs, comprising hsa-miR-200b-3p, hsa-miR-200c-3p, and hsa-miR-137. SEPHS2 was able to predict 30 miRNAs, such as hsa-miR-19a-3p, hsa-miR-186-5p, and hsa-miR-19b-3p. Additionally, RAD54B predicted one miRNA, specifically hsa-miR-186-5p, but no lncRNAs were identified. In contrast, TPRXL and WT1 did not predict any miRNAs during this analysis, which precluded their inclusion in the subsequent network construction. Based on the above prediction situations, we successfully constructed an lncRNA-miRNA-mRNA network, which contained 87 nodes and 98 edges, presenting complex interaction relationships among the nodes and edges (Figure S3A). To understand how chemical substances affect the expression and function of genes, we conducted predictions in the CTD database. The results displayed that the JUN was predicted to be associated with 18 compounds; the RAD54B was predicted to be associated with 7 compounds; the SEPHS2 was predicted to be associated with 6 compounds; the TPRXL was predicted to be associated with 2 compounds; and the WT1 was predicted to be associated with 14 compounds. Among these predicted compounds, Benzo(a)pyrene, Arsenic Trioxide, bisphenol A, titanium dioxide, Valproic Acid, CGP 52608, etc. were found to have a targeting relationship with the prognostic genes (Figure S3B).
The results obtained from the drug sensitivity analysis showed that among the drugs ranked in the top ten in terms of significance, the IC50 for each drug was reduced in the high-risk category. This situation might imply that patientsclassified as high-risk category possess a relatively high intrinsic sensitivity to chemotherapy drugs, which was related to the biological characteristics of the tumor, and might also predict a relatively good short-term prognosis for the patients (Figure S3C).
JUN and RAD54B Associated with Elevated Risk of BC
After a rigorous screening process, it was found that no corresponding eQTL GWAS data could be retrieved for WT1 and TPRXL. In view of this situation, we focused on exploring the possible causal relationships between RAD54B, JUN, and SEPHS2 and BC. The exploration results showed that JUN and RAD54B had significant causal relationships with BC, and both genes were identified as risk factors for BC (Table 2).
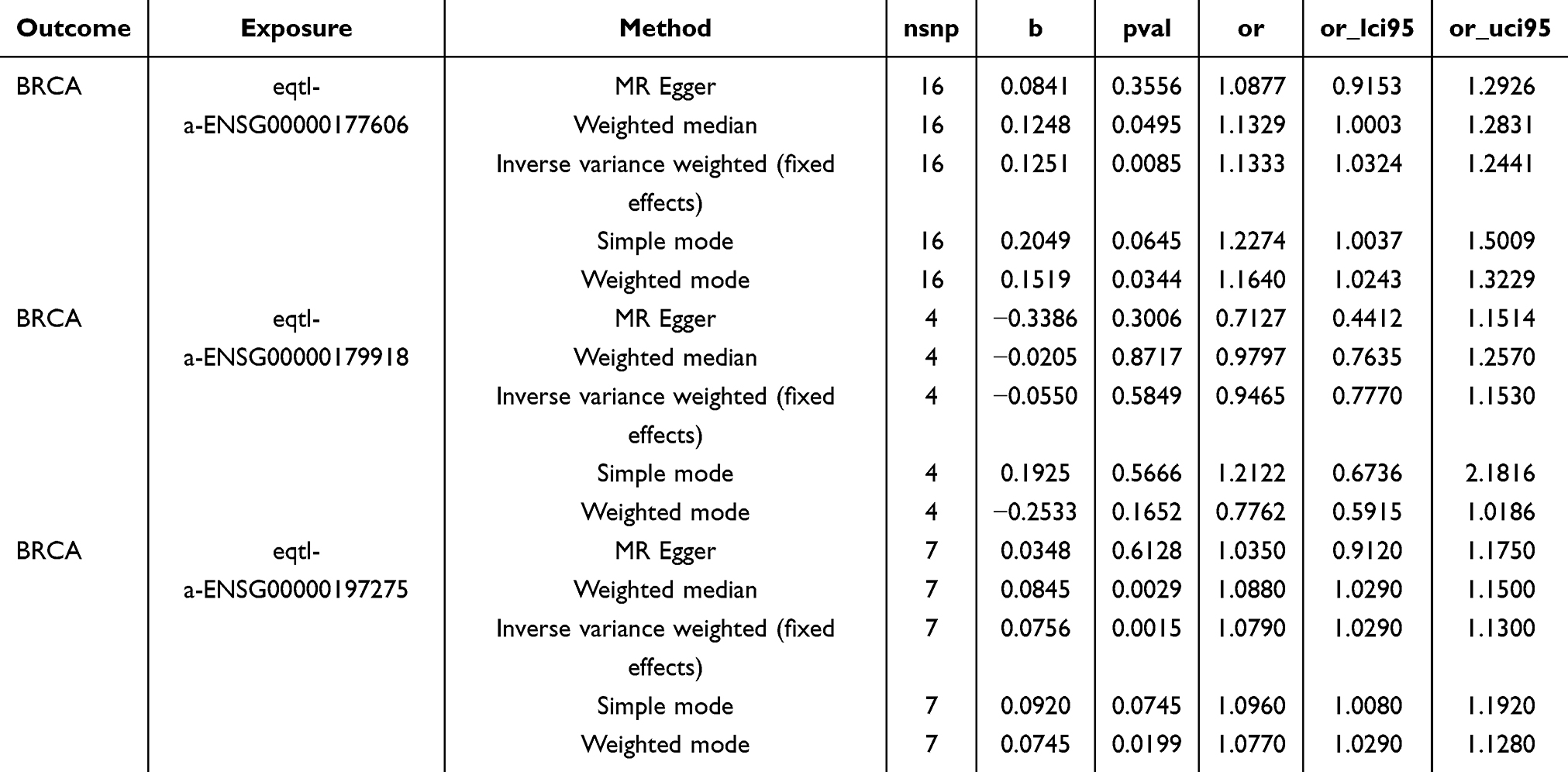 |
Table 2 The Result of MR Analysis for Prognostic Genes and BC |
To enhance the credibility of the aforementioned findings, a sensitivity analysis was conducted. Among them, the Cochran’s Q test showed that there was no heterogeneity in the results (P > 0.05) (Table S1); and horizontal pleiotropy analysis revealed no confounding factors that could affect the outcomes (P > 0.05) (Table S2). Ultimately, the analysis revealed that gradually removing each SNP did not significantly alter the effect of the remaining SNPs on the outcome variable (Figure S4). This result showed that the results obtained from this MR analysis were reliable and had good stability.
