Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Profiling of Microbial Landscape in Lung of Chronic Obstructive Pulmonary Disease Patients Using RNA Sequencing
Authors Shin D ![]() , Kim J
, Kim J ![]() , Lee JH
, Lee JH ![]() , Kim JI
, Kim JI ![]() , Oh YM
, Oh YM ![]()
Received 8 August 2023
Accepted for publication 30 October 2023
Published 10 November 2023 Volume 2023:18 Pages 2531—2542
DOI https://doi.org/10.2147/COPD.S426260
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Dongjin Shin,1,* Juhyun Kim,1,* Jang Ho Lee,2,* Jong-Il Kim,1,3– 5 Yeon-Mok Oh2
1Department of Biomedical Sciences, Seoul National University College of Medicine, Seoul, Republic of Korea; 2Department of Pulmonary and Critical Care Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea; 3Department of Biochemistry and Molecular Biology, Seoul National University College of Medicine, Seoul, Republic of Korea; 4Genomic Medicine Institute, Seoul National University, Seoul, Republic of Korea; 5Seoul National University Cancer Research Institute, Seoul, Republic of Korea
*These authors contributed equally to this work
Correspondence: Jong-Il Kim, Department of Biomedical Sciences, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul, 03080, Republic of Korea, Tel +82-2-740-8246, Email [email protected] Yeon-Mok Oh, Department of Pulmonary and Critical Care Medicine, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-Ro 43-Gil, Songpa-Gu, Seoul, 05505, Republic of Korea, Tel +82-2-3010-3136, Fax +82-2-3010-6968, Email [email protected]
Purpose: The aim of the study was to use RNA sequencing (RNA-seq) data of lung from chronic obstructive pulmonary disease (COPD) patients to identify the bacteria that are most commonly detected. Additionally, the study sought to investigate the differences in these infections between normal lung tissues and those affected by COPD.
Patients and Methods: We re-analyzed RNA-seq data of lung from 99 COPD patients and 93 non-COPD smokers to determine the extent to which the metagenomes differed between the two groups and to assess the reliability of the metagenomes. We used unmapped reads in the RNA-seq data that were not aligned to the human reference genome to identify more common infections in COPD patients.
Results: We identified 18 bacteria that exhibited significant differences between the COPD and non-COPD smoker groups. Among these, Yersinia enterocolitica was found to be more than 30% more abundant in COPD. Additionally, we observed difference in detection rate based on smoking history. To ensure the accuracy of our findings and distinguish them from false positives, we double-check the metagenomic profile using Basic Local Alignment Search Tool (BLAST). We were able to identify and remove specific species that might have been misclassified as other species in Kraken2 but were actually Staphylococcus aureus, as identified by BLAST analysis.
Conclusion: This study highlighted the method of using unmapped reads, which were not typically used in sequencing data, to identify microorganisms present in patients with lung diseases such as COPD. This method expanded our understanding of the microbial landscape in COPD and provided insights into the potential role of microorganisms in disease development and progression.
Keywords: chronic obstructive pulmonary disease, next-generation sequencing, microbiome, metagenomics
Introduction
According to the World Health Organization, chronic obstructive pulmonary disease (COPD) is currently one of the top three leading causes of death worldwide and is incurable. COPD is a heterogeneous lung disease of characterized by chronic respiratory symptoms caused by abnormalities in the bronchus or bronchiole and alveoli, resulting in persistent and often progressive airflow obstruction. 1,2 Several factors can cause the airways to become obstructive and lead to COPD, including tobacco use, occupational exposure to dusts, indoor pollution, a rare genetic condition called alpha-1 antitrypsin deficiency, childhood asthma, and infection by microbiomes.1
The main symptoms of COPD are shortness of breath, cough, and phlegm, which can get progressively worse. It is one of the most important lung diseases because it is chronic and difficult to treat, unlike pneumonia, which can be treated in the acute phase.3
Respiratory viral infections, such as influenza virus and respiratory syncytial virus, can cause inflammation of the airways, increased mucus production, and worsening of COPD symptoms, such as coughing, wheezing, and shortness of breath.4 These viral infections can also damage the epithelial cells lining the airways, making it easier for bacteria to infect the lungs, leading to secondary bacterial infections that can further exacerbate COPD symptoms.5,6 Bacterial infections can cause increased inflammation and mucus production in the airways, leading to worsening of COPD symptoms. In addition, bacterial infections can also cause pneumonia, which can be life-threatening in COPD patients. Streptococcus pneumoniae and Haemophilus influenzae, are also common in COPD patients and can cause acute exacerbations of the disease.7
Ribo-nucleic acid (RNA) sequencing (RNA-seq) is a molecular biology technique used to analyze RNA molecules that are produced during the process of transcription from deoxyribonucleic acid (DNA) to mRNA. RNA-seq allows for accurate measurement of gene expression as it occurs in an organism, and enables quantitative comparison of expressed genes between samples.
Traditionally, the study of bacteria is done by amplifying 16s rRNA. However, in this study, RNA-seq data was used to analyze the data. While it may not be validated as using 16s rRNA in microbiome study, looking at the entire transcriptome can give us clues as to which sequences in the bacteria can infect and cause disease in humans.
However, when sequencing RNA from human samples, non-human reads can be obtained due to a number of factors, including contamination during library construction, actual infection, or the presence of normal microbiome genomes.8 Reads that do not align with the human genome are called unmapped reads, which can range from 10% to 30% depending on the sample.
Even if it is not possible to analyze the entire metagenome, identifying some of the infectious and parasitic microorganisms can be of great significance depending on the disease. There are many tools available to analyze these unmapped reads, and the basic principles and methods are largely similar.9,10
We performed the analysis using RNA-seq unmapped reads from COPD patient lung tissues and non-COPD smokers’ lung tissues that were publicly available for download. There were two problems we wanted to solve. First, how reliable is the metagenome from RNA-seq, and second, if it is reliable, how much contamination or infection occurs in lung tissue, which has a lot of contact with the outside world, and how different is it in COPD and non-COPD smokers’ lung tissue.
