")
Back to Journals » International Journal of General Medicine » Volume 16
Prognostic Analysis and Biomarkers Identification of Immune Infiltration in Early and Late Stage Hepatocellular Carcinoma Based on TCGA Data
Authors Jiang W, Wang Y, Yu C, Sui D, Du G, Li Y
Received 8 May 2023
Accepted for publication 13 June 2023
Published 16 June 2023 Volume 2023:16 Pages 2519—2530
DOI https://doi.org/10.2147/IJGM.S420458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Wenying Jiang,1 Yunxing Wang,1 Changtao Yu,1 Deling Sui,1 Gang Du,2 Youchun Li1
1Department of General Surgery, The Second People’s Hospital of Liaocheng Affiliated to Shandong First Medical University, Liaocheng, Shandong, People’s Republic of China; 2Department of General Surgery, Qilu Hospital of Shandong University, Jinan, Shandong, People’s Republic of China
Correspondence: Youchun Li, Department of General Surgery, The Second People’s Hospital of Liaocheng Affiliated to Shandong First Medical University, No. 306, Health Street, Linqing, Shandong, 252600, People’s Republic of China, Email [email protected]
Background: Hepatocellular carcinoma (HCC) is a major cause of cancer death in the world. The aim of this study was to establish a new model to predict the prognosis of HCC.
Materials and Methods: The mRNA, miRNA and lncRNA expression profiles of early (stage I–II) and late (stage III–IV) stage HCC patients were acquired from The Cancer Genome Atlas (TCGA) database. The differentially expressed mRNAs (DEmRNAs), miRNAs (DEmiRNAs) and lncRNAs (DElncRNAs) were identified between early and late stage HCC. Key molecules associated with the prognosis, and important immune cell types in HCC were identified. The nomogram based on incorporating age, gender, stage, and all important factors was constructed to predict the survival of HCC.
Results: A total of 1516 DEmRNAs, 97 DEmiRNAs and 87 DElncRNAs were identified. A DElncRNA-DEmiRNA-DEmRNA regulatory network including 78 mRNAs, 50 miRNAs and 1 lncRNA was established. Among the regulatory network, 11 molecules were significantly correlated with the prognosis of HCC based on Lasso regression analysis. Then, Preadipocytes and 3 survival-associated DEmRNAs were identified as crucial biomarkers. Subsequently, a nomogram with a differentiation degree of 0.758, including 1 immune cell, 11 mRNAs and 3 miRNAs, was generated.
Conclusion: Our study constructed a model by incorporating clinical information, significant biomarkers and immune cells to predict the survival of HCC, which achieved a good performance.
Keywords: hepatocellular carcinoma, stage, nomogram, TCGA
Background
Primary liver cancer is the most common cancer in the world and a common cause of cancer death.1 Hepatocellular carcinoma (HCC) accounts for 75–85% of primary liver cancer.1 The main risk factors for HCC are chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV).2 Currently, surgical resection and transplantation are the most effective methods for the treatment of liver cancer, but the recurrence rate is still high and the prognosis is poor.3 Surgical resection is the main intervention for HCC patients who are diagnosed in the early stages.4 However, early diagnosis and effective therapy of HCC remain a challenge, and most of HCC cases are diagnosed at an advanced stage, with less opportunity to take curable surgery, thus contributing to the poor survival rate.5 Consequently, it is urgent to reveal the underlying molecular mechanisms of HCC development from early stage to late stage.
In the current study, we downloaded mRNA, miRNA and lncRNA expression profiles of early (stage I–II) and late (stage III–IV) stage HCC patients from The Cancer Genome Atlas (TCGA) database. Then, the differentially expressed mRNAs (DEmRNAs), miRNAs (DEmiRNAs), lncRNAs (DElncRNAs), key molecules associated with the prognosis, and important immune cell types in the late stage HCC were identified. Finally, the nomogram based on age, gender, stage, and all important factors was constructed to predict the survival of HCC.
Methods
Data Acquisition
Clinical data of 377 HCC patients were downloaded from TCGA database (collected before 25 January 2021). The mRNA and lncRNA expression profiles of 305 HCC patients, including 227 early (stage I–II) and 78 late (stage III–IV) stage HCC tissues, and miRNA expression profiles of 302 HCC patients, including 225 early and 77 late stage HCC tissues, were obtained from TCGA as well.