Single-Cell Analysis Revealed Expression of Prognostic Genes
Through a rigorous quality control process, the data was standardized (Figure S5A). Based on this, the top 2000 highly variable genes were carefully chosen from all the genes (Figure S5B). These genes exhibited relatively significant changes in their expression levels and could provide rich and valuable information for subsequent in-depth analysis. Subsequently, in order to effectively reduce the data dimension without losing key information, the principal component analysis method was adopted. Through comprehensive consideration and complex calculations of the data characteristics, the top 30 principal components were finally selected from numerous principal components for further in-depth analysis (Figure S5C). Next, we used the unsupervised clustering analysis method to process the data, and finally obtained 25 cell clusters successfully (Figure 6A). Based on the characteristics of a series of marker genes, these 25 cell clusters were further annotated as 8 cell types, namely T/NK cells, B cells, Endothelial, Epithelial, Fibroblasts, Mononuclear phagocyt, Monocytes, and Pericytes (Figure 6B). Among them, the percentage distribution of Pericytes varied greatly between BC and control groups (Figure 6C). Based on this, we further explored the distribution of prognostic genes in each of the above cell types. After detailed analysis and statistics, we found that the distribution of the JUN on various cells showed a relatively obvious bias, with a relatively large number of distributions on the cells. However no expression of TPRXL was found in the annotated cells (Figure 6D). This finding might imply that the JUN may play a relatively important role in cell-related physiological processes and the mechanism of disease occurrence and development.
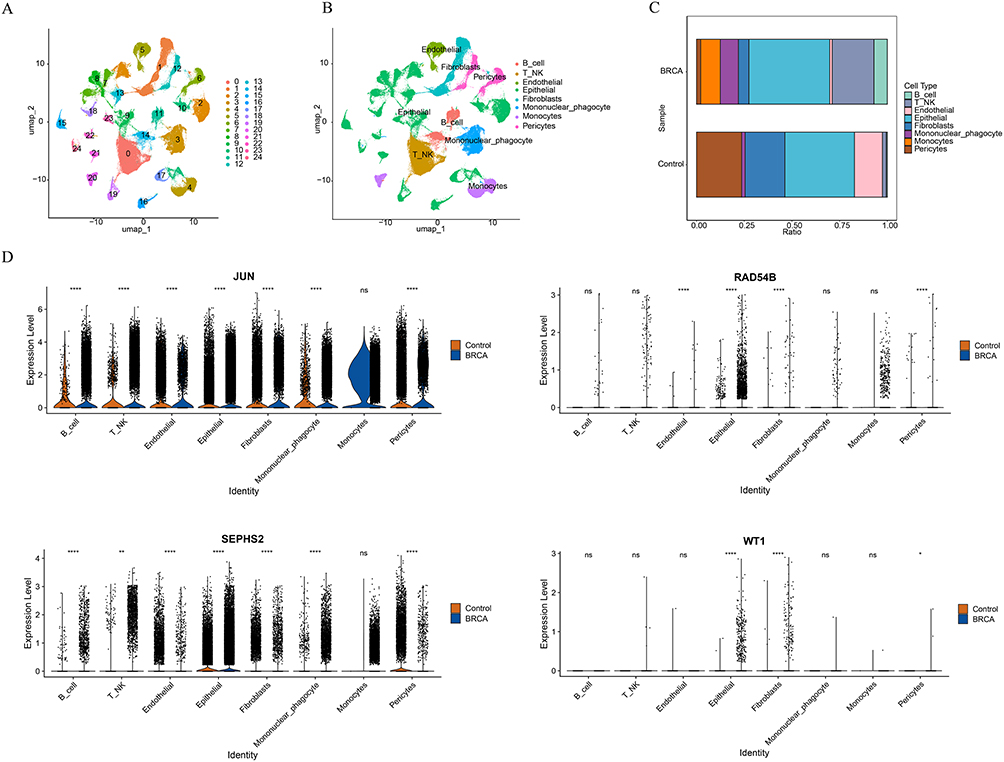 |
Figure 6 Single-cell analysis. (A) UAMP plot of cell annotation after single-cell downscaling clustering. (B) Annotated distribution map of tSNE cells. (C) Plot of the proportion of annotated cells between BC and controls. (D) Expression of prognostic genes in different annotated cells. *P < 0.05, **P < 0.01, ****P < 0.0001, ns P > 0.05. |
In addition, the findings from the cell communication analysis indicated that, in comparison to the control group, the intercellular communication in BC samples showed a significant increase (Figure 7A and B). This enhancement of intercellular communication might have an important impact on many biological behaviors of tumor cells. Finally, through the pseudotemporal analysis, we also discovered an interesting phenomenon. In BC samples, most of the samples were mainly distributed in the late stage of pericytes differentiation (Figure 7C). Moreover, the JUN was expressed both in the early and late stages of pericytes differentiation (Figure 7D). This phenomenon suggested that the JUN gene may be involved in the whole process of pericytes differentiation.