To address the first question, we decided to conduct a meta-study of the identified microorganisms. If it was a simple sequencing error, it would have already been reported and would look almost identical in most samples. It is unlikely that we will find any species that are particularly sample-specific. The second problem was solved by first determining how much the metagenome varied between samples and between groups, and then comparing the number of reads for each bacterium or virus by heatmap.
Through this study, even though it is not a sequencing platform for analyzing metagenomes, we were able to discover new insights into sequencing data by analyzing reads that might be considered errors, and if we apply it to a larger dataset, we will be able to analyze the already generated data more deeply.
Materials and Methods
Sample and Data
We downloaded sample FASTQ files from the National Center for Biotechnology Information (NCBI) Genome Expression Omnibus (GEO) under accession number GSE57148 and received additional two non-COPD smokers’ samples and one COPD sample. Additional three samples were not stored in GSE57148, because these samples were collected after dataset upload. In the end, we analyzed 99 COPD and 93 non-COPD smokers’ samples, and the demographics of the samples are as presented in Table 1. In subsequent analyses, non-COPD smokers are referred to as the normal group for convenience.
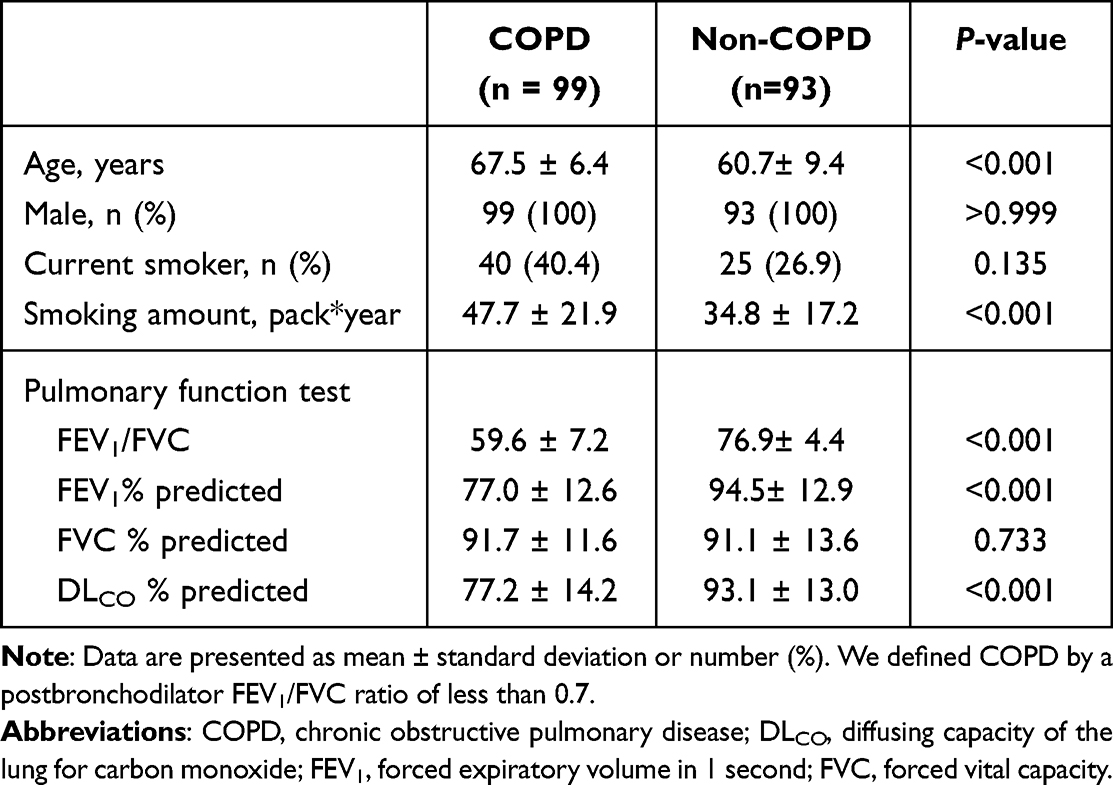 |
Table 1 Baseline Characteristics of Study Population |
Preprocessing RNA-Seq Data
The downloaded raw FASTQ was trimmed using Trimmomatic (v0.39).11 We created a human reference that had CHM13 (v1.0) + GRCh38 Y as host genome with STAR aligner (v2.7.7a)12 with --outFilterMultimapNmax 20 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --outFilterMismatchNoverLmax 0.04 --alignIntronMin 20 --alignIntronMax 1,000,000 --alignMatesGapMax 1,000,000 --outSAMunmapped Within. And then we removed technical duplicates by Picard MarkDuplicates (Picard v2.23.8).13
The aforementioned analysis was done using the computing server at the Genomic Medicine Institute Research Service Center.
Pathogen Read Detection
We utilized the Samtools14 software suite to extract unmapped reads from sequencing data, which we subsequently employed to generate paired unmapped FASTQ files. We downloaded two pre-built databases in version 12/9/2022, collectively referred to as the Standard Database, containing references for Archaea, Bacteria, Viral, Plasmid, Human, and UniVec_Core. Our analysis pipeline was based on the methodology outlined in the Nature Protocol.15 The unmapped paired reads were processed using Kraken2,16 a taxonomic classification software. We used the “--report-minimizer-data” flag when executing Kraken2 to leverage the KrakenUniq algorithm17 which uses an additional feature of unique k-mer counting for accurate pathogen identification, resulting in the generation of a Kraken report file (.report). To normalize the reads to the value of z-score within the sample and facilitate comparison of pathogen rates between samples, we submitted all the report files from our samples to the Pavian program (v1.2.0).
Alpha and Beta Diversity Analysis
Alpha diversity is used to assess the taxonomic diversity within a sample. As an evaluation metric, the Shannon’s index is used, which means that a higher value indicates more diversity in the sample. The Shannon index is a measure of how diverse the classes of data within a population are. The purpose of the measure is to determine the diversity of biological species (species, classes) within a population, ie, if we know the distribution of animals in regions A/B, we can numerically answer the question of which region has a greater diversity of species.18 It was calculated by excluding homo sapiens reads from Bracken report.