Differential Expression Analysis
First, the difficult-to-detect mRNAs/miRNAs/lncRNAs with a read count of 0 in more than 90% of the samples were filtered and deleted. Based on the read count of each sample, the DEmRNAs, DEmiRNAs and DElncRNAs in the late stage HCC compared to the early stage HCC were determined by edgeR with p-value <0.05. Hierarchical clustering analysis was performed using the R package “pheatmap”. To further analyze the biological functions of DEmRNAs in the late stage HCC, Gene Set Enrichment Analysis (GSEA) was performed by using GSEA (version 4.1.0). The h.all.v7.2.symbols.gmt selected as the reference gene set in the present study. P-value <0.05 was considered statistically significant.
DElncRNA-DEmiRNA-DEmRNA Regulatory Network
Given miRNAs tend to decrease the expression of their target mRNAs, we selected miRNA (down)-mRNA (up) and miRNA (up)-mRNA (down) relationships for further research. DEmiRNA-DEmRNA interaction pairs were obtained with miRwalk 2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2). Similarly, the DElncRNA-DEmiRNA interaction pairs were predicted with NPInter, Starbase and DIANA-LncBase v2. Then, the DElncRNA-DEmiRNA-DEmRNA regulatory network was constructed. Finally, Cytoscape was used to visualize the regulatory network.
Survival Analysis and Nomogram of Key Molecules in DElncRNA-DEmiRNA-DEmRNA Regulatory Network
Univariate Cox regression analysis was performed to identify the prognostic value of all biomarkers in DElncRNA-DEmiRNA-DEmRNA regulatory network. To avoid overfitting of the Cox model, lasso regression was performed on biomarkers significantly associated with prognosis. The receiver operating characteristic (ROC) curves were applied to assess the accuracy of the Cox model. Based on age, gender, stage and biomarkers significantly associated with prognosis, a nomogram was constructed using “rms” package in R. Following that, draw the calibration curve of the nomogram and observe the predictive value of the nomogram.
Immune Correlation Analysis
In order to further explore the molecular mechanism of HCC, we performed the deconvolution analysis with xCell to infer the fraction of immune cell types in HCC. xCell could estimate the abundance scores of 64 cell types based on the single sample Gene Set Enrichment Analysis (ssGSEA) method.6 Then, the immune cells with significant differences in the early and late stage HCC were identified. The patients were stratified into high-xCell score and low-xCell score groups based on the median value of the xCell. Univariate Cox analysis was performed in combination with xCell score of significantly different immune cells and clinical information of patients, followed by multivariate Cox analysis. Then, based on age, gender, stage and survival related immune cells, a nomogram was constructed using “rms” package in R, as well as calibration plot.
Correlation Analysis Between DEmRNAs and Survival Related Immune Cells
Pearson correlation analysis was done for DEmRNAs and survival related immune cells to determine DEmRNAs that were significantly associated with immune cells. Similar to the above process, univariate Cox analysis, Lasso regression analysis and ROC were performed. Then, age, gender, stage, immune cells and immune related DEmRNAs were included to construct the nomogram and calibration plot.
Survival Analysis and Nomograms of All Important Factors
All important factors, including key molecules in DElncRNA-DEmiRNA-DEmRNA regulatory network, survival related immune cells and DEmRNAs significantly associated with immune cells were incorporated into a nomogram.
Real-Time qPCR (RT-qPCR) Validation
Eight tissue samples of early HCC patients (n = 4) and late HCC patients (n = 4) were included in this study. We obtained the written informed consent and the approval from the ethics committee of the hospital. The Trizol reagent was used to separate total RNA from tumor tissues. FastKing cDNA first strand synthesis kit and miRNA first strand cDNA synthesis kit (tailing method) were reverse transcribed to synthesize mRNA and miRNA, respectively. SuperReal PreMix Plus (SYBR Green) and miRNA quantitative PCR kit (dye method) were used to perform RT-qPCR validation of mRNA and miRNA, respectively. The relative quantification of mRNAs and miRNAs was normalized to GAPDH and ACTB or U6 with the 2−ΔΔCT method.
