")
Back to Journals » OncoTargets and Therapy » Volume 9
Profile of panitumumab as first-line treatment in patients with wild-type KRAS metastatic colorectal cancer
Authors Patel S, Gill D , Garrido-Laguna I
Received 7 May 2015
Accepted for publication 27 October 2015
Published 30 December 2015 Volume 2016:9 Pages 75—86
DOI https://doi.org/10.2147/OTT.S68558
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Daniele Santini
Shiven B Patel,* David Gill,* Ignacio Garrido-Laguna
Department of Internal Medicine, Oncology Division and Center for Investigational Therapeutics, Huntsman Cancer Institute, University of Utah, Salt Lake City, UT, USA
*These authors contributed equally to this work
Abstract: Targeted therapies against EGFR, vascular endothelial growth factor, and vascular endothelial growth factor receptor have expanded treatment options for patients with metastatic colorectal cancer (mCRC). Unfortunately, biomarkers to identify patients that are most likely to derive benefit from targeted therapies in this disease are still needed. Indeed, only RAS mutations have been identified as predictive of lack of benefit from monoclonal antibodies against EGFR in patients with mCRC. Panitumumab is a fully humanized monoclonal antibody against EGFR. In this study, we review data to support the use of panitumumab in combination with a chemotherapy backbone, in the first line setting in patients with RAS wild-type mCRC. Ongoing efforts are aimed at identifying smaller subsets of patients within the RAS wild-type group that will derive the largest benefit from anti-EGFR therapy. In the meantime, treatment with anti-EGFR therapy should be reserved for patients with RAS wild-type mCRC.
Keywords: panitumumab, metastatic colorectal cancer, first-line, RAS
Introduction
Colorectal cancer (CRC) is the second most common cause of cancer death in the United States and Europe,1,2 although its incidence is steadily decreasing in the US.3 This is likely secondary to increased screening (colonoscopy) and resection of premalignant lesions (adenomatous polyps).4 Both in the US and Europe, survival is also steadily increasing.3,5 Palliative chemotherapy is the backbone of treatment for patients with unresectable metastatic colorectal cancer (mCRC). In these patients, 5-fluorouracil or its derivatives have been the standard cytotoxic treatment for nearly 50 years.6 Modulated 5-fluorouracil resulted in improved response rate (RR) with modest increments in survival. In the last decade, the addition of new cytotoxic agents (oxaliplatin, irinotecan) and, more recently, targeted therapies such as antiangiogenic agents (bevacizumab, aflibercept, regorafenib) or monoclonal antibodies against EGFR ( cetuximab or panitumumab) have contributed to improving the outcomes of patients with newly diagnosed unresectable mCRC. Currently, treatment strategies for these patients are based on a continuum care paradigm whereby patients are exposed throughout the course of their disease to different active drugs, their treatment is personalized according to the need for rapid response and the burden of disease and RAS status, drugs are often reintroduced if they showed activity in a previous line of therapy, and finally, intervals of maintenance chemotherapy are considered.7 This strategy has recently provided survival figures above 30 months for patients with unresectable disease.8,9 Here, we review available data for the use of panitumumab, a monoclonal antibody against EGFR, as the first-line treatment in patients with KRAS exon 2 wild-type mCRC.