Beta diversity was calculated through the Bray–Curtis dissimilarity matrix. The Bray–Curtis dissimilarity is a statistical method named after J. Roger Bray and John T. Curtis, which is utilized to measure the degree of compositional dissimilarity between two distinct sites based on the counts of each element present in them.19 After combining the kraken report, the sum of the read counts for each species was excluded because it was calculated as an outlier. Both alpha and beta diversity was calculated using scripts included in KrakenTools (https://github.com/jenniferlu717/KrakenTools).
To determine the distance between samples, probabilistic Count Matrix Factorization (pCMF) method was used, and the r package pCMF was used. pCMF is one of the dimension reduction methods. This is based on the Gamma-Poisson hierarchical factor model and is suitable for application zero-inflated counts data.20
Validation the Pathogen Reads
To confirm the infection of each pathogen in samples, we extract the next-generation sequencing (NGS) reads from unmapped FASTQ files using extract_kraken_reads.py in the KrakenTools module with –include-children flag.
The extracted reads were identified using Nucleotide BLAST (BLASTN), where the database used the standard nucleotide collection and the organism was limited to bacteria (taxid:2). The program was optimized with highly similar sequences (megablast option was applied).21
The genome assembly of the identified species was downloaded from NCBI nucleotide database in FASTQ format and bowtie indexed (bowtie2-build -f bacterial_genome output_name). After that, re-alignment was performed using bowtie2 (bowtie2 -x reference_dir -f -p 1 −1 unmapped_fasta_read1 −2 unmapped_fasta_read2 --local -S SAM_output).
For visualization, I converted the output from Sequence Alignment Map (SAM) format to Binary Alignment Map (BAM) format, sorted and indexed using samtools, and put the BAM file and BAM index file as input to the alignment viewer of Pavian Shiny app. (https://fbreitwieser.shinyapps.io/pavian/)
Results
Optimizing Pipeline for Searching Metagenome Reads in RNA-Seq
We perform pathogen detection analysis of 99 COPD tissues with 93 control tissues of the Korean cohort. The raw RNA-seq data were obtained from the GEO read and applying pipeline, as shown in Figure 1 (A: Preprocessing step; B: Microbiome analysis step; and C: Pathogen identification analysis step).
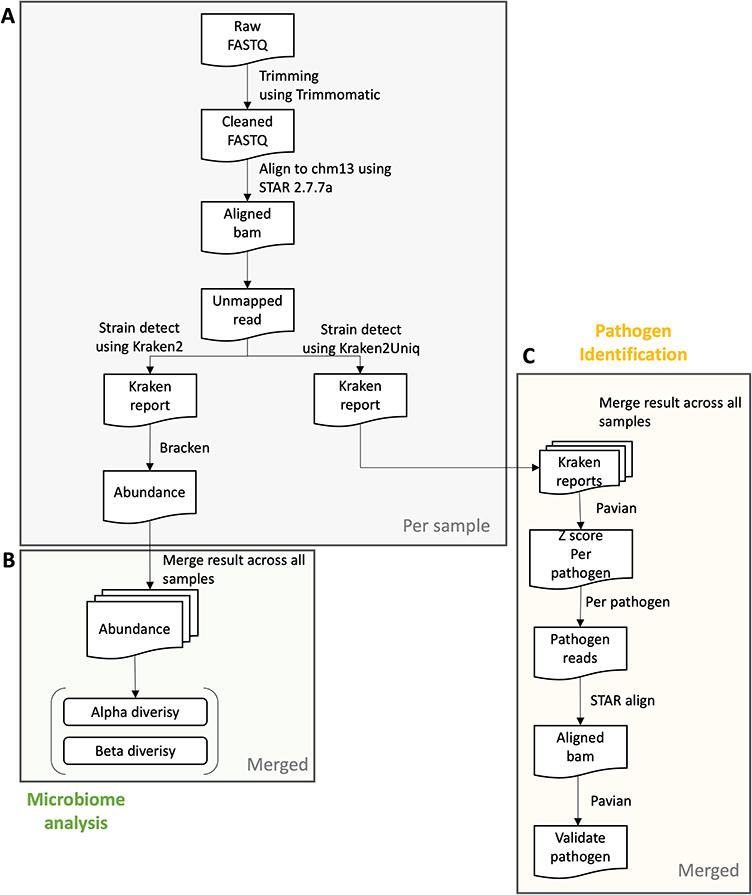 |
Figure 1 Pipeline overview. A preview of (A) the preprocessing step per sample, (B) microbiome analysis step, and (C) the pathogen Identification analysis step in integrated samples. In the preprocessing step, we start with a FASTQ file and trim the reads according to the quality of each base and adapter sequences. Then, we align the cleaned reads to CHM13 reference using STAR aligner and extract the reads that do not align with the reference. Alpha and beta diversity can be calculated from abundance determined using Kraken2 and Bracken. To conduct the pathogen identification step, we assign the unmapped reads to the species of Kraken2 database which includes archaea, bacteria, viral, plasmid, etc. Normalized detection amounts are obtained from the z-score from the raw detected count in each sample. The top pathogen in each sample may be a real infected pathogen of the sample. Abbreviations: COPD, chronic obstructive pulmonary disease. |
The obtained raw reads were trimmed based on their base quality and adapter sequence similarity. The cleaned reads were processed pathogen identification step consisted of (1) removal of host DNA from microbial read; (2) classification of the remaining microbial reads; (3) comparison of sample reads against control samples; and (4) validation of pathogen classifications Kraken protocol.15 We used the CHM13 (v.1.0) reference during removing host DNA by aligning the cleaned reads to the reference, because CHM13 is a full human reference without gaps22 to remove as many host reads as possible. However, at the time we conducted the analysis, there were no full Y chromosome sequences in CHM13. We concatenated the Y chromosome of GRCh38 to the CHM13 autosome, X chromosome and mitochondrial chromosome to make a reference (CHM13+Y_GRCh38). We retrieved all of the unmapped reads to CHM13+Y_GRCh38 based on the flag of reads. These unmapped paired reads are an input into Kraken2 software and assigned to each microbiome species (Figure 1A). To determine the inter-sample and inter-group diversity of the species found, we calculated alpha and beta diversity by merging all the resulting files (Figure 1B). The Kraken reports files are uploaded to the Pavian Shiny App, which compiles all of the read counts per species and allows between-sample visualization and comparison by calculating the z-scores between read counts, with higher z-scores meaning evidence of infection for each sample. Finally, pathogen identification can be validated using KrakenTools by extract classified reads to the species in sample and double-confirm the reads really come from the species by using BLAST and aligning to the species using Bowtie (Figure 1C).
