Back to Journals » Infection and Drug Resistance » Volume 13
Profiling the mRNA and miRNA in Peripheral Blood Mononuclear Cells in Subjects with Active Tuberculosis
Authors Cao D ![]() , Wang J, Ji Z, Shangguan Y, Guo W, Feng X
, Wang J, Ji Z, Shangguan Y, Guo W, Feng X ![]() , Xu K, Yang J
, Xu K, Yang J ![]()
Received 25 August 2020
Accepted for publication 4 November 2020
Published 23 November 2020 Volume 2020:13 Pages 4223—4234
DOI https://doi.org/10.2147/IDR.S278705
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Dan Cao,* Ju Wang,* Zhongkang Ji, Yanwan Shangguan, Wanru Guo, Xuewen Feng, Kaijin Xu, Jiezuan Yang
State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiezuan Yang
State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qingchun Road, Hangzhou 310003 Zhejiang, People’s Republic of China
Tel +86 571 87236756
Fax +86 571 87236423
Email [email protected]
Purpose: To identify candidate hub genes and miRNAs associated with active tuberculosis (ATB) and reveal the potential molecular mechanisms of disease progression.
Patients and Methods: The expression of mRNA and miRNA was evaluated in peripheral blood mononuclear cells (PBMC) from 4 ATB patients and 4 healthy donors (HD) using high throughput sequencing (HTS) and bioinformatics analysis. Moreover, differentially expressed miRNAs were validated with 35 ATB patients and 35 HDs using reverse transcription quantitative real-time PCR (RT-qPCR).
Results: A total of 2658 significantly differentially expressed genes (DEG) including 1415 up-regulated genes and 1243 down-regulated genes were identified in the ATB group compared with HDs, and the DEGs enriched in immune-related pathways, especially in TNF signaling pathway, cytokine–cytokine receptor interaction, mitogen-activated protein kinase (MAPK) signaling pathways and tuberculosis. Additionally, 10 hub genes were acquired according to protein–protein interaction (PPI) analysis of DEGs. Moreover, 26 differentially expressed miRNAs were found in ATB group compared with HDs. Furthermore, RT-qPCR results showed that hsa-miR-23a-5p (P=0.0106), hsa-miR-183-5p (P=0.0027), hsa-miR-193a-5p (P=0.0021) and hsa-miR-941(P=0.0001) were significantly increased in the ATB patients compared with HD group, and the hsa-miR-16-1-3p was significantly decreased (P=0.0032).
Conclusion: Our research provided a characteristic profile of mRNAs and miRNAs expressed in ATB subjects, and 10 hub genes related with ATB were found, which will contribute to explore the role of miRNAs and hub genes in the pathogenesis of ATB, and improve the ability of differential diagnosis and treatment for the disease.
Keywords: mRNA, miRNA, active tuberculosis, high throughput sequencing, RT-qPCR
Introduction
Tuberculosis (TB) is a worldwide disease mainly caused by Mycobacterium tuberculosis (Mtb), which is one of the top ten causes of death in the world.1 According to the report of the World Health Organization (WHO) in 2019, there are 10 million people falling ill with TB each year, and the morbidity is more than 1.33‰, the annual death toll for Mtb infection is up to 1.5 million,2 which lead to serious health problems and socioeconomic burdens. Therefore, it is particularly important to conduct early diagnosis and treatment for TB. In recent decades, some progress has been made in the diagnosis of Mtb infection; however, there are few reliable, simple, and effective ways to definitively diagnose tuberculosis with the exception of T-SPOT.TB assay.3 Although the combined usage, such as rifampicin (RFP), isoniazid (INH), pyrazinamide (PZA), and other medicine could effectively treat TB, resistant tuberculosis has appeared and gradually threatened TB control efforts.4
Additionally, T-SPOT.TB assay could determine whether the subjects were currently infected with Mtb with high specificity and sensitivity, and the test results are not influenced by BCG vaccination and host immune function.5 Our previous research found that characteristic T cell receptor beta variable (TCRBV) in the peripheral blood T cell repertoire could be used in the differential diagnosis of the subject with active tuberculosis (ATB) or latent TB infection (LTBI).6 However, the molecular mechanism of the occurrence and development of Mtb infection has not been fully understood, besides the diagnosis and differential diagnosis of ATB.
MicroRNA (miRNA) is widely found in various eukaryotes such as animal, plant, parasite, and so on,7 miRNA is small, evolutionarily conserved, noncoding RNA with length (~22 nts).8 Studies have shown that microRNA regulation of life activities, including regulating the release of cytokines, immune cell signal transduction, cell apoptosis and autophagy and so on.9 It is a regulator of post-transcriptional regulation of gene expression in many eukaryotes.10 Studies have found that some miRNAs can be more stable in the serum of tuberculosis patients,11 and some of them will be up-regulated or down-regulated, which is an important surrogate (biomarker) of the diagnosis for TB, or target of effective treatment for TB. Kathirvel et al indicated that changes in circulating miRNA expression levels could be involved in host immune response disorders to tuberculosis.12 Although some miRNAs have been found to be associated with diagnostic biomarker of ATB, there are few reports about the role of miRNA–mRNA interaction in the pathogenesis of ATB, and about the miRNA related signaling pathways involved in.
In the present study, we used high throughput sequencing (HTS) technique to evaluate the expression profiles of mRNA and miRNA in peripheral blood mononuclear cells (PBMC) from Chinese ATB patients, and analyze the correlation between the miRNA and ATB infection status. Moreover, we constructed the miRNA–mRNA co-regulatory network for ATB patients by combing the miRNAs and mRNAs that expressed significant difference compared with that of healthy donors. Therefore, the results will contribute to study the role of miRNAs and mRNA in the pathogenesis of ATB, and improve the diagnosis and treatment for ATB.
Materials and Methods
Sample Collection
Thirty-nine subjects with active tuberculosis (ATB) and 39 healthy donors (HD) were enrolled in the Infectious Disease Department (IDD) and Health Examination Center (HEC) of the First Affiliated Hospital, Zhejiang University School of Medicine. ATB was identified by sputum or effusion smear and T-SPOT.TB assay, and confirmed on the basis of clinical syndromes and radiological findings, the more detailed diagnostic criteria could be found in previous reports.13 Additionally, subjects were excluded if they were co-infected with other infectious diseases, such as cytomegalovirus (CMV) infection, or human immunodeficiency virus (HIV), more exclusion criteria were shown in our previous report.6 Healthy donors were individuals who were excluded from LTBI by T-SPOT and other clinical diagnostic indicators. Moreover, individuals with cancer, diabetes, immune-compromised conditions, and allergic diseases were excluded. The additional characteristics of the enrolled subjects at the time of the study are shown in Table 1.