Epithelial growth factor signaling pathway in CRC
The EGFR family, or ErbB family, includes transmembrane glycoproteins with an intracellular tyrosine kinase domain, a transmembrane domain, and an extracellular ligand-binding domain.10 There are four transmembrane receptors in this family: HER1 (EGFR), HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4).11 These receptors can form homo- or heterodimers once activated. HER3 is the only member of this family that lacks a functional kinase domain and, therefore, can only be activated by forming heterodimers.12 EGFR was first identified in 1978 in an A431 squamous cell carcinoma cell line.13 In this A431 cell line, EGF binding resulted in phosphorylation and activation of the receptor.14 EGFR has multiple domains (I–IV) (Figure 1). In its unbound form, EGFR adopts a tethered conformation that prevents its activation. When the tethered conformation is broken, EGFR ligands can bind domain III. This leads to stabilization of the receptor in its extending conformation, which exposes domain II, allowing the receptor to dimerize and initiate downstream signaling (Figure 1).15 Once activated, EGFR will form hetero- or homodimers and activate downstream signaling pathways including MAPK or the PI3K/mTOR pathway, leading to cancer cell proliferation, angiogenesis, migration, and survival.16
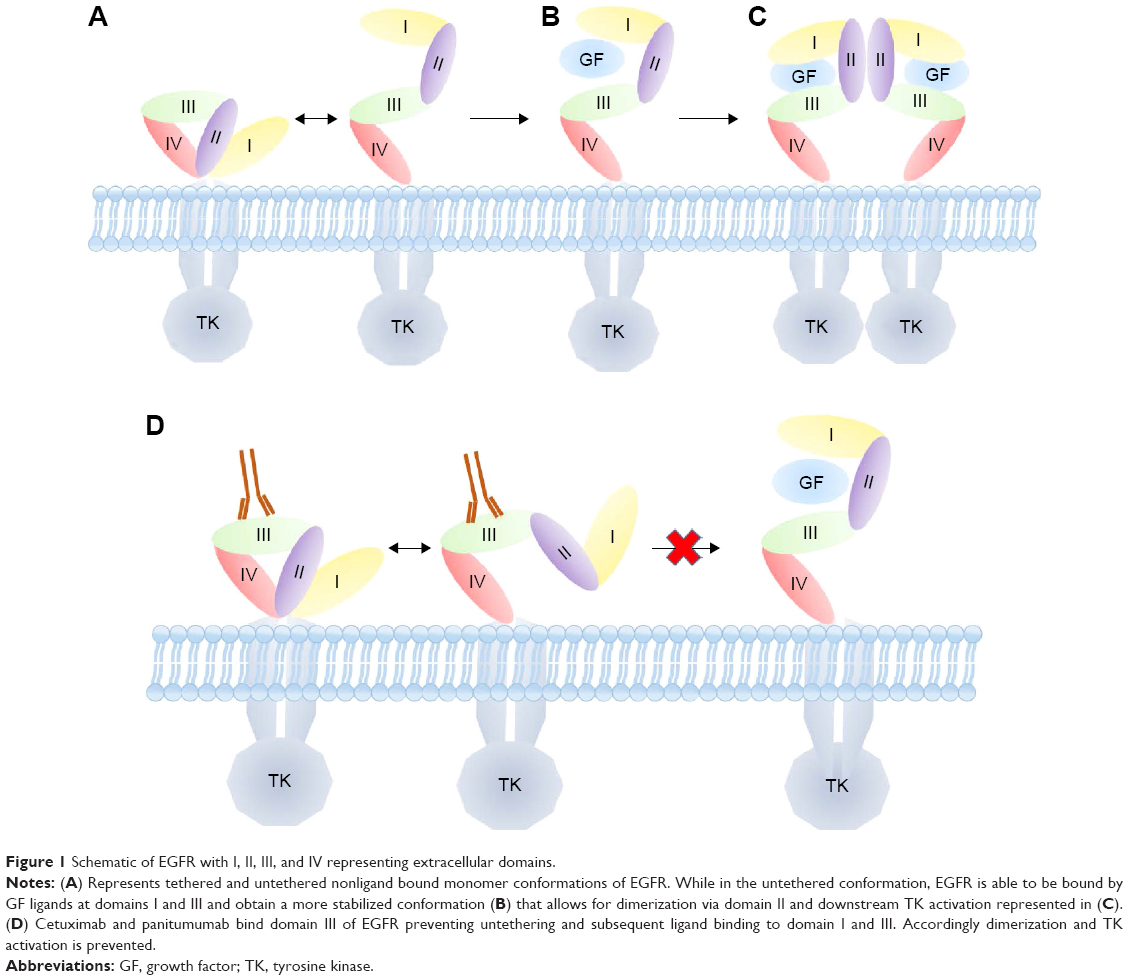 | Figure 1 Schematic of EGFR with I, II, III, and IV representing extracellular domains. |
The EGFR pathway can be deregulated at different levels resulting in increased EGFR ligands, increased EGFR expression and activating EGFR mutations. Activation of EGFR may result from binding to different ligands, including EGF, transforming growth factor α (TGF-α), amphiregulin, and heparin-binding EGF.17–19 EGFR expression in CRC ranges between 20% and 80%.20 However, a correlation between increased EGFR expression and response to monoclonal antibodies against EGFR has not been evidenced in patients with advanced CRC.21,22 Aberrations at the gene level involving EGFR have also been reported in CRC. A smaller subset of CRC patients (8%–12%) have EGFR amplifications defined as >5 gene copies/nucleus.23 A search of The Cancer Genome Atlas (TCGA) data from the cBioPortal for Cancer Genomics (www.cbioportal.org, data accessed on March 30, 2015) identified EGFR missense mutations in 8 (3.7%) patients with CRC (n=212). In addition, EGFR was amplified in one patient (0.4%). A similar search of COSMIC SANGER (www.cancer.sanger.ac.uk/cancergenome, data accessed on March 30, 2015) found EGFR mutations present in 96 (7%) of 1,294 tested samples. Early data suggested that increased EGFR copy number, evaluated by fluorescence in situ hybridization, could predict response to EGFR inhibitors in CRC.24,25 However, results from additional studies have been inconsistent and fail to confirm that hypothesis. In addition, a reproducible cut-off level of amplification that predicts response to anti-EGFR therapy has not been identified in this disease.26