 |
Figure 7 Pseudo-temporal analyses and cellular communication analysis. (A and B) Plot of the number and strength of interactions for each type of cell linkage in BC and control group. (C) Map of pericytes differentiation trajectories. (D) Map of prognostic genes in the distribution of differentiation trajectories. |
Validation of the Expression of Prognostic Genes
To further corroborate the findings of the bioinformatics study, we successfully collected BC patient tissue samples and their adjacent normal tissues. RT-qPCR results demonstrated significant expression differences of five prognostic genes between the BC and control groups (Figure 8A). RAD54B, SEPHS2, and WT1 were highly expressed in the BC group, while TPRXL and JUN exhibited the opposite expression pattern. WB results further confirmed this finding, showing high expression of RAD54B and SEPHS2 in the BC group, while JUN expression was low, with statistically significant differences (Figure 8B). The experimental results aligned with the outcomes of the bioinformatics analysis.
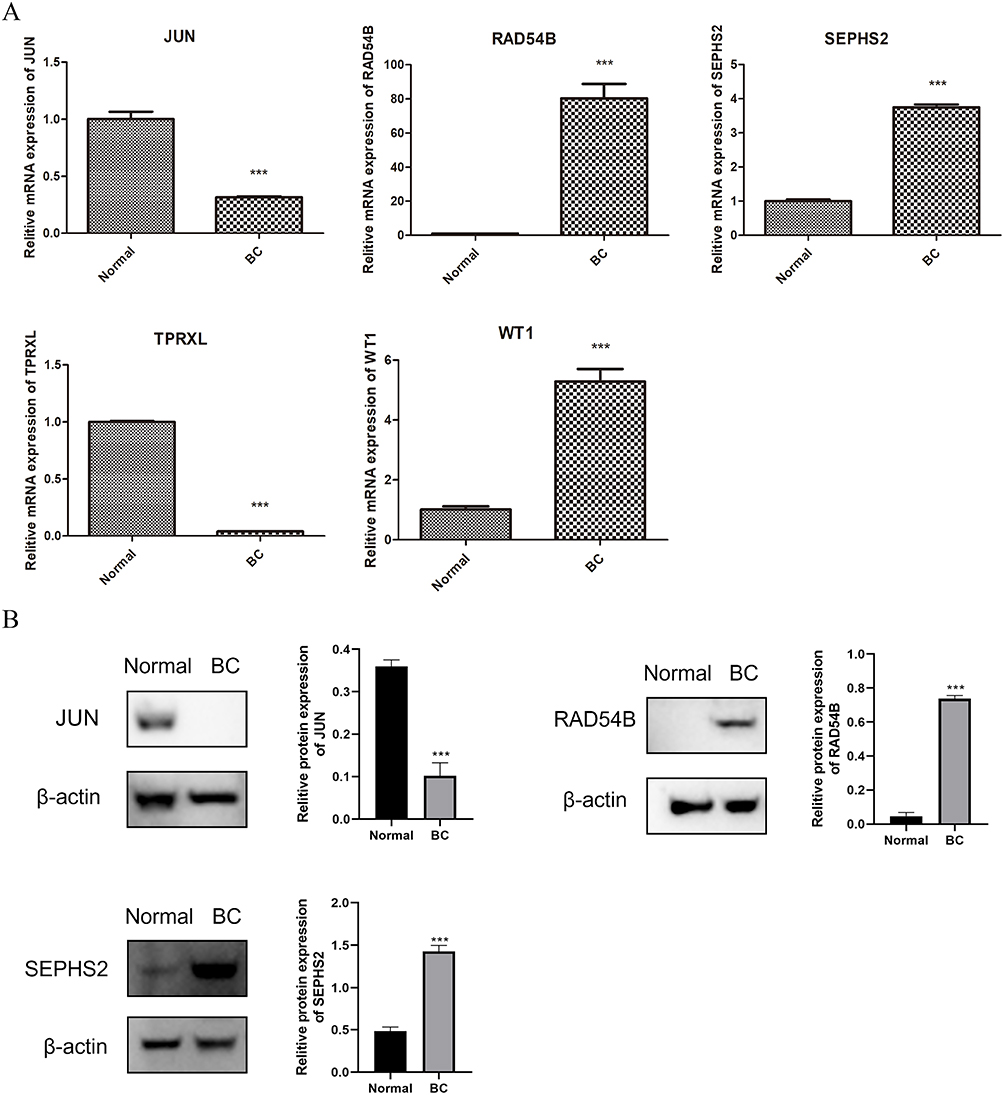 |
Figure 8 The expression of prognostic genes in clinic samples. (A) The result of RT-qPCR. (B) The result of Western blotting. ***P < 0.001. |
Discussion
The development of BC is intricately linked to telomere maintenance mechanisms. Research has indicated that in most BC, telomerase activity is reactivated, enabling cancer cells to acquire unlimited proliferative capacity.39 Furthermore, irregularities in telomere maintenance can act as early markers for forecasting how BC patients will respond to adjuvant therapy and as possible therapeutic targets.40 Research on telomere maintenance mechanisms has provided new directions for BC treatment. For instance, targeting telomerase or alternative lengthening of telomeres (ALT) pathways offers opportunities to develop specific therapeutic strategies for BC.41 These results highlight the essential role of telomere maintenance in the onset and development of BC. However, the specific target genes related to telomere maintenance in BC therapy remain unclear. In this study, we utilized the UCSC Xena database to establish a prognostic model based on TMRGs, providing new insights for further exploration of BC mechanisms and precision treatment strategies.