Validation and Survival Analysis of DEmRNAs in GEO
The gene expression matrix file of GSE14520 was downloaded from the GEO database and annotated according to the annotation file of the GPL3921 platform. Among them, 220 samples (early: late = 170: 50) with stage information were selected for further analysis. The expression of DEmRNAs was validated with the GSE14520 dataset. In addition, survival analysis was performed by using the survival package in R.
Statistical Analysis
R package was used to perform all the statistical analyses. R package edgeR was used for differential expression analysis of mRNA, miRNA and lncRNA. In RT-qPCR, one-way ANOVA was used to evaluate the statistical significance among different groups.
Results
Identification of DEmRNAs, DEmiRNAs and DElncRNAs
A total of 1516 DEmRNAs, 97 DEmiRNAs and 87 DElncRNAs were identified in the late stage HCC, of which 1082 mRNAs (71.4%), 92 miRNAs (94.8%) and 60 lncRNAs (69.0%) were up-regulated while others were down-regulated (Figure 1). The top 10 up- and down-regulated DEmRNAs, DEmiRNAs and DElncRNAs are displayed in Table 1. The GSEA results revealed two gene sets that were significantly up-regulated in the late stage HCC: G2M_checkpoint (p = 0.031) and mitotic_spindle (p = 0.027) (Figure 2).
 |
Table 1 Top 10 Up- and Down-Regulated DEmRNAs, DEmiRNAs and DElncRNAs |
 |
Figure 1 The hierarchical clustering results of top 100 up- and down-regulated DEmRNAs (A), all DEmiRNAs (B) and all DElncRNAs (C) between the early and late stage HCC. Row and column represented DEmRNAs/DEmiRNAs/DElncRNAs and samples, respectively. The color scale represented the expression levels. Orange indicates above the reference channel (high expression genes). Blue indicates below the reference channel (low expression genes). |
 |
Figure 2 GSEA analysis illustrated up-regulated gene sets in the late stage HCC. (A) G2M_checkpoint; (B) mitotic_spindle. |
DElncRNA-DEmiRNA-DEmRNA Regulatory Network and Survival Analysis
A DElncRNA-DEmiRNA-DEmRNA regulatory network including 78 mRNAs, 50 miRNAs and 1 lncRNA was established (Figure 3). Univariate Cox regression analysis performed on all 129 molecules in the above lncRNA-miRNA-mRNA network revealed that 50 molecules were significantly correlated with the prognosis of HCC (Figure S1A). Then, these 50 molecules were incorporated into a multivariate model. Lasso regression analysis indicated that 11 molecules were significantly correlated with the prognosis of HCC (Figure S1B and C). The ROC curve analysis indicated that the 1-, 3- and 5-year area under curve (AUC) values were 0.76, 0.72 and 0.71, respectively (Figure S1D). A nomogram was generated to predict the probability of 1-, 3- and 5-year overall survival (OS), by incorporating age, gender, stage and biomarkers significantly associated with prognosis. As shown in Figure S1E, each factor was assigned points in proportion to its risk contribution to survival. The calibration curve indicated that actual and predicted survival did not match well, and the C-index for the model was 0.559 (Figure S1F).
 |
Figure 3 DElncRNA-DEmiRNA-DEmRNA regulatory network. The rectangle, inverted triangles and elliptical nodes indicate DElncRNAs, DEmiRNAs and DEmRNAs, respectively. Red and blue color represent up- and down-regulation, respectively. |
Immunoinfiltration Analysis
With xCell, 19 significantly different immune cell types were identified between the early and late stage HCC (p-value <0.05) (Figure S2). Univariate Cox analysis performed in combination with xCell score of significantly different immune cells and clinical information of patients identified 11 survival related immune cell types. Then, multivariate Cox analysis of these 11 immune cells indicated that CD4+ Tem, Preadipocytes and Platelets were significantly correlated with survival. Kaplan–Meier curves of these 3 immune cells were presented in Figure S3A–C. The nomogram and calibration plot (C-index = 0.74) are shown in Figure S3D and E.