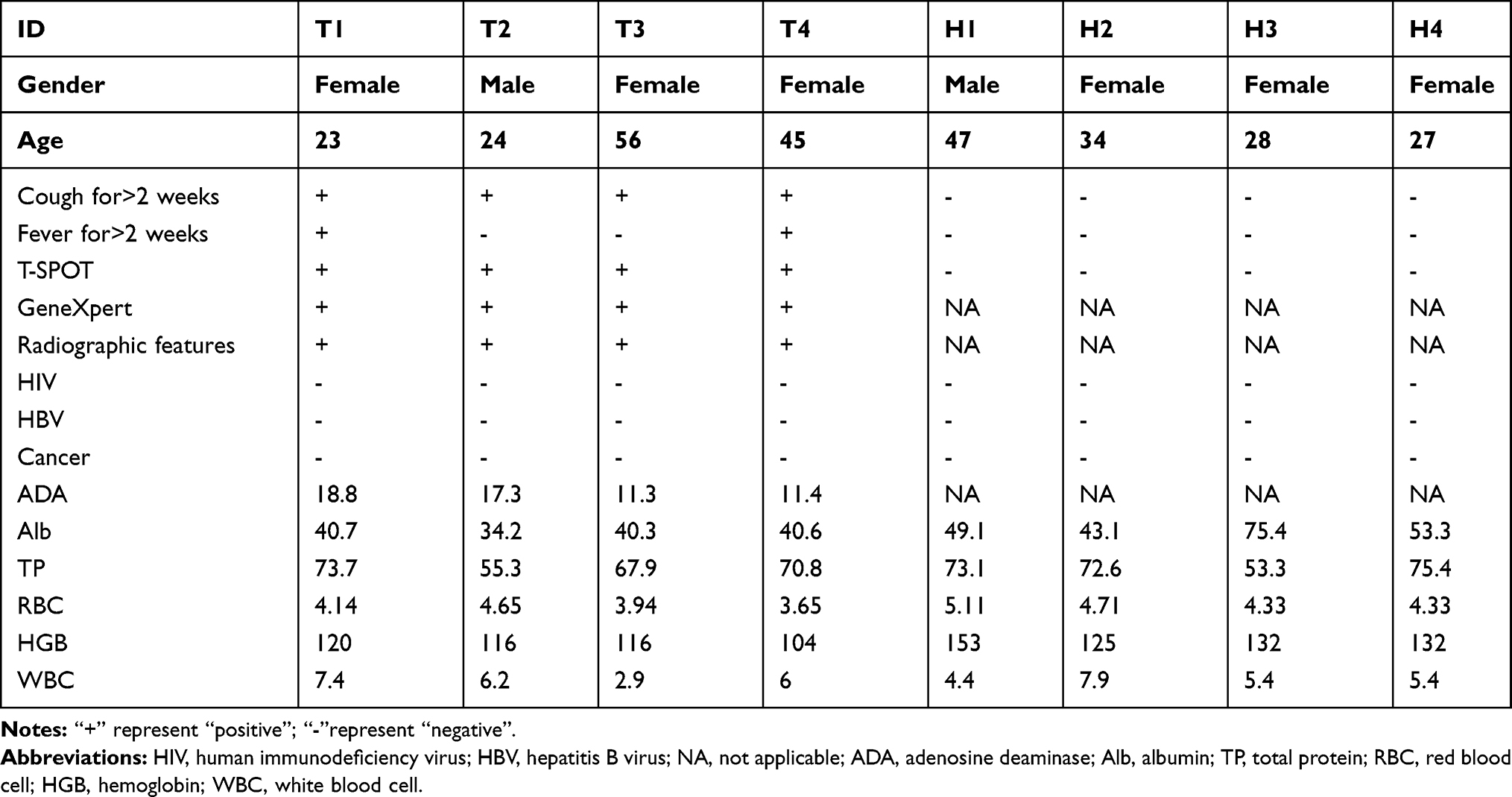 |
Table 1 Clinical Characteristics of the Participants |
This study complies with the declaration of Helsinki, and the protocols involving humans were approved by the ethics committee of the First Affiliated Hospital, Zhejiang University School of Medicine. All participants, including ATB subjects and HDs, agreed to participate in the study and provided written informed consent.
PBMC Preparation
About 5mL of peripheral vein whole blood were obtained from each subject, and the peripheral blood mononuclear cells (PBMCs) were isolated using density gradient centrifugation with Ficoll solution according to operating instructions. Simply, the peripheral vein whole blood was diluted with PBS (×1) in 1: 1 ratio, and the mixture was transferred into a 15mL centrifugal tube pre-filled with 5mL Ficoll. The PBMC were carefully collected in the middle layer (flocculent layer) of the tube following centrifugation for 20 min (500× g). Subsequently, the PBMC were washed and centrifuged with PBS for 6 minutes at 350× g, repeated this step once, and the precipitate was collected into 1.5mL EP tube containing 1.0 mL of TRIzol reagent.
RNA Isolation and Sequencing
Total RNA from PBMCs was extracted using the TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. The extracted total RNA was quantified using a Nanodrop 2000 spectrophotometer (Thermo Fisher, America). The RNA integrity was measured by visualization of the 28S and 18S RNA transcripts on a 1.2% agarose, and the quality of RNA was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Total RNA was reversely transcribed into cDNA, and sequencing library was constructed by Shanghai Majorbio Co. (Shanghai, China) according to the manufacturer’s instructions. Illumina TruSeq Small RNA Library Prep kit and Illumina TruseqTM RNA sample prep kit were used to create small RNA library and mRNA library, respectively. Additionally, the eight sequencing libraries (named T1, T2, T3, T4, H1, H2, H3 and H4) were constructed and sequenced, and the mRNA sequencing was performed on an Illumina Novaseq 6000 machine, the miRNA sequencing was performed on Illumina HiSeq X Ten machine. The miRBase database was used to analyze known miRNAs of ATB and healthy donor groups, and miRDeep2 was used to predict new miRNA. The more detailed protocol was shown in a previous report.14
Identification of DEGs and DEMs
The count data of mRNA and miRNA sequencing were subjected to principal component analysis (PCA) in R software (version 3.6.1, http://www.R-project.org) to detect outliers. Significantly differentially expressed genes (DEGs) and differentially expressed miRNA (DEMs) were screened based on the DESeq2 package in R software. The raw P value was adjusted by the Benjamini and Hochberg method (BH), and the |log2FC| ≥ 1 and adjusted P value <0.05 were regarded as the cutoff criteria and significantly.