We conducted the first systematic study of TMRGs associated with BC prognosis. Using data from public databases, we identified five prognostic genes (WT1, TPRXL, RAD54B, JUN, and SEPHS2) to construct a risk model. WT1 is a classical tumor suppressor gene closely associated with Wilms tumor, a pediatric kidney cancer. It encodes a zinc finger transcription factor involved in regulating various biological processes.42 Jablonowski et al found that the rate-limiting step of telomerase activation is the transcription of TERT, and the knockdown and knockout of WT1 expression are associated with the reduction of TERT transcription.43 In recent years, the function of WT1 and its potential mechanisms in BC have garnered significant attention. Research has indicated that WT1 expression is closely associated with increased invasiveness and poor prognosis in BC.44 A histopathological analysis further verified that WT1 protein levels are notably elevated in breast tumor tissues than in normal tissues,45 which is consistent with our results. Additionally, overexpression of WT1 in BC cells significantly promotes cell proliferation and invasion, suggesting that WT1 may drive cancer cell growth via modulating cell cycle-associated genes such as Cyclin D1.46 Notably, WT1 expression is also closely associated with molecular subtypes of BC. Research has demonstrated that WT1 overexpression occurs across all molecular subtypes of BC and is significantly linked to poorer survival outcomes.47 Moreover, the methylation state of the WT1 promoter region could be crucial in the onset and development of BC. Studies have indicated that this methylation event is common during the early stages of BC.48 In summary, the multifaceted roles of WT1 in BC suggest that it could be an important therapeutic target with significant potential.
As a significant member of the AP-1 family, JUN is a transcription factor essential for cell proliferation, differentiation, and apoptosis.49 In cancer research, abnormal expression of JUN has been closely linked to the initiation and advancement of multiple cancers. Studies have shown that JUN may indirectly affect telomere stability by influencing DNA repair and signaling pathways, such as JNK signaling.50 In BC, the mechanisms involving JUN have become a key focus of research. Studies have shown that aberrant expression of JUN is strongly correlated with BC development.51 Zhang et al reported that overexpression of JUN may contribute to BC metastasis.52 It is worth noting that, as shown in this study, the expression level of JUN in BC tissues was lower than that in adjacent tissues. This might be because the expression of the JUN gene is regulated by multiple factors, including post-transcriptional regulation, post-translational modification, and the influence of the cellular microenvironment. Recent studies have indicated that miR-10b can up-regulate the expression of c-Jun by regulating the RhoC/NF1 pathway. If the expression level of miR-10b in the patient sample decreases, it will lead to a decrease in the level of c-Jun.53 Furthermore, although most literature reports that c-Jun is highly expressed in BC, some studies have observed that the expression of JNK (C-Jun N-terminal kinase) in BC tissues is lower than that in adjacent normal tissues.54 Therefore, in the future, it is necessary to further verify the expression pattern of JUN and its exact biological function in larger sample cohorts by combining different subtypes of BC. RAD54B is a key gene in the homologous recombination repair pathway, and its mutations can lead to defects in DNA repair functions, thereby promoting tumorigenesis. DNA damage repair mechanisms have attracted significant attention in studies on radiosensitization of BC. As a core protein in homologous recombination repair, dysfunction of RAD54B may significantly affect the sensitivity of BC cells to radiotherapy. Earlier research has shown that reducing RAD54B expression can increase the radiosensitivity of BC cells, leading to better treatment results.55 Furthermore, RAD54B shows unusual expression levels in different types of cancer. Studies indicate that RAD54B is overexpressed in hepatocellular carcinoma cell lines and is linked to unfavorable outcomes. Suppression of RAD54B expression not only significantly inhibits the proliferation and colony formation of hepatocellular carcinoma cells but also impacts tumor progression by modulating the Wnt/β-catenin signaling pathway.56,57 The telomeres of Rad54-deficient mice are significantly shorter than those of wild-type controls, indicating that Rad54 activity plays a crucial role in maintaining telomere length in mammals.58 In our study, experimental validation revealed high expression levels of RAD54B in BC tissues, suggesting that it may participate in BC development through similar mechanisms. SEPHS2 is a selenoprotein involved in a critical step of tRNA aminoacylation.59 Studies have indicated that this gene is highly expressed in triple-negative BC (TNBC),60 which aligns with our findings. However, its specific role and mechanisms in BC remain to be further elucidated. On the other hand, research on TPRXL in BC remains limited. Qian et al found that this gene is closely associated with the prognosis of BC patients,61 corroborating our findings. Additionally, we observed low expression levels of TPRXL in BC. However, its specific function and underlying mechanisms require further investigation. At present, there are relatively few studies on SEPHS2 and TPRXL in the field of telomere maintenance, which also provides a direction for our future exploration. In summary, these prognostic genes may play significant roles in the initiation and progression of BC and merit further exploration for their potential clinical applications as therapeutic targets.