Two monoclonal antibodies against EGFR have gained regulatory approval for treating mCRC. Cetuximab was the first targeted therapy to gain approval in mCRC. Cetuximab is a chimeric IgG1 immunoglobulin, which binds EGFR with high affinity. In cetuximab, the antigen-binding regions (Fv) of mouse antibody are combined with human IgG constant domains, which can lead to infusion reactions in up to 5% of patients.27 According to the cetuximab label, premedication with antihistaminic drugs is recommended with the first infusion.28 Panitumumab, unlike cetuximab, is a fully humanized IgG2 monoclonal antibody. It was generated in transgenic strains of mouse and modified to express human immunoglobulin genes (XenoMouse). Panitumumab was developed by immunizing the XenoMouse IgG2 strain with cells from the human cervical epidermal carcinoma cell line A431. Neutralizing antibodies to this cell line were isolated and sorted by binding affinity to EGFR.29 In vivo studies in xenografts from the A431 cell line showed that panitumumab blocked activation of EGFR, prevented tumor formation, and eradicated established tumors.30 Since panitumumab is fully humanized, premedication is not recommended per US Food and Drug Administration (FDA) label since infusion reactions rarely occur (1%–5%).31 Similar to cetuximab, proposed mechanisms of growth inhibition include induction of cell cycle arrest, promotion of apoptosis, and EGFR downregulation.32
Safety data
The safety of panitumumab has been evaluated in different dose escalation studies. In the first study, four different dose levels of panitumumab (from 1 to 2.5 mg/kg weekly) were evaluated in 88 patients with advanced kidney cancer.33 Panitumumab was well tolerated in these patients. The most common adverse event (AE) was rash, which was usually most severe between the 3rd and the 5th week. At a 2.5 mg/kg weekly dose, 100% of patients developed skin toxicity. The rash was typically distributed in the facial, periorificial regions, and the upper trunk. For most patients, rash intensity decreased despite treatment continuation. Responses were rare, but there was a trend toward improved progression-free survival (PFS) in patients with severe rash. No human antibodies were identified. A Phase I trial tested different schedules of panitumumab including: weekly panitumumab from 0.01 to 5 mg/kg, 6 mg/kg every 2 weeks, and 9 mg/kg every 3 weeks in 96 patients with different tumor types, most notably CRC (41%).34 Treatment was well tolerated. The most common treatment-related AEs were rash (60%) and dry skin (19%). Most skin-related toxicities were grade 1–2. Seven percent of patients across all cohorts developed grade 3 skin toxicity (17% in the every 3-week cohort). Median time for skin toxicity onset of any grade was 1 week. The maximum tolerated dose was not identified. Two patients in the 1 mg/kg weekly cohort had dose-limiting toxicities of severe maculopapular rash. One patient, at 2.5 mg/kg weekly, had a dose-limiting toxicity of fatigue, angina pectoris, and dyspnea. None of the patients experienced infusion reactions. Responses were seen in 13% of patients with CRC. In addition, 23% of CRC patients had a best