Functional Gene Enrichment
To further understand the function of identified DEGs, Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed. The Goatools software (https://github.com/tanghaibao/goatools) was used to perform GO enrichment analysis, and the R program to perform KEGG pathway enrichment analysis. P value<0.05 was considered statistically significant.
Construction of PPI Network and Module Analysis
The STRING database (version: 11.0, https://string-db.org) is a biological database designed to analyze the protein–protein interaction (PPI) network. The brief steps were the DEGs were mapped to STRING to evaluate the interactive relationships, and the cutoff criterion was defined as more than 0.4 (medium confidence). Finally, Cytoscape (version: 3.7.1, www.cytoscape.org) was used for network construction, and Hub genes were ranked by the CytoHubba plugin. The sub-network modules in the PPI network were obtained by the plug-in unit (Molecular Complex Detection, MCODE). An MCODE score >4 and a number of nodes >5 were used as the criteria to define a module. The default parameters were set as node score cutoff=0.2, degree cutoff=2, k-core=2 and max depth=100.
Construction of miRNA–mRNA Network
To explore the potential functions of the significantly differentially expressed miRNAs (DEMs), the Miranda (http://www.miranda.org/) and RNAhybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/) algorithms were used to predict their target genes. We selected significant DEMs and their corresponding target genes, which were also identically expressed in our test of mRNA expression (DEGs), to construct the miRNA–mRNA interactive network by Cytoscape. A group of up-regulated miRNAs and corresponding down-regulated target mRNAs and another group of down-regulated miRNAs and corresponding up-regulated target mRNAs were determined. Finally, we constructed the regulatory network based on the negative correlation of miRNA and its target gene.
RT-qPCR for miRNA Verification
The RNA of PBMC from 35 ATB patients and 35 HDs were used to verify the DEMs with RT-qPCR. The simple operation is as follows, cDNA was synthesized using the PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara, Dalian, China). RT-qPCR analysis was performed using SYBR Premix Ex Taq (Takara) on the Applied Biosystems 7500 (Applied Biosystems, CA, USA). RT-qPCR reactions were performed on the Applied Biosystems 7500 (Applied Biosystems, Foster City, CA). The expression of miRNAs was determined using the Bulge-LoopTM miRNA RT-qPCR primer kit (RiBoBio, Guangzhou, China). U6 was used as reference gene. 2-ΔΔCt method was used to quantify qPCR data.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 7 software (GraphPad Software, La Jolla, CA, USA). The nonparametric Mann–Whitney test was used to evaluate the differences between groups. The miRNA expression value in the subject with healthy donors was normalized to 1. P value<0.05 was considered statistically significant.
Results
Sequencing Information and Quality Control
There are two sets of high throughput sequencing (HTS) data (mRNA, and miRNA) obtained at the present study. Sequencing readings of each sample and their profile of mapping to the genome were summarized in Table S1 and S2. Moreover, the results of principal component analysis (PCA) for the sequencing results are shown in Figure S1.
Identification of DEGs
According to the above results, the 8 samples were eligible for differential expression analysis. Differentially expressed genes (DEGs) were screened with fold change (|log2FoldChange| > 1) and adjusted P (P value < 0.05) determined by DESeq2 analysis. In comparison with the HD group, we identified 1415 up-regulated DEGs (uDEGs) and 1243 down-regulated DEGs (dDEGs) in the ATB individuals. The volcano plot of DEGs is shown in Figure 1. Additionally, the gene expression of ATB and HD samples were significantly distinguished in the heat-map clustering of DEGs (Figure S2).
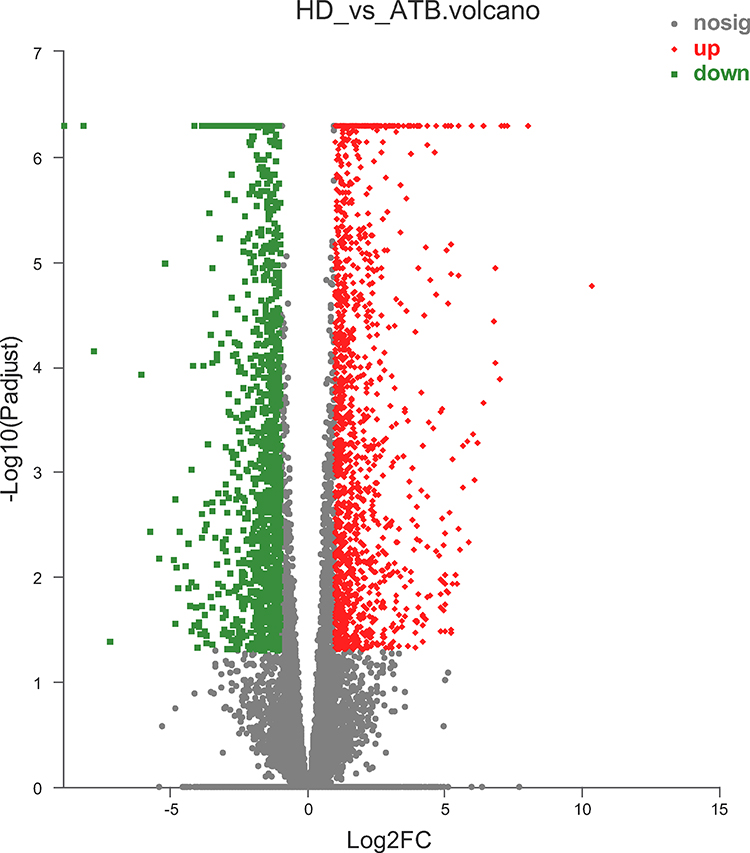 |
Figure 1 Volcano plot of differentially expressed genes (DEGs). Red, significantly up-regulated gene; green, significantly down-regulated gene; gray, non-significantly differently expressed gene. |
Gene Ontology and KEGG Analysis of DEGs
Top 20 Gene Ontology including biological process, cellular component and molecular function were analyzed. Go terms with adjusted P value <0.05 were selected and ranked by adjusted P value. The GO analysis results showed that up-regulated DEGs were predominantly enriched in the following biological processes (BP): negative regulation of leukocyte activation, cellular response to extracellular stimulus and negative regulation of secretion. The down-regulated DEGs were mainly involved in DNA deamination and small molecule metabolic process in biological processes. As for cellular component (CC), the down-regulated DEGs were enriched in intracellular organelle and intracellular membrane-bounded organelle, but there were no up-regulated DEGs enriched in this subgroup. When regarding molecular function (MF), the down-regulated DEGs were enriched in transcription regulator activity, DNA-binding transcription factor activity and DNA binding. Similarly, no up-regulated DEGs were enriched in this subgroup either (Figure 2A and B).