Statistics of Analyzed Unmapped Reads
The amount of metagenome detectable in the RNA-seq data was significantly less. The goal of this study was to identify reads that provided evidence of infection in the unmapped reads. First, the unmapped reads were average 2 million reads in COPD patients and normal, and the classified reads were 95% to each species including human genome, bacterial, and virus. Microbial reads accounted for approximately 10% of the reads, while viral reads accounted for only 0.01%. Both of these percentages are considered to be low. All statistical information is organized in Supplementary Table 1.
Comparison Diversity Between COPD and Normal Groups
First, we calculated alpha diversity for each sample, which describes the species richness within a sample, and found that the COPD group had significantly higher alpha diversity (t-test, p-value < 0.05) However, there was a difference in the distribution, with alpha diversity in the normal group being widely spread out, while in the COPD group it tended to cluster around a Shannon index of 2.4. This suggests that there is a greater diversity of species but with less heterogeneity between samples in the COPD group compared to the normal group (Figure 2A).
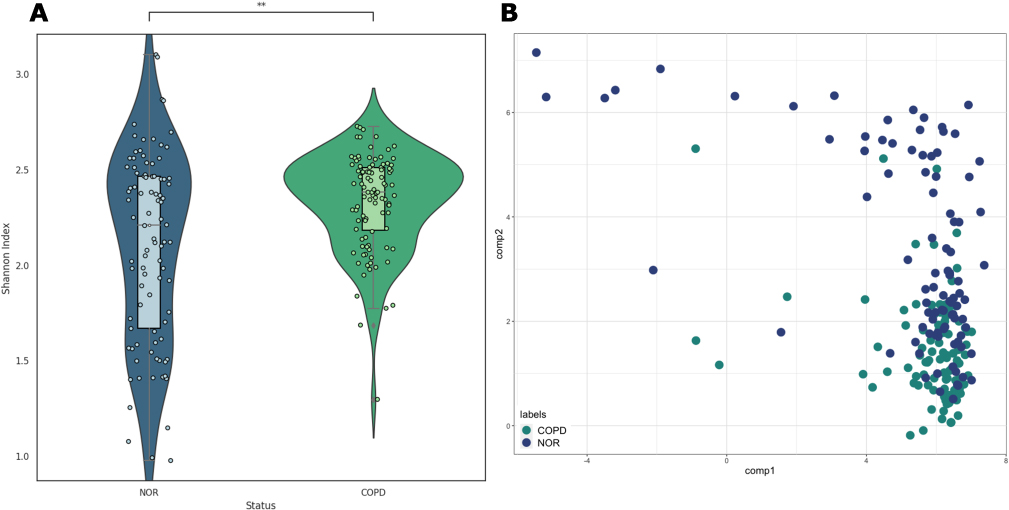 |
Figure 2 Alpha and beta diversity. (A) alpha diversity (Shannon Index) of normal and COPD groups; the Mann–Whitney U test resulted in a p-value of 2.185e-03. (B) Probabilistic count matrix factorization (pCMF) results using raw read counts. Bray–Curtis dissimilarity (beta diversity) is 0.610. Abbreviation: COPD, chronic obstructive pulmonary disease. |
Beta diversity can refer to the difference in diversity between communities and the difference in diversity between samples, and to check both, we used the pCMF method based on the Gamma-Poisson hierarchical factor model. This methodology is best suited for analyzing very sparse matrices like the data in this study. The results show that there is no significant difference between the normal and COPD groups, but rather different diversity among the samples (Figure 2B). According to the Bray–Curtis dissimilarity calculation, the beta diversity is 0.610, where 0 means that all species are equally distributed and 1 means that they are completely different, so about 40% have the same species.
Specific Bacteriome in COPD Patients
By selecting only those species whose calculated rates differed by more than 10 between the two groups, as shown in Figure 3A, we were able to identify a total of 18 species. Based on the assigned for each species, we calculated the infection rate between the COPD group and the normal group in Figure 3B. Of these, 5 species were found to be more prevalent in the COPD group and 13 species were found to be more prevalent in the normal group. Of these, for Yersinia enterocolitica, a higher number of reads were found in the COPD samples (Figure 3C). This can be seen more intuitively when comparing the normalized read counts observed in the COPD and Normal groups. The normalized counts were found to be higher in COPD, with a p-value of 0.029 (Figure 3D).