Correlation Analysis Between DEmRNAs and Survival Related Immune Cells
The correlation analysis between 3 immune cells and 1516 DEmRNAs was performed, showing that 82 DEmRNAs were significantly associated with Preadipocytes (|pcor| > 0.5 and p-value <0.05). Univariate Cox analysis further indicated that 70 DEmRNAs were significantly associated with survival. Then, Lasso regression analysis identified that 3 DEmRNAs (CFHR4, CYP2C9 and SAPCD2) were crucial biomarkers significantly associated with survival (Figure S4A and B). The ROC curve analysis indicated that the 1-, 3- and 5-year AUC values were 0.75, 0.74 and 0.71, respectively (Figure S4C). A nomogram was generated to predict the probability of 1-, 3- and 5-year OS, by incorporating age, gender, stage, xCell score of Preadipocytes and 3 DEmRNAs expression levels (CFHR4, CYP2C9 and SAPCD2) (Figure S4D). The calibration plot indicated that the C-index for the model was 0.73 (Figure S4E).
Survival Analysis and Nomograms of All Important Factors
Combined with the above analysis, all important factors, including xCell score of Preadipocytes and expression levels of SLC1A2, TECTB, CTNNA3, TRIM55, TRPM8, RANBP3L, RAMP1, PON1, hsa-miR-5581-3p, hsa-miR-7-5p, hsa-miR-767-5p, CFHR4, CYP2C9, and SAPCD2, were integrated into a Cox model. The nomogram is shown in Figure 4A. The calibration plot indicated that the C-index for the model was 0.758 (Figure 4B).
 |
Figure 4 Nomogram (A) and calibration curve (B) in HCC. |
RT-qPCR Validation
Two DEmRNAs (TRPM8 and TECTB) and four DEmiRNAs (hsa-miR-5581-3p, hsa-miR-7-5p, hsa-miR-767-3p and hsa-miR-490-3p) were selected to use for RT-qPCR analysis. Hsa-miR-5581-3p, hsa-miR-7-5p and hsa-miR-767-3p were up-regulated trend while TRPM8, TECTB and hsa-miR-490-3p were down-regulated trend in the late stage HCC. It is noted that the RT-qPCR results were in line with our integrated analysis (Figure 5). However, we found that RT-qPCR results lacked significance, which may be caused by the small sample size.
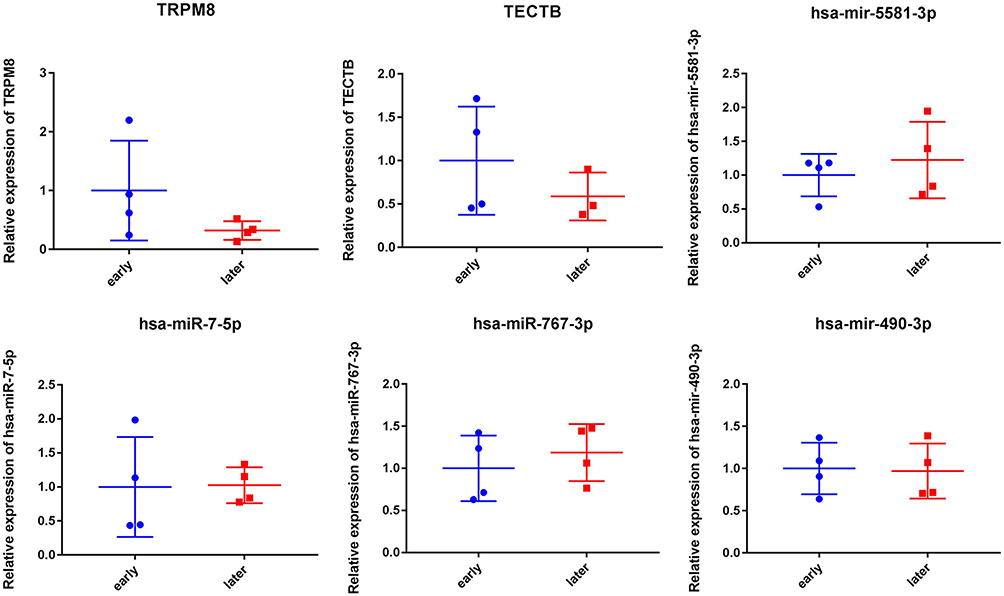 |
Figure 5 RT-qPCR results of DEmRNAs and DEmiRNAs. |
Validation and Survival Analysis of DEmRNAs in GSE14520
The expression patterns of seven DEmRNAs (CFHR4, CTNNA3, CYP2C9, PON1, RAMP1, SLC1A2, and TRPM8) were validated in GSE14520 (Figure 6). The results revealed that except for CTNNA3 and RAMP1, the other five mRNAs were significantly down-regulated in the late stage HCC. Even so, CTNNA3 and RAMP1 showed the down-regulated trend in the late stage HCC. These findings were consistent with the results of our integrated analysis. In addition, survival analysis displayed that the expression of all these seven mRNAs was significantly correlated with the overall survival time of HCC patients (Figure S5).