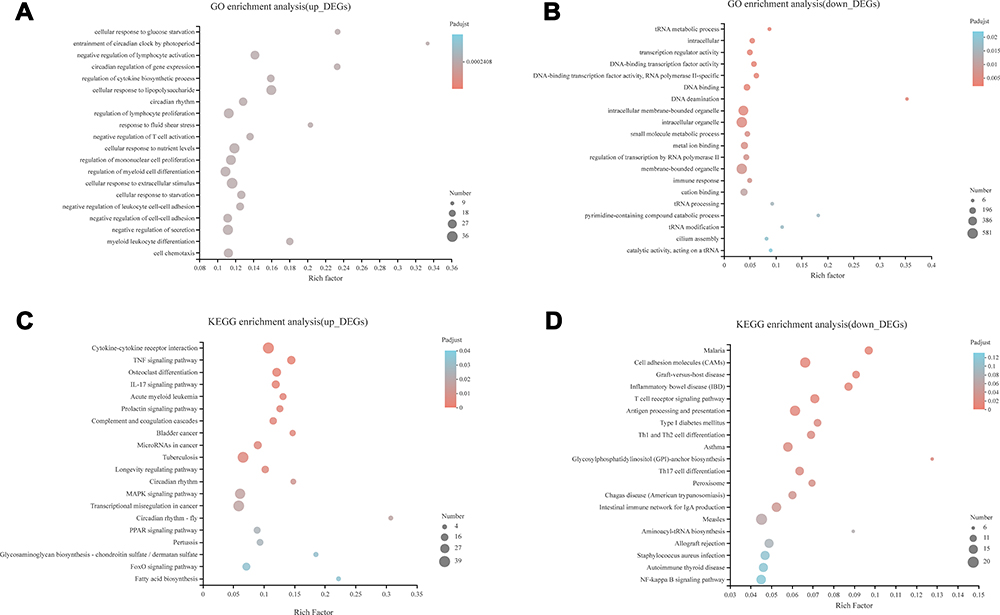 |
Figure 2 Bubble diagram of top 20 enriched GO terms and KEGG pathways for significantly differentially expressed genes (DEGs) between ATB and HD. (A) Top 20 ranked GO terms of up-regulated DEGs. (B) Top 20 ranked GO terms of down-regulated DEGs. (C) Top 20 pathways of up-regulated DEGs. (D) Top 20 pathways of down-regulated DEGs. Rich factor: the ratio of genes enriched in the GO term (KEGG) to the number of annotation genes, the enrichment degree was stronger with a bigger Rich factor. The size of dots indicates the number of genes in the GO term (KEGG). |
The results of KEGG pathway analysis showed that the up-regulated DEGs were significantly enriched in MAPK signaling pathway, cytokine–cytokine receptor interaction, TNF signaling pathway, TB (tuberculosis) and peroxisome proliferator-activated receptor (PPAR) signaling pathway. Most of the down-regulated DEGs were related to cell adhesion molecules (CAMs), antigen processing and presentation, and T cell receptor signaling pathway (Figure 2C and D).
PPI Generation and Module Analysis
Top 1000 up-regulated DEGs and top 1000 down-regulated were added to the PPI network, including 1429 nodes and 7134 edges. Hub genes were ranked by cytoHubba of Cytoscape, and found CUL3, KLHL2, KLHL22, SPSB2, KBTBD8, FBXO7, KCTD7, FBXO40, FBXL16 and FBXL8 as hub genes. Cytoscape MCODE was applied to screen the modules of the PPI network. Besides, top 2 primary modules from the PPI network were selected for functional enrichment analysis. Module 1 consists of 20 nodes and 190 edges, and Module 2 comprises 16 nodes and 96 edges (Figure 3). Enrichment analyses for Module 1 demonstrated that the pathways were markedly enriched in ubiquitin-mediated proteolysis and Hedgehog signaling pathway. The most significantly enriched BP in module 1 included protein poly-ubiquitination, ubiquitin-dependent protein catabolic process and modification-dependent macromolecule catabolic process (Figure S3). Additionally, the KEGG pathways in Module 2 were mainly associated with Chemokine signaling pathway, cytokine–cytokine receptor interaction and Neuroactive ligand–receptor interaction. The most significantly enriched BPs in Module 2 was response to lipopolysaccharide, response to molecule of bacterial origin and regulation of signaling (Figure S4).