 |
Figure 3 Comparison of overall metagenomic profile between normal and COPD (A). Top 18 species that differ between normal and COPD by z-score. (B) Positive rate detected in Kraken2 Standard database. The rank was sorted by the number of differences between COPD and the normal groups. (C)The percentage of detected reads in each sample. It was sorted by the amount of detected percentage and by the color description of the group. Black: COPD, white: normal (D) Violin plot of Yersinia enterocolitica z-score by normal and COPD (*1.00e-02 < p ≤ 5.00e-02). Abbreviation: COPD, chronic obstructive pulmonary disease. |
In the case of Yersinia enterocolitica, primarily inhabit numerous mammals, birds, cold-blooded animals, and even terrestrial and aquatic crevices.23,24 Lung infections of Yersinia enterocolitica have been known for a long time, and it is a known bacteria that causes pneumonia.25–30 Moraxella osloensis has also been found in the human respiratory tract and has been reported to cause severe pneumonia in lung cancer patients.31–34 It was found more in normal tissues, but given that both samples were COPD lesion tissue and normal tissue from cancer patients, they could be sufficiently infected. Micrococcus luteus has been well studied in relation to asthma, with a recent study showing that extracellular vesicles derived from Micrococcus luteus inhibit interleukin (IL)-1β production by regulating miRNAs in airway epithelial cells.35 Gardnerella vaginalis is usually found in the vagina of women, but rare infections in the lungs of men have been reported.36 We have also seen it observed in pneumonia patients through case reports.37 Staphylococcus haemolyticus is well known for infections in the lungs and has been reported to increase survival after first line treatment, especially in patients with non-small cell lung cancer.38,39 It was also found in a study of the lung microbiota in patients with idiopathic pulmonary fibrosis.40 Despite its well-recognized role in the lungs, its function has been poorly understood, with lung cancer being the most studied. Pseudomonas sp. OE 28.3 is a member of the Pseudomonas genus, which includes Pseudomonas aeruginosa, Pseudomonas oryzihabitans, and Pseudomonas plecoglossicida, all of which have well-documented roles as pathogens, especially Pseudomonas aeruginosa, which has been found in the respiratory tract of patients with cystic fibrosis.41 Cutibacterium acnes is primarily a skin colonizer, but very rarely extracutaneous infections are found, and pleural infections have been reported in COPD patients.42 It has also been reported as the causative agent of lung abscesses in patients with COPD.43 There have also been reports of infections following skin biopsies in patients with metastatic lung cancer, resulting in pericarditis and empyema.44
Although Xanthomonas campestris, Pseudomonas ficuserectae, Rhizobium oryzihabitans, Schlegelella aquatica, Pseudomonas yamanorum, and Rubrobacter xylanophilus exhibited significant differences between the COPD and normal groups, the six bacteria are not human normal flora and there was no report as pathogens in human.
This shows that even NGS data can contain metagenomic reads and that this is not something to be ignored. Unmapped reads can also contain infected bacteria or viruses. In particular, lung tissue is one of the organs that is in frequent contact with the outside world, so the probability of exposure to various microorganisms is high, which makes such an analysis meaningful.
Validation of Detected Reads
To eliminate false positives due to contamination, extracted reads were assigned to each species and ran them through BLAST, and then removed the mismatches between the two results from different databases that were regarded as false positives.
We could re-validate these by checking how they aligned with the reference genome of each identified bacteria. Since the data in this study are not targeted to 16S rRNA, alignment distribution was expected to be uniformly spread across genome. We visualized these results using Pavian Shiny app.
As a result of the validation of 44 differential species between the COPD and normal groups, 26 were judged as false positives, and the BLAST results showed that the extracted reads were indeterminate to other species. The results are summarized in Supplementary Table 2. Interestingly, 19 of the 26 cases were identified as Staphylococcus aureus strain WH9628 in BLAST databases. Staphylococcus aureus is a bacterium that is primarily present on the skin,45 indicating that it may have been contaminated during sample preparation or library preparation. Despite the use of paired-end data, we found that reads 1 and 2 were irregularly aligned to the reference genome of the strain (Figure 4A).
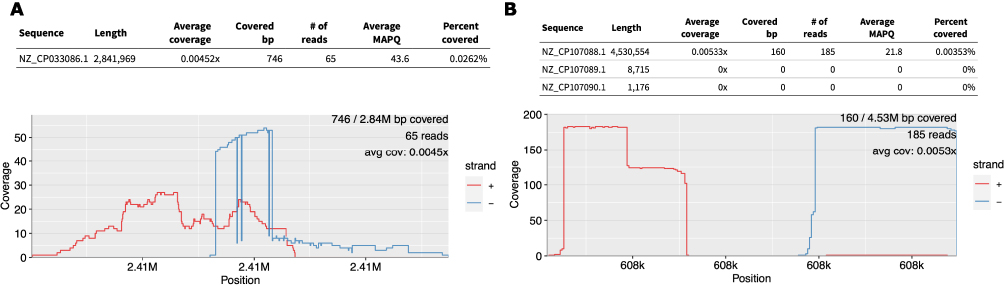 |
Figure 4 Re-alignment results for each species shown by Pavian Shiny app. (A) Case of false positive, detected as Saccharopolyspora rosea, but actually aligned to the Staphylococcus aureus genome. (B) Result of Yersinia enterocolitica. |
However, in the case of Yersinia enterocolitica, which showed the largest difference between the COPD and normal groups, BLAST showed that it was strain GP200, and alignment to the corresponding genome confirmed that the extracted read1 and read2 were aligned normally (Figure 4B).
Discussion
In this study, we aimed to investigate the microbiological differences between COPD and normal tissues using RNA-seq. To achieve this, we employed a multifaceted approach to identify and validate evidence of infection, which included using Kraken1, Kraken2, and BLAST. In the final step, we re-aligned assigned reads with each reference genome of the strain to ensure that the assigned reads originated from the strain.
However, there are some controversial points in analyzing metagenomics using NGS data. This is because the RNA of eukaryotes is significantly different from that of prokaryotes, starting with the way it is produced. In prokaryotes, transcription and translation are coupled by a single RNA polymerase, occur rapidly and produce half of the transcriptome without a poly A tail. On the other hand, eukaryotes require three RNA polymerases to produce different types of RNA, mostly with a poly A tail. However, many previous studies have found evidence of infections caused by various viruses and bacteria, which motivated us to conduct metagenomic analysis using RNA-seq data.46–50
The alpha diversity values were similar between COPD and normal tissues, but the normal tissue samples had diverse values of alpha diversity, indicating a wider but restricted range of microbiome communities. This suggests that COPD tissues may have a distinct microbiome community compared to normal tissues. Based on our analysis, we hypothesize that Micrococcus luteus and Yersinia enterocolitica might be related to development of COPD, rather than a false positive. Here, we would like to suggest that Micrococcus luteus and Yersinia enterocolitica could be a potential causative agent of COPD development.