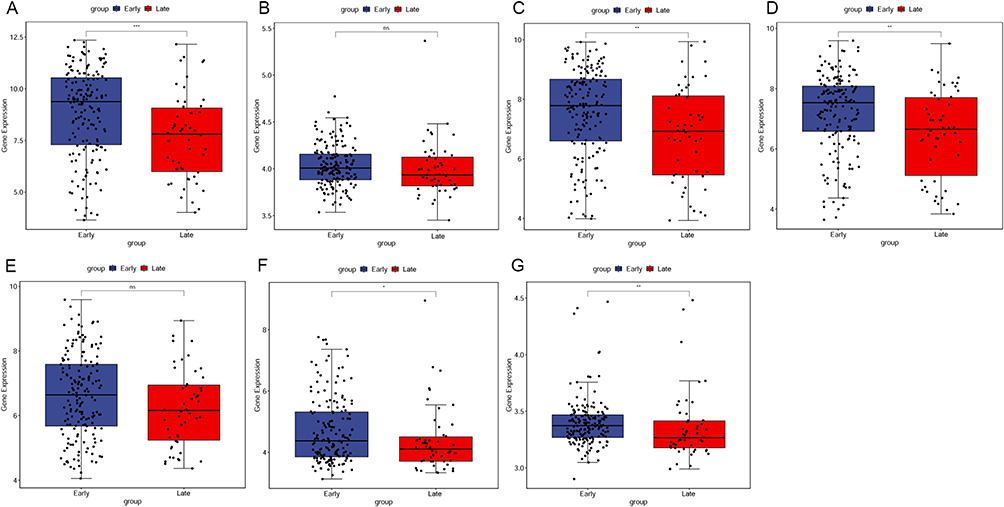 |
Figure 6 Validation of DEmRNAs in GSE14520. (A) CFHR4, (B) CTNNA3, (C) CYP2C9, (D) PON1, (E) RAMP1, (F) SLC1A2, (G) TRPM8. |
Discussion
At present, four different nomograms were constructed by different methods. Firstly, we constructed the DElncRNA-DEmiRNA-DEmRNA regulatory network, which included 78 mRNAs, 50 miRNAs and 1 lncRNA. Univariate Cox regression analysis and Lasso regression analysis further identified 11 key molecules associated with the prognosis, and a nomogram by incorporating age, gender, stage and 11 molecules was generated, which performed worse (C-index = 0.559). Secondly, 3 important immune cell types were identified by Univariate Cox analysis and multivariate Cox analysis to construct nomogram (C-index = 0.74). Thirdly, correlation analysis between DEmRNAs and survival related immune cells was performed, and Preadipocytes and 3 DEmRNAs expression levels (CFHR4, CYP2C9 and SAPCD2) were selected to construct nomogram (C-index = 0.73). Lastly, by incorporating all important factors identified above, a nomogram, including 1 immune cell, 11 mRNAs and 3 miRNAs, was generated to predict the survival of HCC, which achieved a good performance (C-index = 0.758) than the model constructed using only single factor.
Tripartite motif containing 55 (TRIM55) was suggested to act as a potential tumor suppressor to inhibit the malignant biological behavior of lung adenocarcinoma cells.7 A model of immune-related-gene-signature including TRIM55 was constructed to predict survival outcome and reflect the immune status of lung squamous cell carcinoma.8 It has been demonstrated that TRIM55 was decreased in HCC tissues, and its overexpression inhibited migration and invasion of HCC cells.9 Ding et al suggested that serum paraoxonase 1 (PON1) could potentially be used to diagnose microvascular invasion and to guide more personalized treatment strategy in HCC.10 It has been reported that low expression of PON1 was associated with poor survival in HCC patients, which may be helpful for the diagnosis of HCC.11 Both TRIM55 and PON1 were detected to be decreased in the late stage HCC, suggesting their important roles in the development of HCC.