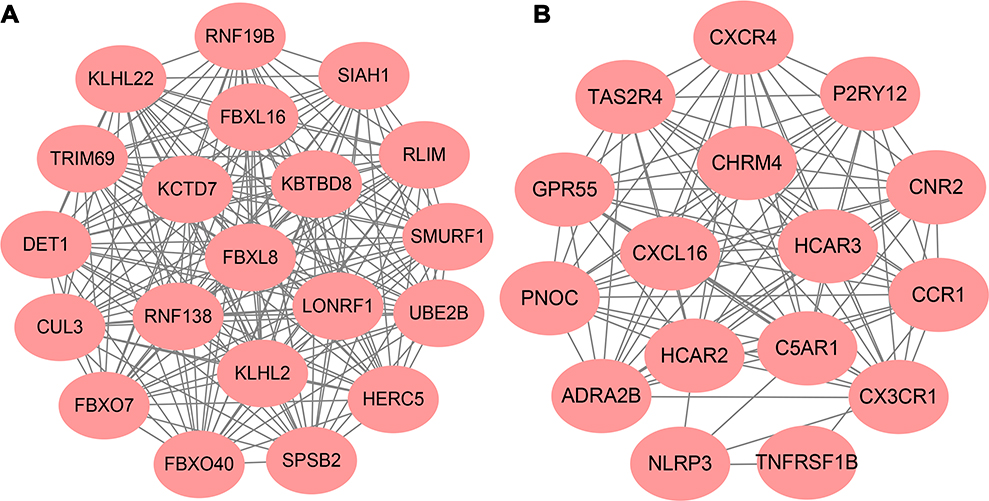 |
Figure 3 Top 2 modules of PPI sub-network by MCODE in Cytoscape software. (A) module 1; (B) module 2. |
Significant Difference of miRNA Expression
There are 9 up-regulated miRNAs and 17 down-regulated miRNAs in ATB compared with HD group with differential criteria (|log2FC|>1, adjusted P value<0.05). Among the 26 DEMs, 22 miRNAs are known, and 4 miRNAs are novel (Table 2). Additionally, 428 target genes were identified for 9 up-regulated miRNAs, and 55 target genes were identified for 8 down-regulated miRNAs (no target gene found for another 9 down-regulated miRNAs) by Miranda and RNAhybrid algorithms.
 |
Table 2 Differentially Expressed miRNAs (DEM) in ATB Compared with HDs |
miRNAs–mRNAs Network
The intersection genes between DEGs (mRNA) and the target gene of DEMs (miRNA) were determined by the Venn diagram, and found 4 up-regulated genes, and 19 down-regulated genes (Figure 4). Subsequently, the DEMs and intersection genes were used to construct the miRNA-mRNA network, and there were 5 up-regulated DEMs (miRNA) corresponding to 19 down-regulated DEG (mRNAs), and 1 down-regulated DEMs (miRNA) corresponding to 4 up-regulated DEG (mRNAs) (Figure 5).
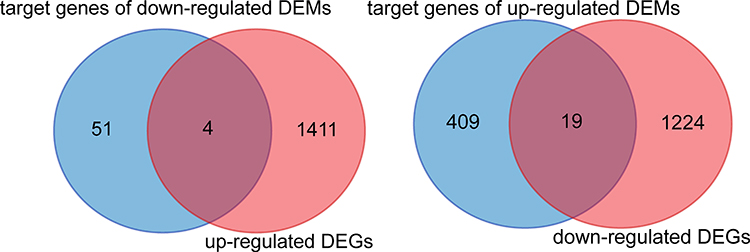 |
Figure 4 Intersection genes between DEGs and target gene of DEMs. DEGs, significantly differentially expressed genes (mRNA) between ATB and HD; DEMs, significantly differentially expressed genes (miRNA) between ATB and HD. |
 |
Figure 5 MiRNA-mRNA regulatory network was analyzed using Cytoscape software. Diamond-shaped represents miRNAs, and circular nodes represent target genes. Red, up-regulated miRNA or mRNA; green, down-regulated miRNA or mRNA in active tuberculosis subjects. |
Validation of DEMs by RT-qPCR
To confirm the DEMs resulted from HTS and Bioinformatics analysis, 5 DEMs (hsa-miR-23a-5p, hsa-miR-193a-5p, hsa-miR-941, hsa-miR-183-5p, hsa-miR-16-1-3p) were selected and validated by RT-qPCR analysis with samples of 35 ATB patients and 35 healthy donors. The results were consistent with the prediction, the expression of 4 out of the 5 miRNAs were significantly increased in the ATB patients compared to healthy donors; however, the another one (hsa-miR-16-1-3p) was significantly decreased (P=0.0026) (Figure 6).
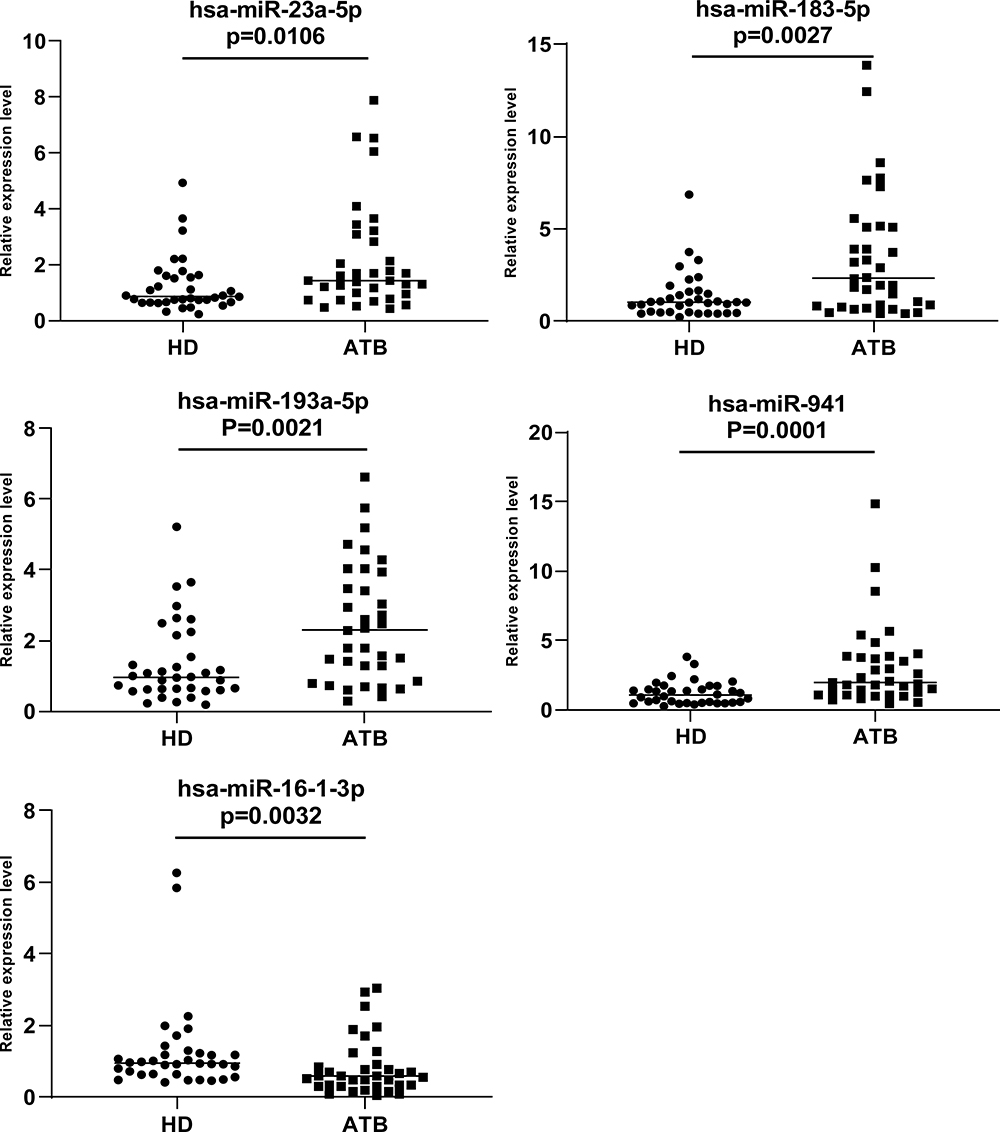 |
Figure 6 We analyzed the expression of five miRNAs (hsa-miR-23a-5p, hsa-miR-183-5p, hsa-miR-193a-5p and hsa-miR-941 and hsa-miR-16-1-3p) in 35 active tuberculosis patients versus 35 healthy donors. U6 was used as reference gene. 2-ΔΔCt method was used to quantify RT-qPCR data. Mann–Whitney test was used to evaluate the differences between groups. P value<0.05 was considered statistically significant. |
Discussion
Mycobacterium tuberculosis (Mtb) infection is a serious health problem on worldwide among the young, elderly, and immunocompromised population.15 The transcriptional profile of PBMC in the subjects with Mtb infection are complicated and could be due to multiple factors, such as age and genetic background.16 In the present study, we examined the transcriptomes of ATB patients and healthy individuals (HDs) and uncovered specific molecular features and pathways associated with ATB.