response of stable disease. Drug exposure was similar with weekly doses at 2.5 mg/kg compared to 6 mg/kg every 2 weeks or 9 mg/kg every 3 weeks. A different Phase I study tested a dose of 6 mg/kg every 2 weeks given as a 60-minute infusion, or a 30-minute infusion if the first dose was well tolerated, or 9 mg/kg every 3 weeks as a 60-minute infusion.35 The study enrolled 84 patients with solid tumors (CRC =10). The incidence of grade 3 AEs was higher in the 9 mg q3 week cohort (62% vs 37%) although overall, the safety profile was similar across different schedules. Partial responses were seen in patients with CRC (18%). In addition, 45% of CRC patients attained stable disease. These initial studies showing preliminary evidence of activity in patients with CRC fueled interest in developing panitumumab for this disease.
Panitumumab and oxaliplatin schedules in first line mCRC
The PRIME study (Panitumumab Randomized trial In combination with chemotherapy for Metastatic colorectal cancer to determine Efficacy) showed improved PFS when panitumumab was added to FOLFOX (folinic acid, 5-flurouracil, oxaliplatin) in the first-line treatment in exon 2 wild-type KRAS mCRC. This randomized Phase III study allocated 1,183 patients in a 1:1 fashion to FOLFOX4 plus panitumumab (6 mg/kg every 2 weeks) vs FOLFOX4 alone. The study was initially designed to test treatment effect in all randomized patients. However, once KRAS was established as a predictive biomarker, the study was amended to compare outcomes according to KRAS status. KRAS status was available for 93% of the patients enrolled in the study.36 The primary endpoint was PFS. The final analysis, 30 months after the last patient was enrolled, showed improved PFS for exon 2 wild-type KRAS patients enrolled in the FOLFOX4 plus panitumumab arm (10 vs 8.6 months, hazard ratio [HR] =0.80; 95% confidence interval [CI]: 0.67–0.95, P=0.01) (Table 1). A detrimental effect in PFS was seen in the subgroup of patients with exon 2 mutations enrolled in the FOLFOX4 plus panitumumab arm (7.4 vs 9.2 months, HR =1.27, 95% CI: 1.04–1.55, P=0.02). An exploratory analysis conducted after >80% patients had an overall survival (OS) event also showed an improvement in OS in the experimental arm (23.8 vs 19.4 months, HR =0.83, 95% CI: 0.70–0.98, P=0.03) in exon 2 wild-type KRAS patients. Adding panitumumab also increased RR in exon 2 wild-type KRAS patients (57% vs 48%, odds ratio =1.47, 95% CI: 1.07–2.04, P=0.02). However, no differences in complete resection rate were seen in exon 2 wild-type KRAS patients with the addition of panitumumab (10% vs 8%). A subset analysis, limited to patients with liver-only disease, showed an increased rate of complete resection for patients in the FOLFOX plus panitumumab arm, but it was not statistically significant (27.9 vs 17.5, difference in rates =10.3, 95% CI: −6.2 to 25.9).
 | Table 1 Summary of efficacy data from PRIME36 and PEAK37 studies stratified by RAS molecular subtypes |
Results from extended RAS analysis from this trial are discussed in the “Predictive biomarkers: patient stratification” section.