The advancement of BC is affected by multiple factors. In this research, we created a strong risk model that accurately assesses the survival risk and prognosis for BC patients. Similarly, Tian et al constructed a prognostic risk model based on extracellular matrix-related genes in BC using a comparable approach.62 Regarding the association between clinical characteristics and risk scores, our research identified notable variations in risk scores among BC patients at different T stages, suggesting a possible connection between risk scores and both tumor size and local invasion. T stage reflects the extent of tumor progression; higher T stages are associated with increased invasiveness and metastatic potential.63 These findings suggest that the risk model may capture tumor biological behavior and aggressiveness. In summary, the risk model developed in this study provides new perspectives and tools for BC prognosis assessment. However, further optimization of the model, validation with larger cohorts, and exploration of interaction mechanisms among various factors are needed to better guide clinical practice in the future.
Additionally, we conducted a comprehensive analysis of the immune microenvironment in BC patients across various risk levels. The results revealed that the infiltration levels of activated CD4 T cells, central memory CD8 T cells, and γδ T cells were significantly elevated in the high-risk group. This may reflect tumor-induced metabolic reprogramming or secretion of pro-inflammatory factors, leading to T cell exhaustion or a shift toward pro-tumor functions.64,65 Furthermore, ESTIMATE analysis showed that Stromal Score and Estimate Score were significantly higher in the low-risk group. This indicates that the tumor stromal environment in low-risk patients may be more conducive to immune defense against tumors. These results imply that the tumor microenvironment in the low-risk group might prevent tumor growth by encouraging immune cell infiltration or preserving antigen presentation functions.66 On the other hand, the expression levels of 19 immune checkpoint molecules were significantly elevated in the high-risk group. This likely reflects an immunosuppressive characteristic of the tumor microenvironment in high-risk patients. Overexpression of immune checkpoint molecules is often considered a key mechanism by which tumors evade immune surveillance.
Single-cell analysis revealed the expression patterns of prognostic genes within the BC microenvironment and their distribution across specific cell types. We annotated eight major cell types, among which pericytes showed significant differences in proportional distribution between BC and normal control groups, suggesting their critical role in the tumor microenvironment. Pericytes are essential contributors to angiogenesis, and their dysfunction is closely associated with disrupted vascular homeostasis, invasion, and metastasis.67 Further analysis revealed that JUN, a prognostic gene identified in our study, was expressed across multiple cell types and throughout all stages of pericyte differentiation from early to late phases. This indicates that JUN may regulate pericyte differentiation or function to facilitate tumor angiogenesis and thereby promote BC progression. Pseudotime analysis demonstrated that pericytes in BC samples were predominantly at late differentiation stages, potentially linked to abnormal angiogenesis and enhanced tumor-supporting invasive capabilities. Moreover, significantly enhanced intercellular communication observed in BC samples highlighted the complexity of the tumor microenvironment and its critical impact on disease progression. These enhanced communications likely involve key signaling pathways such as TGF-β, VEGF, and Notch pathways, which have been shown to play pivotal roles in angiogenesis, immune evasion, and drug resistance in BC.68–70 This study underscores the central role of pericytes within the BC microenvironment and provides potential directions for targeted interventions. However, we acknowledge certain limitations in our research. A robust multi-gene signature model requires external validation across independent cohorts; thus, our proposed model needs further verification through multicenter clinical trials and prospective studies. Additionally, experimental validation at the biological level is necessary for genes included in our model to establish a foundation for clinical application.
Conclusion
In conclusion, we systematically analyzed telomere maintenance-related prognostic genes in BC and developed a robust risk model. This model not only demonstrated a significant association with immune infiltration but also provided an effective screening tool for the diagnosis and prognosis of BC. Furthermore, this study offers a novel approach to exploring the relationship between BC and telomere maintenance mechanisms, laying a foundation for future research and precision therapy.
Data Sharing Statement
The datasets generated during and/or analysed during the current study are available in the UCSC Xena database (https://xenabrowser.net/datapages/) and the GEO database (https://www.ncbi.nlm.nih.gov/gds), accession number: [GSE20685 and GSE161529]. The GWAS dataset of BC (ebi-a-GCST90018799) and the eQTL GWAS data were obtained from the IEU OpenGWAS database (https://gwas.mrcieu.ac.uk/).
Ethics Declarations
The guidelines outlined in the Declaration of Helsinki were followed. The study was approved by the Medical Ethics Committee of General Hospital of Taiyuan Iron and Steel (Group) Co., Ltd. (Ethics ID: k202502).