Mtb infection results in the complex intracellular bacterial infection, and the control of Mtb infection requires innate and adaptive immune responses.17,18 The cell-mediated immune response is considered to play a vital role in controlling Mtb.17 When immune cells are activated by Mtb-specific antigens, they can release proinflammatory cytokines, such as TNF-α, IFN-γ and IL-17. Interestingly, in the study, the KEGG pathway analysis showed that DEGs were enriched in TNF signaling pathway, cytokine–cytokine receptor interaction, IL-17 and MAPK signaling pathway. In previous research, TNF-α was found to initiate innate cytokine response and phagocyte activation in Mtb infection.19 In addition to controlling Mtb infection, TNF-α can also cause serious tissue damage.20 IL-17 is essential for survival following Mycobacterium tuberculosis infection.21 Recently, Choi demonstrated that the expansion of IFN-γ/IL-17-producing cells in the lung is of great significance for the protective effect in the tuberculosis subunit vaccine.22 Su et al found that Rv1016c protein inhibits the Th17 cell differentiation normally polarized by BCG-infected DCs and decreased the production of IL-17. As for the function of the MAPK signaling pathway, some studies have suggested that MMPs induce lung tissue remodeling and regulate granuloma formation after Mtb infection.23,24 Various MMP inhibitors have been used to study the immunomodulatory role of MMPs in Mtb infection and showed promising effect.25,26 For example, Xu et al reported that inhibition of matrix metalloproteinase (MMP) enhances the in vivo potency of isoniazid and rifampicin.27 In summary, our results revealed a series of processes and pathways related to Mtb infection, which provide a new basis for further explore the pathogenesis of ATB.
According to PPI analysis of DEGs, CUL3, KLHL2, KLHL22, SPSB2, KBTBD8, FBXO7, KCTD7, FBXO40, FBXL16 and FBXL8 were identified as hub genes and most of them are related to the process of ubiquitination. Ubiquitination is very important in regulating eukaryotic cell processes, including innate immune system responses and cell cycle progression.28,29 Some findings also reveal the close connection between ubiquitination and the biological process of tuberculosis. Cheng et al indicated that the host cytosolic RNA sensing pathway stimulates ICAM-1 expression through the inhibition of CRL4COP1/DET1, an E3 ubiquitin ligase, and promotes T Lymphocyte-mediated mycobacterial killing in macrophages.30 Jia et al found that Gal9 can be recruited by impaired lysosomes to regulate ubiquitination and contribute to autophagic control during Mtb infection.31 Additionally, Chai et al demonstrated that Mtb surface protein Rv1468c could recruit ubiquitin to trigger host xenophagy.32 Furthermore, miR-325-3p was found to target the ubiquitin ligase LNX1, and leads to abnormal accumulation of NEK6, thus inhibiting the cell apoptosis of Mycobacterium tuberculosis.33 Our results are consistent with other’s reports. In the study, we recognized some important genes related to ubiquitin which may regulate the biological process in Mtb infection.