The PEAK study (Panitumumab Efficacy in combination with mFOLFOX6 Against bevacizumab plus mFOLFOX6 in mCRC subjects with wild-type KRAS tumors) was designed to address whether to include anti-EGFR vs anti-vascular endothelial growth factor (VEGF) monoclonal antibodies in the first-line treatment of patients with mCRC.37 In this Phase II, multicenter, randomized two-arm study, patients (n=285) were randomized 1:1 to each treatment arm. The primary objective was PFS, although no formal hypothesis testing was planned. A prespecified secondary objective was to evaluate PFS and OS in RAS wild-type patients (expanded RAS testing included exons 2, 3, and 4 of KRAS and NRAS). The study was negative for its primary objective since no statistically significant difference was detected in PFS in patients with exon 2 wild-type KRAS mCRC (PFS 10.9 vs 10.1 months, HR =0.87, P=0.35) (Table 1). An early OS analysis (46% deaths reported) showed, in the panitumumab arm, an improvement in OS (34.2 vs 24.3 months, HR =0.62, P=0.009). However, a definitive separation between survival curves was not seen until approximately 15 months, suggesting that second-line therapies may have impacted these results. Indeed, patients with exon 2 wild-type KRAS mCRC/ other mutations in RAS in the panitumumab arm were more likely to receive bevacizumab beyond progression (50% vs 7%) as well as cytotoxic agents (83% vs 56%). Alternatively, it is possible that anti-EGFR therapies may have a greater impact on OS rather than on PFS as these results are consistent with results recently reported from the Phase III FIRE-3 clinical trial.8 The analysis of secondary endpoints from the PEAK trial also provided valuable information to understand the benefit of adding anti-EGFR in different molecular subtypes. These results are discussed in the “Predictive biomarkers: patient stratification” section.
Panitumumab and irinotecan schedules in first line mCRC
No data are available from Phase III randomized clinical trials testing panitumumab in combination with irinotecan schedules in the first-line setting. However, this regimen is included as one of the treatment options for KRAS/NRAS wild-type patients in the National Comprehensive Cancer Network (NCCN) and European Society for Medical Oncology guidelines. This recommendation is based on extrapolating data from study 2005-0181 in the second-line setting. In this study, the addition of panitumumab to FOLFIRI in KRAS exon 2 wild-type resulted in an improved RR (36% vs 10%, P<0.0001) and PFS (6.7 vs 4.9 months, HR =0.82, P=0.023). A benefit in OS was not seen, possibly due to the impact of therapy beyond progression, as treatment cross-over was allowed in this study, and 90 of 119 patients (76%) in the best supportive care (BSC) group received panitumumab postprogression.38
Two Phase II studies have tested panitumumab in combination with irinotecan schedules in the first line. The first study tested panitumumab at 2.5 mg/kg weekly in combination with either IFL (irinotecan, bolus 5FU, and leucovorin) or FOLFIRI. Of all patients in the IFL arm, 58% had grade 3–4 diarrhea vs 25% in the FOLFIRI arm. No differences were seen in RRs; however, patients in the FOLFIRI arm had a trend toward increased PFS and OS.
A second single-arm, Phase II study combined panitumumab (6 mg/kg) with FOLFIRI every 2 weeks.39 Mutations in KRAS exon 2 were assessed in 94% of the patients. Overall, with the caveat that this was not a randomized trial, objective responses (56% vs 38%), median duration of response (13 vs 7.4 months), and R0 resection (8% vs 5%) were higher in the KRAS exon 2 wild-type subset.