Cytochrome P2C (CYP2C) subfamily members are known to participate in clinical drug metabolism.12 Cytochrome P450 family 2 subfamily C member 9 (CYP2C9) is a member of the CYP2C subfamily that is localized in a single gene locus on chromosome 10.13 Previous studies have revealed that dysregulation of CYP2C9 may participate in the development of HCC. An integrated bioinformatics analysis indicated that CYP2C9 was strongly associated with the risk of liver cancer development and progression in obese individuals.14 It has been reported that CYP2C9 is involved in the regulation of iron homeostasis and associated with the prognosis of HCC.15 Mu et al found that CYP2C9 was associated with poor OS in HCC patients.16 Suppression of CYP2C9 by hsa-miR-128-3p results in tumor cell invasion in HCC.12 In this analysis, CYP2C9 was markedly reduced in the late stage HCC, indicating that CYP2C9 may exert momentous roles in the progression of HCC.
Aberrant expression of suppressor APC domain containing 2 (SAPCD2) has been indicated in a variety of cancers, including HCC. SAPCD2 level was correlated to the tumor size, Tumor Node Metastasis (TNM) stage and lymph node metastasis, and knocking down SAPCD2 inhibited migration and invasiveness ability of breast cancer cells.17 The expression of SAPCD2 was significantly increased in fibrosarcoma tissues, and silencing SAPCD2 inhibited the proliferation and lung metastasis of fibrosarcoma cells.18 It has been suggested that overexpression of SAPCD2 correlates with proliferation and invasion of colorectal carcinoma cells.19 Silencing SAPCD2 suppressed the proliferation, migration, and invasiveness of prostate cancer cells, suggesting that SAPCD2 may serve as an oncogene in prostate cancer.20 A prognostic gene model consisting of four genes, including SAPCD2, was constructed to predict the OS of HCC patients.21 SAPCD2 exhibited increased expression in late stage HCC, which added to the evidence that SAPCD2 performs a critical function in HCC.
Hsa-miR-7-5p has been reported to suppress proliferation, migration and promote apoptosis in HCC cell lines by inhibiting SPC24 expression.22 It has been reported that hsa-miR-7-5p had independent prognostic significance for the OS of HCC.23 Hsa-miR-767-3p was reported to be inhibited by hsa_circ_0000673 in the malignant progression of HCC, which evidenced the expected tumor-suppressive effect of miR-767-3p.24 In addition, the up-regulation of miR-5581-3p can significantly enhance the viability and proliferation of cancer cells, and enhance the migration and invasion of cancer cells, suggesting that miR-5581-3p may play a promoting role in the progression of HCC by regulating cancer cells.25 In this study, these three miRNAs were identified as significant signatures and included in the model to predict survival in HCC patients.
Studies have found that lncRNA X inactive specific transcript (XIST) participates in HCC development by targeting multiple miRNA/mRNA axes. It has been reported that lncRNA XIST accelerates HCC cell growth by inhibiting miR-488.26 Wang et al suggested that lncRNA XIST up-regulates TRIM25 by targeting and binding to miR-192 to accelerate the occurrence and development of HCC.27 Liu et al indicated that XIST could enhance the cell growth ability of HCC by targeting miR-200b-3p, which suggested that XIST may be a potential therapeutic target in HCC.28 Similarly, hsa-miR-490-3p has been linked to HCC by different lnRNA/hsa-miR-490-3p/mRNA axes. For instance, BCYRN1/miR-490-3p/POU3F2 was suggested to promote tumor cell growth and metastasis of HCC.29 SNHG15/miR-490-3p/HDAC2 axis was demonstrated to promote HCC progression.30 In this analysis, XIST/hsa-miR-490-3p/VHLL axis was detected in the DElncRNA-DEmiRNA-DEmRNA regulatory network, suggesting its potential role in the progression of HCC.