Module analysis of the PPI network indicated that ATB infection was related to ubiquitin-mediated proteolysis, hedgehog signaling pathway, cytokine–cytokine receptor interaction, chemokine signaling pathway and neuroactive ligand–receptor interaction. Among them, ubiquitin-mediated proteolysis and cytokine–cytokine receptor interaction pathway have been discussed above. In addition, the chemokine signaling pathway, neuroactive ligand–receptor interaction and hedgehog signaling pathway are important pathways that should be concerned about. Chemokines play a central role in cell recruitment to the site of Mycobacterium tuberculosis infection and within the granuloma.34 Interestingly, a study reported that Mtb Heat-Shock Protein 16.3 promotes macrophages to M2 phenotype via CCRL2/CX3CR1.35 In an in vitro model of central nervous system tuberculosis, the Hedgehog pathway was down-regulated and impaired blood-brain barrier function.36 Neuroactive ligand–receptor interaction has been reported to be associated with various cancers including lung cancer by pathway analysis.37
The important function of microRNA is to regulate the expression of target genes.38 Each miRNA can have multiple target genes and affect the synthesis of multiple proteins. Several miRNAs can also jointly regulate the same target gene to promote or inhibit the process of translation. There are two main modes of action: one is that the target gene is not completely complementary to inhibit the expression of the target gene without affecting its stability; the other is that it is completely complementary with the target gene, leading to the degradation of the target gene.39 Therefore, the analysis of candidate genes and pathways related to ATB can provide a cognitive basis for the occurrence of diseases. Moreover, miRNAs are key in both adaptive and innate immunity by regulating the differentiation of various immune cell subsets as well as their immunological functions.40 In the present study, we identified 26 miRNAs significantly different between ATB and HD samples. Function of differentially expressed miRNAs (DEMs) are still not well understood, such as miR-12,316 and miR-1537. However, some miRNAs, such as hsa-miR-23a-5p, have been connected to specific signaling pathways.41 In response to Mtb infection, miR-23a-5p regulates Mtb survival and autophagy via TLR2/MyD88/NF-κB pathway by targeting TLR2.42 Consistent with our sequencing results, hsa-miR-101 was significantly decreased in the serum of pulmonary TB patients compared with healthy controls.43 High miR-183 level was found to be correlated with macrophage activity in the serum of TB patients.44 In addition, Yi et al reported that hsa-mir-19b-2 was significantly down-regulated in TB group compared with controls.45 The expression of has-mir-16 in serum was significantly down-regulated in the ATB group compared with the BCG-inoculated group.46 In this study we validated 5 significantly different miRNA (hsa-miR-23a-5p, hsa-miR-193a-5p, hsa-miR-941, hsa-miR-183-5p and hsa-miR-16-1-3p) using RT-qPCR analysis. To our knowledge, hsa-miR-193a-5p and hsa-miR-941were first reported to be significantly elevated in ATB compared with that of HD control. Furthermore, we constructed the miRNA–mRNA regulatory network to explore the function of DEMs. The result indicated that hsa-miR-23a-5p/AIFM2, hsa-miR-193a-5p/ZSCAN20, hsa-miR-941/OLFM2 regulatory axes pay a key role in ATB onset and progression, although, which should be further confirmed in the future with more cases.
Conclusion
In summary, the characteristic profiles of mRNA and miRNA in PBMCs were determined in patients with ATB infection, and some predicted targets of characteristic miRNA are identical to our determined genes, although this should be confirmed in the future with more cases. Our results could provide a theoretical basis for studying the role of miRNAs in the pathogenesis of ATB and indicate the different mRNAs and miRNAs contribute to improve the diagnosis and treatment for ATB infection and distinguish the different clinical status of Mtb infection.
Acknowledgments
We sincerely thank all of the subjects participating in the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81671557), the Zhejiang Provincial Natural Science Foundation of China (LY19H190004), and the National Science and Technology Major Project (2018ZX10101-001).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bao H, Liu K, Wu Z, et al. Tuberculosis among full-time teachers in Southeast China, 2005–2016. Int J Environ Res Public Health. 2018;15(9).
2. World Health Organization. Global tuberculosis report; 2019. Available from: https://www.who.int/tb/publications/global_report/en/.
3. Furin J, Cox H, Pai M. Tuberculosis. Lancet. 2019;393(10181):1642–1656. doi:10.1016/S0140-6736(19)30308-3
4. Tiberi S, Du Plessis N, Walzl G, et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect Dis. 2018;18(7):e183–e198. doi:10.1016/S1473-3099(18)30110-5
5. Wen A, Qu XH, Zhang KN, Leng EL, Ren Y, Wu XM. Evaluation of interferon-gamma release assays in extrasanguinous body fluids for diagnosing tuberculosis: a systematic review and meta-analysis. Life Sci. 2018;197:140–146. doi:10.1016/j.lfs.2018.02.004
6. Yang J, He J, Huang H, et al. Molecular characterization of T cell receptor beta variable in the peripheral blood T cell repertoire in subjects with active tuberculosis or latent tuberculosis infection. BMC Infect Dis. 2013;13:423. doi:10.1186/1471-2334-13-423
7. Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. doi:10.1126/science.1064921
8. Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi:10.1038/nature02871
9. KK GA F, Jan C, Lewis BP, et al. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi:10.1126/science.1121158
10. Kumar M, Sahu SK, Kumar R, et al. MicroRNA let-7 modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-kappaB pathway. Cell Host Microbe. 2015;17(3):345–356. doi:10.1016/j.chom.2015.01.007
11. Qi Y, Cui L, Ge Y, et al. Altered serum microRNAs as biomarkers for the early diagnosis of pulmonary tuberculosis infection. BMC Infect Dis. 2012;12:384. doi:10.1186/1471-2334-12-384
12. Kathirvel M, Saranya S, Mahadevan S. Expression levels of candidate circulating microRNAs in pediatric tuberculosis. Pathog Glob Health. 2020;114(5):262–270. doi:10.1080/20477724.2020.1761140
13. Migliori GB, Sotgiu G, Rosales-Klintz S, et al. ERS/ECDC statement: European Union standards for tuberculosis care, 2017 update. Eur Respir J. 2018;51(5).
14. Liao R, Lv Y, Zhu L, Lin Y. Altered expression of miRNAs and mRNAs reveals the potential regulatory role of miRNAs in the developmental process of early weaned goats. PLoS One. 2019;14(8):e0220907.
15. Houben RM, Glynn JR, Mallard K, et al. Human immunodeficiency virus increases the risk of tuberculosis due to recent re-infection in individuals with latent infection. Int J Tuberc Lung Dis. 2010;14:909–915.
16. Wang C, Yang S, Sun G, et al. Comparative miRNA expression profiles in individuals with latent and active tuberculosis. PLoS One. 2011;6(10):e25832. doi:10.1371/journal.pone.0025832
17. Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422. doi:10.1146/annurev.immunol.021908.132703
18. Natarajan K, Kundu M, Sharma P, Basu J. Innate immune responses to M. tuberculosis infection. Tuberculosis (Edinb). 2011;91(5):427–431. doi:10.1016/j.tube.2011.04.003
19. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol Spectr. 2016;4(5). doi:10.1128/microbiolspec.TBTB2-0018-2016
20. Amaral EP, Lasunskaia EB, D’Imperio-Lima MR. Innate immunity in tuberculosis: how the sensing of mycobacteria and tissue damage modulates macrophage death. Microbes Infect. 2016;18(1):11–20. doi:10.1016/j.micinf.2015.09.005
21. Shen HB, Chen ZW. The crucial roles of Th17-related cytokines/signal pathways in M. tuberculosis infection. Cell Mol Immunol. 2018;15(3):216–225. doi:10.1038/cmi.2017.128
22. Choi HG, Kwon KW, Choi S, et al. Antigen-specific IFN-gamma/IL-17-co-producing CD4(+) T-cells are the determinants for protective efficacy of tuberculosis subunit vaccine. Vaccines (Basel). 2020;8(2).