One of the distinct characteristics of microbiome in COPD was decreased microbiome diversity.51 This characteristic suggested that deficiency of specific bacterium could be related to the development of COPD. In our study, Micrococcus luteus was more commonly detected in normal group, and this result was in line with the result of previous study.52 Sim et al reported that Micrococcus luteus-derived extracellular vesicles reduced neutrophilic airway inflammation by inhibiting reducing IL-1β and IL-17 production by regulating miRNAs in airway epithelial cells.33 Because IL-1β and IL-17 levels are elevated in some COPD patients, it is possible that colonization of Micrococcus luteus in lung tissue has a protective effect against the development of COPD.53–55
COPD is characterized by inflammation, excessive mucus secretion, and bronchial mucosal epithelial lesions, and the release of large amounts of inflammatory factors have all been implicated in the development of COPD.56
Yersinia enterocolitica has been linked to lung infections in several case reports, with symptoms including pneumonia, lung abscesses, mediastinal adenopathy, and other lung diseases.25,57,58 However, these findings have not been observed at the cohort level. At the molecular level, Yersinia enterocolitica infection has been shown to promote highly inflammatory responses by affecting T-cell function,59,60 with IL-12 playing a role in preventing this. Interestingly, previous studies have also linked IL-27, a member of the IL-12 family, to an increased risk of COPD.61 In particular, it has been shown to increase the inflammatory response during lung infections, and IL-12 plays a role in preventing this.27 Our study presented that Yersinia enterocolitica is more frequently found in COPD lung tissue than in normal lung tissue at the cohort level. When we analyzed the association with clinical information based on the detection status of Yersinia enterocolitica, we found that the detection rate increased with smoking history, but it was not enough to find an association with lung function. We could only speculate that the detection rate of Yersinia enterocolitica could be influenced by smoking (Supplementary Figure 1). These findings suggest that further study might be required to demonstrate association between Yersinia enterocolitica infections and COPD development.
Including two forementioned bacteria, 18 species presented different lung colonization rate between the COPD and normal groups. Interestingly, these species were rarely reported in previous studies about microbiome in COPD patients. One of the reasons is that we utilized resected lung tissue for this study. Most of the studies related to lung microbiome utilized sputum sample or bronchoalveolar lavage fluid.62 Intraindividual difference in lung microbiome according to sample type was well known.63 Among various sample types, lung tissue could provide direct information related to microbiome in lung parenchyma.62 Because it was difficult to directly investigate microbiome of lung tissue and there was only limited study, the result of this study might be valuable and trigger further studies.
Of course, it is very difficult to identify the causative agent because microbial infections vary depending on the region, climate, diet, etc. Therefore, it is thought that more diverse sequencing techniques should be applied for COPD-specific metagenomics or metataxonomic in East Asia. In particular, the metagenomic reads contained in the unmapped reads of the RNA-seq data found in this study are not considered as simple contamination, but rather can provide clues to know more about the symbiotic relationship between bacteria and humans. Therefore, expanding the resources for metagenomic research means that more diverse research methods are possible, which will be very helpful in researching how to distinguish between symbiosis and parasitism and how to understand the infection mechanism of pathogens.
Conclusion
Through this study, we identified the microorganisms that are commonly detected in COPD patients and confirmed their distinct populations compared to normal lung tissue. While RNA-seq data analysis is typically limited to gene expression profiling, we demonstrated the potential of metagenomic studies utilizing unmapped reads through various methodologies. This allowed us to explore the utilization of diverse NGS data in the analysis of lung disease patient data. The results of this study contribute to a broader understanding of the microbial component of COPD and demonstrate the value of integrating metagenomics into respiratory research.
Abbreviations
COPD, chronic obstructive pulmonary disease; NGS, next-generation sequencing; RNA, ribonucleic acid; DNA, deoxyribonucleic acid; RNA-seq, RNA sequencing; SAM, Sequence Alignment Map; BAM, Binary Alignment Map; IL, interleukin; pCMF, probabilistic Count Matrix Factorization; BLAST, Basic Local Alignment Search Tool; BLASTN, Nucleotide BLAST; FEV1, forced expiratory volume in 1 second.
Data Sharing Statement
The data used in this study are all publicly available in the Gene Expression Omnibus (GEO). The accession number is GSE57148. The additional three samples (1 COPD sample, 2 normal samples) are available from the respective authors upon reasonable request.
Ethics Approval and Informed Consent
The study protocol was approved by the Institutional Review Board of the Asan Medical Center (IRB no. 2021-1337). The board waived the requirement for obtaining patient informed consent because of the nature of the analysis. All provided data from the GlaxoSmithKline were anonymized, and this study did not present any identifiable and private information.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2020R1A6A1A03047972), as well as the Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2021R1A6C101A445).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Global strategy for prevention, diagnosis and management of COPD: 2023 report. 2023. Available from: https://goldcopd.org/2023-gold-report-2/.
2. Agustí A, Celli BR, Criner GJ, et al. Global initiative for chronic obstructive lung disease 2023 report: GOLD executive summary. Eur Respir J. 2023;61(4):2300239. doi:10.1183/13993003.00239-2023
3. Miravitlles M, Ribera A. Understanding the impact of symptoms on the burden of COPD. Respir Res. 2017;18(1):67. doi:10.1186/s12931-017-0548-3
4. Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. The Lancet. 2007;370(9589):786–796. doi:10.1016/S0140-6736(07)61382-8
5. Ghosh B, Gaike AH, Pyasi K, et al. Bacterial load and defective monocyte-derived macrophage bacterial phagocytosis in biomass smoke-related COPD. Eur Respir J. 2019;53(2):1702273. doi:10.1183/13993003.02273-2017
6. Singh R, Mackay AJ, Patel AR, et al. Inflammatory thresholds and the species-specific effects of colonising bacteria in stable chronic obstructive pulmonary disease. Respir Res. 2014;15(1):114. doi:10.1186/s12931-014-0114-1
7. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. New England Journal of Medicine. 2008;359(22):2355–2365. doi:10.1056/NEJMra0800353
8. Strong MJ, Xu G, Morici L, et al. Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLoS Pathog. 2014;10(11):e1004437. doi:10.1371/journal.ppat.1004437
9. Xu G, Strong MJ, Lacey MR, Baribault C, Flemington EK, Taylor CM. RNA CoMPASS: a dual approach for pathogen and host transcriptome analysis of RNA-seq datasets. PLoS One. 2014;9(2):e89445. doi:10.1371/journal.pone.0089445
10. Liebhoff A-M, Menden K, Laschtowitz A, Franke A, Schramm C, Bonn S. Pathogen detection in RNA-seq data with Pathonoia. BMC Bioinformatics. 2023;24(1). doi:10.1186/s12859-023-05144-z
11. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
12. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi:10.1093/bioinformatics/bts635