Panitumumab plus bevacizumab in the first line
Dual inhibition of EGFR and VEGF with cetuximab and bevacizumab, in combination with irinotecan, resulted in an improved RR and PFS compared to cetuximab and bevacizumab in the BOND-2 study.40 This was a randomized Phase II clinical trial for patients (N=83) who had previously progressed on irinotecan. The primary objective of the study was to evaluate the safety of the combination; therefore, efficacy analysis was only exploratory. Interestingly, both arms did better than historical controls, which strongly suggest a high degree of patient selection. However, large randomized Phase III trials have failed to validate this strategy in the first-line setting. The PACCE study (Panitumumab Advanced Colon Cancer Evaluation trial) assessed dual inhibition of both EGFR and VEGF in combination with fluoropyrimidines and either oxaliplatin or irinotecan in the first-line treatment of 1,053 patients with mCRC.41 The primary endpoint was PFS. Regardless of KRAS status, the addition of panitumumab was detrimental in this study in regards to both PFS (10 vs 11.4 months, HR =1.27; 95% CI: 1.06–1.52) and toxicity. Grade 3 and 4 AEs were more frequent in the panitumumab arm (90% vs 77% in oxaliplatin cohort and 90% vs 63% in irinotecan cohort). Skin toxicity, diarrhea, infections, pulmonary emboli, and deep vein thrombosis were all more common in the panitumumab arm.41 A detrimental effect from adding cetuximab to CAPOX-bevacizumab was also observed in the Dutch Colorectal Cancer Group CAIRO-2 study.42
Predictive biomarkers: patient stratification
Efforts in drug development have increasingly focused on identifying predictive biomarkers of response to enrich studies with subsets of patients most likely to benefit from targeted therapy. This strategy has, in recent years, led to expedited regulatory approval of novel targeted therapies used to treat different solid tumors.43
Unfortunately, in CRC, advances in personalizing therapy have been scarce. Only RAS mutations have been identified as a negative predictive biomarker and incorporated in the clinical decision-making process.
EGFR expression
EGFR expression was initially hypothesized to be a predictive biomarker of response to EGFR monoclonal antibodies. To that end, the initial randomized trials leading to approval of these drugs in mCRC required EGFR expression by immunohistochemistry (IHC) as part of the eligibility criteria for enrollment. However, in the BOND study, a randomized clinical trial comparing cetuximab monotherapy vs cetuximab in combination with irinotecan in irinotecan refractory patients with mCRC, no correlation was found between EGFR expression in the tumor and efficacy of cetuximab. Subsequently, responses to cetuximab were seen in patients lacking EGFR expression.21 Therefore, it was believed that EGFR expression was not predictive of response to anti-EGFR therapies in mCRC.
KRAS exon 2 mutations
The first evidence to support the role of KRAS exon 2 mutations as a negative predictor of response in mCRC came from a randomized Phase III clinical trial testing panitumumab vs BSC after progression following treatment with cytotoxic agents.22 In this study, 427 out of 463 (92%) enrolled patients underwent KRAS exon 2 (codon 12 and 13) testing by real-time polymerase chain reaction (RT-PCR) in paraffin-embedded tumor sections. KRAS exon 2 mutations were found in 43% of patients. The primary study endpoint was PFS. In KRAS exon 2 wild-type patients, panitumumab resulted in improved PFS (12.3 vs 7.3 weeks, HR =0.45, P<0.0001). There were no differences in OS. However, crossover at the time of progression may have contributed to diluting benefit in OS. A detrimental effect from panitumumab in the KRAS mutant subset was not seen in this study. Within the population enrolled in this study, KRAS exon 2 mutations were a prognostic biomarker, as survival for this molecular subtype was shorter irrespective of treatment arm.
In the C0.17 clinical trial, 572 patients with mCRC who progressed on standard fluoropyrimidines, oxaliplatin, and irinotecan were randomized to cetuximab vs BSC.44 KRAS exon 2 mutations were evaluated by PCR in paraffin-embedded tumor samples from 394 (69%) of these patients.45 Mutations in KRAS exon 2 were found in 42% of the patients evaluated. The primary endpoint of the study was OS. In KRAS exon 2 wild-type patients, cetuximab improved OS (9.5 vs 4.8 months, HR =0.55, P<0.001) as well as PFS (3.7 vs 1.9 months, HR =0.40, P<0.001). Importantly, 7% of patients randomized to the BSC arm crossed over and received cetuximab therapy.44 Similar to the panitumumab trial, cetuximab had no negative impact on PFS/OS in the KRAS mutant subset. Different from that trial, in C0.17, KRAS mutations were not found to be prognostic of poor outcomes in the BSC group.