However, there are some limitations in this study. Firstly, the lack of significance of RT-qPCR results may be caused by the small sample size, so it is necessary to collect samples for further study. However, the validation in GEO database increased the reliability of our analysis. Secondly, the detailed molecular mechanisms of the key DEmRNAs, DEmiRNAs and DElncRNAs identified remain unclear and require further study. Thirdly, additional prospective validation is required before the prognosis model can be considered for clinical application. The reliability of this model needs to be iteratively improved in long-term clinical applications with larger sample sizes. To further validate these findings and evaluate prognostic value and clinical applicability of the model, a multicenter prospective study with more patients and a longer follow-up period in the real world is required.
Conclusion
In summary, this study identified DEmRNAs/DEmiRNAs/DElncRNAs in the early and late stage HCC that may serve as factors related to the progression of HCC. Meanwhile, immune cell types related to the progression of HCC were obtained by immune infiltration analysis with ssGSEA. The Cox survival model was constructed to screen out the key factors significantly related to the prognosis of HCC. Finally, a Cox model with a differentiation degree of 0.758 including 14 molecules and 1 type of immune cells was obtained. This study is the first attempt to use immune cells as a variable to predict survival in HCC patients, providing new ideas for future research. In addition, the identification of new biomarkers provides potential targets for the diagnosis and management of early stage HCC.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article. We searched for HCC public gene expression data and complete clinical annotations from TCGA (https://tcga-data.nci.nih.gov/tcga/) database. The accession numbers is TCGA: HCC (Platform: Illumina RNAseq), respectively. The raw data of RT-qPCR validation has been deposited in the Harvard Dataverse database (https://doi.org/10.7910/DVN/I8L6QI).
Ethics Approval Statement
Approval was obtained from the ethics committee of The Second People’s Hospital of Liaocheng Affiliated to Shandong First Medical University (2020-9). The procedures used in this study adhere to the tenets of the Declaration of Helsinki. Written informed consent about the use of these samples was obtained from each patient.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no conflict of interest.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi:10.3322/caac.21660
2. Yang JD, Hainaut P, Gores GJ, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589–604. doi:10.1038/s41575-019-0186-y
3. Galle PR, Forner A, Llovet JM, et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182–236.
4. Villanueva A, Longo DL. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi:10.1056/NEJMra1713263
5. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi:10.1016/S0140-6736(18)30010-2
6. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi:10.1186/s13059-017-1349-1
7. Guo T, Zhang Z, Zhu L, et al. TRIM55 suppresses malignant biological behavior of lung adenocarcinoma cells by increasing protein degradation of Snail1. Cancer Biol Ther. 2022;2022:1–10.
8. Li N, Wang J, Zhan X. Identification of immune-related gene signatures in lung adenocarcinoma and lung squamous cell carcinoma. Front Immunol. 2021;12:752643. doi:10.3389/fimmu.2021.752643
9. Li X, Huang L, Gao W. Overexpression of tripartite motif containing 55 (TRIM55) inhibits migration and invasion of hepatocellular carcinoma (HCC) cells via epithelial-mesenchymal transition and matrix metalloproteinase-2 (MMP2). Med Sci Monit. 2019;25:771–777. doi:10.12659/MSM.910984
10. Ding GY, Zhu XD, Ji Y, et al. Serum PON1 as a biomarker for the estimation of microvascular invasion in hepatocellular carcinoma. Ann Tran Med. 2020;8:204. doi:10.21037/atm.2020.01.44
11. Zhang Y, Ying X, Zhao Q, et al. Identification of protein expression changes in hepatocellular carcinoma through iTRAQ. Dis Markers. 2020;2020:2632716. doi:10.1155/2020/2632716