Acknowledgments
We are grateful to UCSC Xena database (https://xenabrowser.net/datapages/), GEO database (https://www.ncbi.nlm.nih.gov/gds), and IEU OpenGWAS database (https://gwas.mrcieu.ac.uk/) for providing data support, which had played a significant role in our research. We also thank General Hospital of Taiyuan Iron and Steel (Group) Co., Ltd. for providing clinical samples, which helped us complete the experimental part of this study. Without these valuable resources, this research could not have been carried out smoothly.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Nadia Harbeck, Frédérique Penault-Llorca, Javier Cortes, et al. Breast cancer. Nat Rev Dis Primers. 2019;5(1):67. doi:10.1038/s41572-019-0122-z
2. Nicholson WK, Silverstein M, Wong JB, et al; US Preventive Services Task Force. Screening for breast cancer: us preventive services task force recommendation statement. JAMA. 2024;331(22):1918. doi:10.1001/jama.2024.5534.
3. Cardoso MJ, Poortmans P, Senkus E, Gentilini OD, Houssami N. Breast cancer highlights from 2023: knowledge to guide practice and future research. Breast. 2024;74:103674. doi:10.1016/j.breast.2024.103674
4. De Boniface J, Filtenborg Tvedskov T, Rydén L, et al. Omitting axillary dissection in breast cancer with sentinel-node metastases. N Engl J Med. 2024;390(13):1163–1175. doi:10.1056/NEJMoa2313487
5. Sommer A, Royle NJ. ALT: a multi-faceted phenomenon. Genes. 2020;11(2):133. doi:10.3390/genes11020133
6. Hou K, Yu Y, Li D, et al. Alternative lengthening of telomeres and mediated telomere synthesis. Cancers. 2022;14(9):2194. doi:10.3390/cancers14092194
7. Fan HC, Chang FW, Tsai JD, et al. Telomeres and Cancer. Life. 2021;11(12):1405. doi:10.3390/life11121405
8. De Vitis M, Berardinelli F, Sgura A. Telomere length maintenance in cancer: at the crossroad between telomerase and alternative lengthening of telomeres (ALT). Int J Mol Sci. 2018;19(2):606. doi:10.3390/ijms19020606
9. Min J, Kim JY, Choi JY, Kong ID. Association between physical activity and telomere length in women with breast cancer: a systematic review. J Clin Med. 2022;11(9):2527. doi:10.3390/jcm11092527
10. Murillo-Ortiz BO, García-Corrales K, Martínez-Garza S, et al. Association of hTERT expression, Her2Neu, estrogen receptors, progesterone receptors, with telomere length before and at the end of treatment in breast cancer patients. Front Med. 2024;11:1450147. doi:10.3389/fmed.2024.1450147
11. Ennour-Idrissi K, Maunsell E, Diorio C. Telomere length and breast cancer prognosis: a systematic review. Cancer Epidemiol Biomark Prev. 2017;26(1):3–10. doi:10.1158/1055-9965.Epi-16-0343
12. Gao J, Pickett HA. Targeting telomeres: advances in telomere maintenance mechanism-specific cancer therapies. Nat Rev Cancer. 2022;22(9):515–532. doi:10.1038/s41568-022-00490-1
13. Falcinelli M, Dell’Omo G, Grassi E, et al. Colorectal cancer patient-derived organoids and cell lines harboring ATRX and/or DAXX mutations lack alternative lengthening of telomeres (ALT). Cell Death Dis. 2023;14(2):96. doi:10.1038/s41419-023-05640-3
14. Subhawong AP, Heaphy CM, Argani P, et al. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod Pathol. 2009;22(11):1423–1431. doi:10.1038/modpathol.2009.125
15. Heaphy CM, Subhawong AP, Hong SM, et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol. 2011;179(4):1608–1615. doi:10.1016/j.ajpath.2011.06.018
16. Poremba C, Heine B, Diallo R, et al. Telomerase as a prognostic marker in breast cancer: high-throughput tissue microarray analysis of hTERT and hTR. J Pathol. 2002;198(2):181–189. doi:10.1002/path.1191
17. Zou J, Chu S, Bao Q, Zhang Y. Telomere maintenance genes-derived prognosis signature characterizes immune landscape and predicts prognosis of head and neck squamous cell carcinoma. Medicine. 2023;102(31):e34586. doi:10.1097/MD.0000000000034586
18. Holysz H, Lipinska N, Paszel-Jaworska A, Rubis B. Telomerase as a useful target in cancer fighting—the breast cancer case. Tumor Biol. 2013;34(3):1371–1380. doi:10.1007/s13277-013-0757-4
19. Shen A, Garrett A, Chao CC, et al. A comprehensive meta-analysis of tissue resident memory T cells and their roles in shaping immune microenvironment and patient prognosis in non-small cell lung cancer. Front Immunol. 2024;15:1416751. doi:10.3389/fimmu.2024.1416751
20. Yan M, Zhang Z, Wang L, et al. Cross-talk of three molecular subtypes of telomere maintenance defines clinical characteristics and tumor microenvironment in gastric cancer. J Cancer. 2024;15(10):3227–3241. doi:10.7150/jca.92207
21. Mao J, Zhang Q, Wang Y, et al. TERT activates endogenous retroviruses to promote an immunosuppressive tumour microenvironment. EMBO Rep. 2022;23(4):e52984. doi:10.15252/embr.202152984
22. Moura T, Laranjeira P, Caramelo O, Gil AM, Paiva A. Breast cancer and tumor microenvironment: the crucial role of immune cells. Curr Oncol. 2025;32(3):143. doi:10.3390/curroncol32030143
23. Zhang J, Xia X, He S. Deciphering the causal association and underlying transcriptional mechanisms between telomere length and abdominal aortic aneurysm. Front Immunol. 2024;15:1438838. doi:10.3389/fimmu.2024.1438838
24. Armendáriz-Castillo I, López-Cortés A, García-Cárdenas J, et al. TCGA pan-cancer genomic analysis of alternative lengthening of telomeres (ALT) related genes. Genes. 2020;11(7):834. doi:10.3390/genes11070834