13. Broad Institute. Picard toolkit. Broad Institute; 2019. Available from: https://broadinstitute.github.io/picard/.
14. Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10(2). doi:10.1093/gigascience/giab008
15. Lu J, Rincon N, Wood DE, et al. Metagenome analysis using the Kraken software suite. Nat Protoc. 2022;17(12):2815–2839. doi:10.1038/s41596-022-00738-y
16. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20(1):257. doi:10.1186/s13059-019-1891-0
17. Breitwieser FP, Baker DN, Salzberg SL. KrakenUniq: confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 2018;19(1):198. doi:10.1186/s13059-018-1568-0
18. Shannon CE. A mathematical theory of communication. The Bell System Technical Journal. 1948;27(3):379–423. doi:10.1002/j.1538-7305.1948.tb01338.x
19. Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecological Monographs. 1957;27(4):325–349. doi:10.2307/1942268
20. Durif G, Modolo L, Mold JE, Lambert-Lacroix S, Picard F. Probabilistic count matrix factorization for single cell expression data analysis. Bioinformatics. 2019;35(20):4011–4019. doi:10.1093/bioinformatics/btz177
21. Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schaffer AA. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24(16):1757–1764. doi:10.1093/bioinformatics/btn322
22. Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science. 2022;376:6588):44–53. doi:10.1126/science.abj6987
23. Robins‐Browne RM. Yersinia enterocolitica. Food Microbiol. 2012;2012:339–376.
24. Fàbrega A, Vila J. Yersinia enterocolitica: pathogenesis, virulence and antimicrobial resistance. Enfermed Infecc Y Microbiol Clin. 2012;30(1):24–32. doi:10.1016/j.eimc.2011.07.017
25. Bigler RD, Atkins RR, Wing EJ. Yersinia enterocolitica lung infection. Arch Intern Med. 1981;141(11):1529–1530. doi:10.1001/archinte.1981.00340120137029
26. Cropp AJ, Gaylord SF, Watanakunakorn C. Cavitary pneumonia due to Yersinia-enterocolitica in a healthy man. Am J Med Sci. 1984;288(3):130–132. doi:10.1097/00000441-198410000-00007
27. Gutierrez JG, Valdez SR, Di Genaro S, Gomez NN. Interleukin-12p40 contributes to protection against lung injury after oral Yersinia enterocolitica infection. Inflamm Res. 2008;57(11):504–511. doi:10.1007/s00011-008-7162-2
28. Wong KK, Fistek M, Watkins RR. Community-acquired pneumonia caused by Yersinia enterocolitica in an immunocompetent patient. J Med Microbiol. Apr. 2013;62(Pt 4):650–651. doi:10.1099/jmm.0.053488-0
29. Greene JN, Herndon P, Nadler JP, Sandin RL. Case report: Yersinia enterocolitica necrotizing pneumonia in an immunocompromised patient. Am J Med Sci. 1993;305(3):171–173. doi:10.1097/00000441-199303000-00008
30. Dempsey T, Kalra S. Yersinia enterocolitica pneumonia: a rare presentation of a common organism. B52 BACTERIAL INFECTION CASE REPORTS. Am Thora Soc. 2018;2018:A3586–A3586.
31. Vandamme P, Gillis M, Vancanneyt M, Hoste B, Kersters K, Falsen E. Moraxella lincolnii sp. nov, isolated from the human respiratory-tract, and reevaluation of the taxonomic position of Moraxella osloensis. Int J Syst Bacteriol. 1993;43(3):474–481. doi:10.1099/00207713-43-3-474
32. Lee WS, Hsueh PR, Yu FL, Chen FL, Hsieh TC, Ou TY. Moraxella osloensis bacteremia complicating with severe pneumonia in a patient with lung cancer. J Microbiol Immunol. 2017;50(3):395–396. doi:10.1016/j.jmii.2015.03.005
33. Bilyk V, Ali O, Moghrabi A. Moraxella osloensis bacteremia with pneumonia: first reported case in Israel. Harefuah. 2020;159(3):163–165.
34. Koleri J, Petkar HM, Husain AA, Almaslamani MA, Omrani AS. Moraxella osloensis bacteremia, a case series and review of the literature. IDCases. 2022;e01450. doi:10.1016/j.idcr.2022.e01450
35. Sim S, Lee DH, Kim KS, et al. Micrococcus luteus-derived extracellular vesicles attenuate neutrophilic asthma by regulating miRNAs in airway epithelial cells. Exper Mol Med. 2023;55(1):196–204. doi:10.1038/s12276-022-00910-0
36. Wu SM, Hu WH, Xiao W, Li YX, Huang Y, Zhang X. Metagenomic next-generation sequencing assists in the diagnosis of gardnerella vaginalis in males with pleural effusion and lung infection: a case report and literature review. Infect Drug Resist. 2021;14:5253–5259. doi:10.2147/Idr.S337248
37. Souhami L, Feld R, Tuffnell PG, Feller T. Micrococcus luteus pneumonia: a case report and review of the literature. Medical and Pediatric Oncology. 1979;7(4):309–314. doi:10.1002/mpo.2950070404
38. Zhang M, Zhang Y, Han Y, Zhao X, Sun Y. Characteristics of pathogenic microbes in lung microenvironment of lung cancer patients without respiratory infection. J BUON. 2021;26:1862–1870.
39. Zhang M, Zhang Y, Sun Y, Wang S, Liang H, Han Y. Intratumoral microbiota impacts the first-line treatment efficacy and survival in non-small cell lung cancer patients free of lung infection. J Healthc Eng. 2022;2022(5466853). doi:10.1155/2022/5466853
40. D’Alessandro-Gabazza CN, Yasuma T, Kobayashi T, et al. Inhibition of lung microbiota-derived proapoptotic peptides ameliorates acute exacerbation of pulmonary fibrosis. Nat Commun. 2022;13(1):1558. doi:10.1038/s41467-022-29064-3
41. Woods DE, Bass JA, Johanson WG, Straus DC. Role of adherence in the pathogenesis of Pseudomonas aeruginosa lung infection in cystic fibrosis patients. Infect Immun. 1980;30(3):694–699. doi:10.1128/iai.30.3.694-699.1980
42. Cobo F, Borrego J, Rodriguez-Granger J, Sampedro A, Navarro-Mari JM. A rare case of pleural infection due to Propionibacterium acnes (Cutibacterium acnes). Rev Esp Quim. 2018;31(2):173–174.