23. Sabir N, Hussain T, Mangi MH, Zhao D, Zhou X. Matrix metalloproteinases: expression, regulation and role in the immunopathology of tuberculosis. Cell Prolif. 2019;52(4):e12649. doi:10.1111/cpr.12649
24. Parasa VR, Muvva JR, Rose JF, Braian C, Brighenti S, Lerm M. Inhibition of tissue matrix metalloproteinases interferes with Mycobacterium tuberculosis-induced granuloma formation and reduces bacterial load in a human lung tissue model. Front Microbiol. 2017;8:2370. doi:10.3389/fmicb.2017.02370
25. Walker NF, Clark SO, Oni T, et al. Doxycycline and HIV infection suppress tuberculosis-induced matrix metalloproteinases. Am J Respir Crit Care Med. 2012;185(9):989–997. doi:10.1164/rccm.201110-1769OC
26. Ordonez AA, Pokkali S, Kim S, et al. Adjunct antibody administration with standard treatment reduces relapse rates in a murine tuberculosis model of necrotic granulomas. PLoS One. 2018;13(5):e0197474. doi:10.1371/journal.pone.0197474
27. Xu Y, Wang L, Zimmerman MD, et al. Matrix metalloproteinase inhibitors enhance the efficacy of frontline drugs against Mycobacterium tuberculosis. PLoS Pathog. 2018;14(4):e1006974. doi:10.1371/journal.ppat.1006974
28. Ciechanover A, Schwartz AL. The ubiquitin system: pathogenesis of human diseases and drug targeting. Biochim Biophys Acta. 2004;1695(1–3):3–17. doi:10.1016/j.bbamcr.2004.09.018
29. Liu X, Wang Q, Chen W, Wang C. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev. 2013;24(6):559–570. doi:10.1016/j.cytogfr.2013.07.002
30. Cheng Y, Kiene NJ, Tatarian A, Eix EF, Schorey JS, Hawn TR. Host cytosolic RNA sensing pathway promotes T Lymphocyte-mediated mycobacterial killing in macrophages. PLoS Pathog. 2020;16(5):e1008569. doi:10.1371/journal.ppat.1008569
31. Jia J, Bissa B, Brecht L, et al. AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell. 2020;77(5):951–969 e959. doi:10.1016/j.molcel.2019.12.028
32. Chai Q, Wang X, Qiang L, et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat Commun. 2019;10(1):1973. doi:10.1038/s41467-019-09955-8
33. Fu B, Xue W, Zhang H, et al. MicroRNA-325-3p facilitates immune escape of Mycobacterium tuberculosis through targeting LNX1 via NEK6 accumulation to promote anti-apoptotic STAT3 signaling. mBio. 2020;11(3). doi:10.1128/mBio.00557-20.
34. Monin L, Khader SA. Chemokines in tuberculosis: the good, the bad and the ugly. Semin Immunol. 2014;26(6):552–558. doi:10.1016/j.smim.2014.09.004
35. Zhang Y, Li S, Liu Q, et al. Mycobacterium tuberculosis heat-shock protein 16.3 induces macrophage M2 polarization through CCRL2/CX3CR1. Inflammation. 2020;43(2):487–506. doi:10.1007/s10753-019-01132-9
36. Brilha S, Ong CWM, Weksler B, Romero N, Couraud PO, Friedland JS. Matrix metalloproteinase-9 activity and a downregulated Hedgehog pathway impair blood-brain barrier function in an in vitro model of CNS tuberculosis. Sci Rep. 2017;7(1):16031. doi:10.1038/s41598-017-16250-3
37. Shi K, Li N, Yang M, Li W. Identification of key genes and pathways in female lung cancer patients who never smoked by a bioinformatics analysis. J Cancer. 2019;10(1):51–60. doi:10.7150/jca.26908
38. Guo L, Zhao J, Qu Y, et al. microRNA-20a inhibits autophagic process by targeting ATG7 and ATG16L1 and favors mycobacterial survival in macrophage cells. Front Cell Infect Microbiol. 2016;6:134. doi:10.3389/fcimb.2016.00134
39. Eulalio A, Huntzinger E, Izaurralde E. Getting to the root of miRNA-mediated gene silencing. Cell. 2008;132(1):9–14. doi:10.1016/j.cell.2007.12.024
40. Rebane A, Akdis CA. MicroRNAs: essential players in the regulation of inflammation. J Allergy Clin Immunol. 2013;132(1):15–26. doi:10.1016/j.jaci.2013.04.011
41. Zhang YB, Zhang XL, Zhao ZY, Zheng YL, Xiao Z, Li F. Integrated bioinformatics analysis and validation revealed potential immune-regulatory miR-892b, miR-199b-5p and miR-582-5p as diagnostic biomarkers in active tuberculosis. Microb Pathogen. 2019;134:103563. doi:10.1016/j.micpath.2019.103563
42. Gu X, Gao Y, Mu DG, Fu EQ. MiR-23a-5p modulates mycobacterial survival and autophagy during mycobacterium tuberculosis infection through TLR2/MyD88/NF-kappaB pathway by targeting TLR2. Exp Cell Res. 2017;354(2):71–77. doi:10.1016/j.yexcr.2017.03.039
43. Zhang X, Guo J, Fan S, et al. Screening and identification of six serum microRNAs as novel potential combination biomarkers for pulmonary tuberculosis diagnosis. PLoS One. 2013;8(12):e81076. doi:10.1371/journal.pone.0081076
44. Zhang XGJ, Fan S, Li Y, et al. High serum miR-183 level is associated with the bioactivity of macrophage derived from tuberculosis patients. Int J Clin Exp Pathol. 2015;8(1):655–659.
45. Yi Z, Fu Y, Ji R, Li R, Guan Z, Goletti D. Altered microRNA signatures in sputum of patients with active pulmonary tuberculosis. PLoS One. 2012;7(8):e43184. doi:10.1371/journal.pone.0043184
46. Zhang H, Sun Z, Wei W, et al. Identification of serum microRNA biomarkers for tuberculosis using RNA-seq. PLoS One. 2014;9(2):e88909. doi:10.1371/journal.pone.0088909
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.