25. Liu Z, Liu X. Prognostic model of osteosarcoma based on telomere-related genes and analysis of immune characteristics. Int Immunopharmacol. 2025;151:114198. doi:10.1016/j.intimp.2025.114198
26. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
27. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
28. Li Y, Lu F, Yin Y. Applying logistic LASSO regression for the diagnosis of atypical Crohn’s disease. Sci Rep. 2022;12(1):11340. doi:10.1038/s41598-022-15609-5
29. Blanche P, Dartigues JF, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. 2013;32(30):5381–5397. doi:10.1002/sim.5958
30. Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4(1):2612. doi:10.1038/ncomms3612
31. Maeser D, Gruener RF, Huang RS. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021;22(6):bbab260. doi:10.1093/bib/bbab260
32. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587.e29. doi:10.1016/j.cell.2021.04.048
33. Qiu X, Hill A, Packer J, Lin D, Ma YA, Trapnell C. Single-cell mRNA quantification and differential analysis with Census. Nat Meth. 2017;14(3):309–315. doi:10.1038/nmeth.4150
34. Jin S, Guerrero-Juarez CF, Zhang L, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12(1):1088. doi:10.1038/s41467-021-21246-9
35. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. JAMA. 2021;326(16):1614. doi:10.1001/jama.2021.18236
36. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408. doi:10.7554/eLife.34408
37. Gustavsson EK, Zhang D, Reynolds RH, Garcia-Ruiz S, Ryten M. ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics. 2022;38(15):3844–3846. doi:10.1093/bioinformatics/btac409
38. Gu Z, Hübschmann D. Make interactive complex heatmaps in R. Bioinformatics. 2022;38(5):1460–1462. doi:10.1093/bioinformatics/btab806
39. Hannen R, Bartsch JW. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018;592(12):2023–2031. doi:10.1002/1873-3468.13084
40. Zhang Y, Luo S, Zhang X, Jia Y. Telomere maintenance mechanism dysregulation serves as an early predictor of adjuvant therapy response and a potential therapeutic target in human cancers. Int J Cancer. 2022;151(2):313–327. doi:10.1002/ijc.34007
41. Yildirim İ, Biray Avci Ç. Telomerase inhibition in breast cancer and breast cancer stem cells: a brief review. Med Oncol. 2024;42(1):14. doi:10.1007/s12032-024-02562-8
42. Zhang Y, Yan WT, Yang ZY, et al. The role of WT1 in breast cancer: clinical implications, biological effects and molecular mechanism. Int J Biol Sci. 2020;16(8):1474–1480. doi:10.7150/ijbs.39958
43. Jablonowski CM, Gil HJ, Pinto EM, et al. TERT expression in Wilms tumor is regulated by promoter mutation or hypermethylation, WT1, and N-MYC. Cancers. 2022;14(7):1655. doi:10.3390/cancers14071655
44. Qi XW, Zhang F, Yang XH, et al. High Wilms’ tumor 1 mRNA expression correlates with basal-like and ERBB2 molecular subtypes and poor prognosis of breast cancer. Oncol Rep. 2012;28(4):1231–1236. doi:10.3892/or.2012.1906
45. McGregor RJ, Chau YY, Kendall TJ, Artibani M, Hastie N, Hadoke PWF. WT1 expression in vessels varies with histopathological grade in tumour-bearing and control tissue from patients with breast cancer. Br J Cancer. 2018;119(12):1508–1517. doi:10.1038/s41416-018-0317-1
46. Caldon CE, Lee CSL, Sutherland RL, Musgrove EA. Wilms’ tumor protein 1: an early target of progestin regulation in T-47D breast cancer cells that modulates proliferation and differentiation. Oncogene. 2008;27(1):126–138. doi:10.1038/sj.onc.1210622
47. Ben Haj Othmen H, Othman H, Khamessi O, Bettaieb I, Gara S, Kharrat M. Overexpression of WT1 in all molecular subtypes of breast cancer and its impact on survival: exploring oncogenic and tumor suppressor roles of distinct WT1 isoforms. Mol Biol Rep. 2024;51(1):544. doi:10.1007/s11033-024-09450-4
48. Moelans CB, Verschuur‐Maes AH, Van Diest PJ. Frequent promoter hypermethylation of BRCA2, CDH13, MSH6, PAX5, PAX6 and WT1 in ductal carcinoma in situ and invasive breast cancer. J Pathol. 2011;225(2):222–231. doi:10.1002/path.2930
49. Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem. 2014;57(16):6930–6948. doi:10.1021/jm5004733
50. Kusuma FK, Prabhu A, Tieo G, et al. Signalling inhibition by ponatinib disrupts productive alternative lengthening of telomeres (ALT). Nat Commun. 2023;14(1):1919. doi:10.1038/s41467-023-37633-3
51. Baan B, Pardali E, Ten Dijke P, Van Dam H. In situ proximity ligation detection of c-Jun/AP-1 dimers reveals increased levels of c-Jun/Fra1 complexes in aggressive breast cancer cell lines in vitro and in vivo. Mol Cell Proteomics. 2010;9(9):1982–1990. doi:10.1074/mcp.M110.000943
52. Zhang Y, Pu X, Shi M, et al. Critical role of c-Jun overexpression in liver metastasis of human breast cancer xenograft model. BMC Cancer. 2007;7(1):145. doi:10.1186/1471-2407-7-145
53. Knirsh R, Ben-Dror I, Modai S, Shomron N, Vardimon L. MicroRNA 10b promotes abnormal expression of the proto-oncogene c-Jun in metastatic breast cancer cells. Oncotarget. 2016;7(37):59932–59944. doi:10.18632/oncotarget.11000
54. Huang S, Wang C, Lv Y, Liu Y, Ma J, Wang X. Correlation of expression of WWOX and JNK with clinicopathologic features in human breast carcinoma. Int J Clin Exp Patho. 2018;11(2):695–703.