12. Yu D, Green B, Marrone A, et al. Suppression of CYP2C9 by microRNA hsa-miR-128-3p in human liver cells and association with hepatocellular carcinoma. Sci Rep. 2015;5:8534. doi:10.1038/srep08534
13. Wang X, Yu T, Liao X, et al. The prognostic value of CYP2C subfamily genes in hepatocellular carcinoma. Cancer Med. 2018;7:966–980. doi:10.1002/cam4.1299
14. Ceylan H. Identification of hub genes associated with obesity-induced hepatocellular carcinoma risk based on integrated bioinformatics analysis. Med Oncol. 2021;38:63. doi:10.1007/s12032-021-01510-0
15. Shen H, Wu H, Sun F, et al. A novel four-gene of iron metabolism-related and methylated for prognosis prediction of hepatocellular carcinoma. Bioengineered. 2021;12:240–251. doi:10.1080/21655979.2020.1866303
16. Mu D, Qin F, Li B, et al. Identification of the sixth complement component as potential key genes in hepatocellular carcinoma via bioinformatics analysis. Biomed Res Int. 2020;2020:7042124. doi:10.1155/2020/7042124
17. Zhang Y, Liu JL, Wang J. SAPCD2 promotes invasiveness and migration ability of breast cancer cells via YAP/TAZ. Eur Rev Med Pharmacol Sci. 2020;24:3786–3794. doi:10.26355/eurrev_202004_20844
18. Zhu B, Wu Y, Niu L, et al. Silencing SAPCD2 represses proliferation and lung metastasis of fibrosarcoma by activating hippo signaling pathway. Front Oncol. 2020;10:574383. doi:10.3389/fonc.2020.574383
19. Luo Y, Wang L, Ran W, et al. Overexpression of SAPCD2 correlates with proliferation and invasion of colorectal carcinoma cells. Cancer Cell Int. 2020;20:43. doi:10.1186/s12935-020-1121-6
20. Sun Z, Mao Y, Zhang X, et al. Identification of ARHGEF38, NETO2, GOLM1, and SAPCD2 associated with prostate cancer progression by bioinformatic analysis and experimental validation. Front Cell Develop Bio. 2021;9:718638. doi:10.3389/fcell.2021.718638
21. Li L, Cao Y, Fan Y, et al. Gene signature to predict prognostic survival of hepatocellular carcinoma. Open Med. 2022;17:135–150. doi:10.1515/med-2021-0405
22. Wang Y, Yang H, Zhang G, et al. hsa-miR-7-5p suppresses proliferation, migration and promotes apoptosis in hepatocellular carcinoma cell lines by inhibiting SPC24 expression. Biochem Biophys Res Commun. 2021;561:80–87. doi:10.1016/j.bbrc.2021.05.020
23. Zhang X, Ma L, Zhai L, et al. Construction and validation of a three-microRNA signature as prognostic biomarker in patients with hepatocellular carcinoma. Int J Med Sci. 2021;18:984–999. doi:10.7150/ijms.49126
24. Jiang W, Wen D, Gong L, et al. Circular RNA hsa_circ_0000673 promotes hepatocellular carcinoma malignance by decreasing miR-767-3p targeting SET. Biochem Biophys Res Commun. 2018;500:211–216. doi:10.1016/j.bbrc.2018.04.041
25. Yin J, Liu Q, Chen C, et al. Small regulatory polypeptide of amino acid response negatively relates to poor prognosis and controls hepatocellular carcinoma progression via regulating microRNA-5581-3p/human cardiolipin synthase 1. J Cell Physiol. 2019;234:17589–17599. doi:10.1002/jcp.28383
26. Dong Z, Yang J, Zheng F, et al. The expression of lncRNA XIST in hepatocellular carcinoma cells and its effect on biological function. J BUON. 2020;25:2430–2437.
27. Wang J, Yin G, Bian H, et al. LncRNA XIST upregulates TRIM25 via negatively regulating miR-192 in hepatitis B virus-related hepatocellular carcinoma. Molecul Med. 2021;27:41. doi:10.1186/s10020-021-00278-3
28. Liu WG, Xu Q. Long non-coding RNA XIST promotes hepatocellular carcinoma progression by sponging miR-200b-3p. Eur Rev Med Pharmacol Sci. 2019;23:9857–9862. doi:10.26355/eurrev_201911_19549
29. Ding S, Jin Y, Hao Q, et al. LncRNA BCYRN1/miR-490-3p/POU3F2, served as a ceRNA network, is connected with worse survival rate of hepatocellular carcinoma patients and promotes tumor cell growth and metastasis. Cancer Cell Int. 2020;20:6. doi:10.1186/s12935-019-1081-x
30. Dai W, Dai JL, Tang MH, et al. lncRNA-SNHG15 accelerates the development of hepatocellular carcinoma by targeting miR-490-3p/ histone deacetylase 2 axis. World J Gastroenterol. 2019;25:5789–5799. doi:10.3748/wjg.v25.i38.5789
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.