Some early data suggested that not all KRAS mutations were resistant to anti-EGFR therapy. A retrospective, pooled exploratory analysis of 579 patients treated with cetuximab in different randomized trials showed a survival benefit from cetuximab in the subset of patients with KRAS G13D mCRC (n=45, 17%).46 This is difficult to conciliate with the evidence that KRAS G13D clones are identified in the context of acquired resistance to anti-EGFR therapy.47 Indeed, a randomized Phase II study (ICECREAM) recently compared cetuximab vs cetuximab plus irinotecan in mCRC. The trial included RAS wild-type patients but allowed patients harboring G13D mutations. The study failed to show any benefit from single agent cetuximab in patients with KRAS G13D mCRC.48
Therefore, NCCN guidelines do not recommend treatment with anti-EGFR therapy in patients with KRAS G13D mCRC other than in the context of a prospective clinical trial.
The clinical observation that a large subset of KRAS exon 2 wild-type patients will not respond to anti-EGFR therapy has prompted investigation of additional biomarkers.
Expanded RAS analysis
Archival tissue samples from patients enrolled in the PRIME trial who had KRAS exon 2 wild-type mCRC were recently tested for additional RAS exons. Expanded RAS testing included: KRAS exon 3 (codon 61) or exon 4 (codons 117 or 146); NRAS exon 2 (codon 12 or 13), 3 (codon 61), or 4; or BRAF exon 15 (codon 600).49 Additional RAS mutations were found in 108 of 639 (17%) patients with KRAS exon 2 wild-type mCRC. The survival analysis according to expanded RAS results showed that the benefit from adding panitumumab was even larger in the RAS wild-type subset (Table 1). In this molecular subtype, there was a 2-month improvement in PFS (10.1 vs 7.9 months, HR =0.72, 95% CI: 0.58–0.90, P=0.004) and a 6-month improvement in OS (26 vs 20.2 months, HR =0.78, 95% CI: 0.62–0.99, P=0.04) (Figure 2). However, when panitumumab was added to FOLFOX4 in patients with a wild-type KRAS exon 2 but with a different RAS mutation, there was a nonsignificant decrease in PFS (7.3 vs 8.0 months, HR =1.28, 95% CI: 0.79–2.07, P=0.33) and OS (17.1 vs 18.3 months, HR =1.29, 95% CI: 0.79–2.10, P=0.31). These data prompted the FDA to issue a new label for panitumumab on March 11, 2015, excluding from treatment patients whose tumors had KRAS exon 2 mutations and also those who had any RAS-positive mCRC.31 However, for patients with BRAF-positive mCRC there was still a trend toward increased PFS in patients treated with panitumumab in the PRIME trial (PFS – HR =0.58, 95% CI: 0.29–1.15, OS – HR =0.90). The small number of patients with BRAF-positive mCRC (n=53) may not have been sufficiently robust to evaluate the benefit of adding panitumumab in this subset in the first line. Alternatively, it is possible that adding anti-EGFR therapy in this setting is of no benefit. Indeed, cetuximab, a different EGFR mAb, was detrimental when added to fluoropyrimidines and oxaliplatin in BRAF-positive patients with mCRC treated in the first line in the COIN trial.50 Additionally, two recent meta-analyses showed that there was no improvement in outcomes in patients with BRAF-positive mCRC when either panitumumab or cetuximab is added to standard therapy.51,52 Consistent with this, the NCCN guidelines acknowledge that there are insufficient data to guide the use of anti-EGFR therapy in combination with chemotherapy in the first-line setting in patients with BRAF V600E mCRC. In BRAF V600E mCRC, preclinical data suggest that resistance to BRAF inhibitors is mediated through activation of EGFR.53 For BRAF-positive patients, novel treatment strategies are urgently needed. This may include more intensive therapy or decreasing chemotherapy breaks. In this regard, a subanalysis of the TRIBE study recently showed a trend for increased survival with FOLFOXIRI compared to FOLFIRI (irinotecan with fluorouracil and folinic acid) in BRAF-positive patients, this difference was not statistically significant possibly due to the low prevalence of BRAF-positive CRC.54 Ongoing studies are evaluating BRAF inhibitors in combination with cetuximab and irinotecan or PI3K inhibitors in mCRC patients with these molecular subtypes.55,56 Early results from a dose escalation study with LGX-818 (BRAF inhibitor) plus cetuximab with or without BYL-719 (PI3K inhibitor) in patients with BRAF V600E mCRC were presented at the AACR 2015 Annual Meeting in Philadelphia.57 In this population of patients with very poor prognoses, the RR (30%) and median PFS (4.3 months) hold promise. In addition, a subset of these patients was able to stay on treatment beyond 44 weeks. This suggests that even within the small BRAF V600E, subset heterogeneity is likely to influence response to therapy.