55. Zhou Z, Yang Z, Yu X, Guo X. Highlights on molecular targets for radiosensitization of breast cancer cells: current research status and prospects. Cancer Med. 2018;7(7):3110–3117. doi:10.1002/cam4.1588
56. Wang R, Li Y, Chen Y, et al. Inhibition of RAD54B suppresses proliferation and promotes apoptosis in hepatoma cells. Oncol Rep. 2018;40(3):1233–1242. doi:10.3892/or.2018.6522
57. Feng S, Liu J, Hailiang L, et al. Amplification of RAD54B promotes progression of hepatocellular carcinoma via activating the Wnt/β-catenin signaling. Transl Oncol. 2021;14(8):101124. doi:10.1016/j.tranon.2021.101124
58. Jaco I, Muñoz P, Goytisolo F, et al. Role of Mammalian Rad54 in telomere length maintenance. Mol Cell Biol. 2003;23(16):5572–5580. doi:10.1128/MCB.23.16.5572-5580.2003
59. Vindry C, Guillin O, Wolff P, et al. A homozygous mutation in the human selenocysteine tRNA gene impairs UGA recoding activity and selenoproteome regulation by selenium. Nucleic Acids Res. 2023;51(14):7580–7601. doi:10.1093/nar/gkad482
60. Nunziata C, Polo A, Sorice A, et al. Structural analysis of human SEPHS2 protein, a selenocysteine machinery component, over-expressed in triple negative breast cancer. Sci Rep. 2019;9(1):16131. doi:10.1038/s41598-019-52718-0
61. Qian L, Li L, Li Y, et al. LncRNA HOTAIR as a ceRNA is related to breast cancer risk and prognosis. Breast Cancer Res Treat. 2023;200(3):375–390. doi:10.1007/s10549-023-06982-4
62. Tian Q, Gao H, Ma Y, et al. The regulatory roles of T helper cells in distinct extracellular matrix characterization in breast cancer. Front Immunol. 2022;13:871742. doi:10.3389/fimmu.2022.871742
63. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17(6):1471–1474. doi:10.1245/s10434-010-0985-4
64. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. 2022;19(12):775–790. doi:10.1038/s41571-022-00689-z
65. Ahn T, Bae EA, Seo H. Decoding and overcoming T cell exhaustion: epigenetic and transcriptional dynamics in CAR-T cells against solid tumors. Mol Ther. 2024;32(6):1617–1627. doi:10.1016/j.ymthe.2024.04.004
66. Liu T, Cheng S, Peng B, et al. PD-L2 of tumor-derived exosomes mediates the immune escape of cancer cells via the impaired T cell function. Cell Death Dis. 2024;15(11):800. doi:10.1038/s41419-024-07191-7
67. Singh A, Veeriah V, Xi P, et al. Angiocrine signals regulate quiescence and therapy resistance in bone metastasis. JCI Insight. 2019;4(13). doi:10.1172/jci.insight.125679
68. Jiang X, Shan J, Dai N, et al. Apurinic/apyrimidinic endonuclease 1 regulates angiogenesis in a transforming growth factor β-dependent manner in human osteosarcoma. Cancer Sci. 2015;106(10):1394–1401. doi:10.1111/cas.12763
69. Xu C, Wang Z, Cui R, et al. Co-expression of parathyroid hormone related protein and TGF-beta in breast cancer predicts poor survival outcome. BMC Cancer. 2015;15(1):925. doi:10.1186/s12885-015-1873-x
70. Guo S, Notch G-PRR. IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS One. 2011;6(6):e21467. doi:10.1371/journal.pone.0021467
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Telomere Maintenance-Related Genes are Essential for Prognosis in Breast Cancer

Huang W, Wang W, Dong TZ
Breast Cancer: Targets and Therapy 2025, 17:225-239
Published Date: 24 February 2025