43. Adlakha A, Muppala N. Propionibacterium acnes: an uncommon cause of lung abscess in chronic obstructive pulmonary disease complicated with bullous emphysema. Journal of Osteopathic Medicine. 2022;122(10):493–497. doi:10.1515/jom-2021-0240
44. Steven RD, Papia K, Subhashis M. Bacterial pericarditis and empyema caused by Cutibacterium acnes in a patient with metastatic lung cancer. Anaerobe. 2021;70:102365. doi:10.1016/j.anaerobe.2021.102365
45. Deurenberg RH, Stobberingh EE. The evolution of Staphylococcus aureus. Infection, Genetics and Evolution. 2008;8(6):747–763. doi:10.1016/j.meegid.2008.07.007
46. Selitsky SR, Marron D, Hollern D, et al. Virus expression detection reveals RNA-sequencing contamination in TCGA. BMC Genomics. 2020;21(1):79. doi:10.1186/s12864-020-6483-6
47. Elbasir A, Ye Y, Schaffer DE, et al. A deep learning approach reveals unexplored landscape of viral expression in cancer. Nat Commun. 2023;14(1):785. doi:10.1038/s41467-023-36336-z
48. Zapatka M, Borozan I, Brewer DS, et al. The landscape of viral associations in human cancers. Nat Genet. 2020;52(3):320–330. doi:10.1038/s41588-019-0558-9
49. Croucher NJ, Thomson NR. Studying bacterial transcriptomes using RNA-seq. Curr Opin Microbiol. 2010;13(5):619–624. doi:10.1016/j.mib.2010.09.009
50. Melnick M, Gonzales P, LaRocca TJ, et al. Application of a bioinformatic pipeline to RNA-seq data identifies novel virus-like sequence in human blood. G3 (Bethesda). 2021;11(9). doi:10.1093/g3journal/jkab141
51. Ditz B, Christenson S, Rossen J, et al. Sputum microbiome profiling in COPD: beyond singular pathogen detection. Thorax. 2020;75(4):338–344. doi:10.1136/thoraxjnl-2019-214168
52. Zakharkina T, Heinzel E, Koczulla RA, et al. Analysis of the airway microbiota of healthy individuals and patients with chronic obstructive pulmonary disease by T-RFLP and clone sequencing. PLoS One. 2013;8(7):e68302. doi:10.1371/journal.pone.0068302
53. Zou Y, Chen X, Liu J, et al. Serum IL-1beta and IL-17 levels in patients with COPD: associations with clinical parameters. Int J Chron Obstruct Pulmon Dis. 2017;12:1247–1254. doi:10.2147/COPD.S131877
54. Le Rouzic O, Pichavant M, Frealle E, Guillon A, Si-Tahar M, Gosset P. Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur Respir J. 2017;50(4):1602434. doi:10.1183/13993003.02434-2016
55. Osei ET, Brandsma CA, Timens W, Heijink IH, Hackett TL. Current perspectives on the role of interleukin-1 signalling in the pathogenesis of asthma and COPD. Eur Respir J. 2020;55(2):1900563. doi:10.1183/13993003.00563-2019
56. Guo P, Li R, Piao TH, Wang CL, Wu XL, Cai HY. Pathological mechanism and targeted drugs of COPD. Int J Chron Obstruct Pulmon Dis. 2022;17:1565–1575. doi:10.2147/COPD.S366126
57. Sebes JI, Mabry EH, Rabinowitz JG. Lung abscess and osteomyelitis of rib due to Yersinia enterocolitica. Chest. 1976;69(4):546–548. doi:10.1378/chest.69.4.546
58. Dejima A, Yamamoto N, Hasatani K. Yersinia enterocolitica infection with septic pulmonary embolism and liver and intestinal lymph node abscesses. BMJ Case Rep. 2021;14(4):e242524. doi:10.1136/bcr-2021-242524
59. Echeverry A, Saijo S, Schesser K, Adkins B. Yersinia enterocolitica promotes robust mucosal inflammatory T-cell immunity in murine neonates. Infect Immun. 2010;78(8):3595–3608. doi:10.1128/IAI.01272-09
60. Sugiura Y, Kamdar K, Khakpour S, Young G, Karpus WJ, DePaolo RW. TLR1-induced chemokine production is critical for mucosal immunity against Yersinia enterocolitica. Mucosal Immunol. 2013;6(6):1101–1109. doi:10.1038/mi.2013.5
61. Huang N, Liu L, Wang XZ, Liu D, Yin SY, Yang XD. Association of interleukin (IL)-12 and IL-27 gene polymorphisms with chronic obstructive pulmonary disease in a Chinese population. DNA Cell Biol. 2008;27(9):527–531. doi:10.1089/dna.2007.0715
62. Carney SM, Clemente JC, Cox MJ, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62(3):283–299. doi:10.1165/rcmb.2019-0273TR
63. Cabrera-Rubio R, Garcia-Nunez M, Seto L, et al. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. 2012;50(11):3562–3568. doi:10.1128/JCM.00767-12
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Application of Different Sampling Methods Combined with Metagenomic Next-Generation Sequencing to Detect Pathogens in Children with Severe Pneumonia on Mechanical Ventilation
Nan X, Yu J, Liang Y, Lou P, Pan G
Infection and Drug Resistance 2023, 16:4545-4553
Published Date: 12 July 2023
Clinical Application and Evaluation of Metagenomic Next-Generation Sequencing for Lower Respiratory Tract Infections and Human Tumor Screening

Huang J, Weng H, Ye L, Jiang M, Li Y, Li H
International Journal of General Medicine 2023, 16:5763-5777
Published Date: 7 December 2023