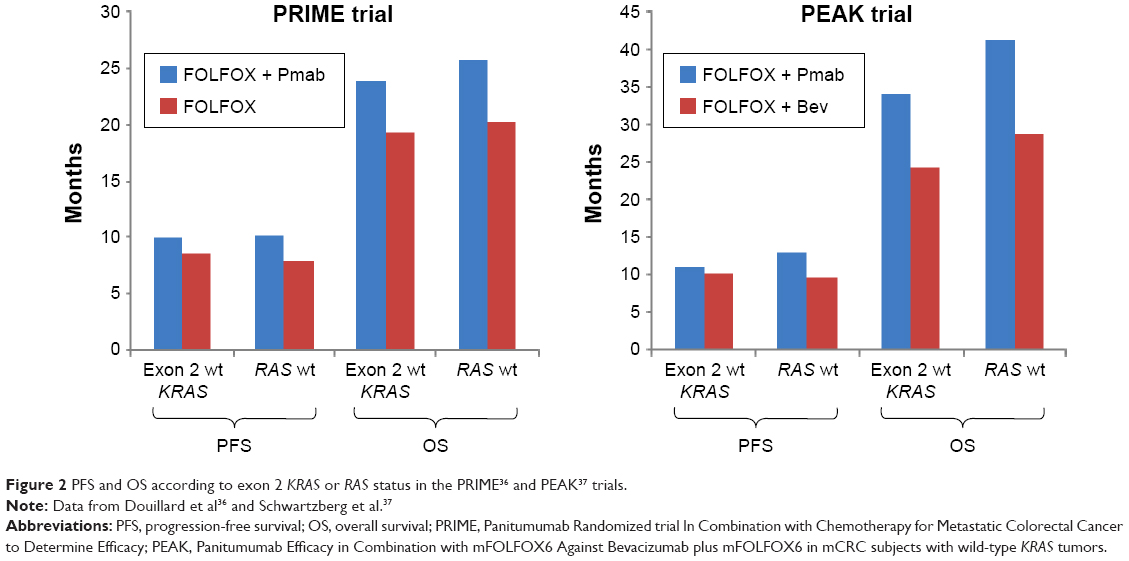 | Figure 2 PFS and OS according to exon 2 KRAS or RAS status in the PRIME36 and PEAK37 trials. |
Similar to the PRIME study, the investigators in the PEAK study also retrospectively carried out additional extended RAS testing. Results from extended RAS testing were available in 233 out of 285 (82%) patients from the PEAK study. Of patients with exon 2 wild-type KRAS mCRC, 23% had additional RAS mutations. In RAS wild-type patients the addition of panitumumab to FOLFOX resulted in improved PFS compared to FOLFOX plus bevacizumab (PFS 13 vs 9.5 months, HR =0.65, P=0.029). Similarly, OS was also increased with the caveats discussed above regarding the potential impact of second-line therapy (OS 41.3 vs 28.9, HR =0.63, P=0.058) (Figure 2). Overall RR was similar in both arms as well as rates of R0 resection (10% vs 8% for panitumumab and bevacizumab, respectively). An exploratory analysis of responses from the PEAK trial limited to patients with RAS wild-type mCRC was recently presented in abstract form.58 Responses occurred earlier (early tumor shrinkage [ETS] at 8 weeks 64% vs 45%, P=0.023), were deeper (65% interquartile range [IQR] 48–67 vs 46%, IQR [29–62] P=0.0007) and lasted longer (11.4 [95% CI 9.7–13.6] vs 8.5 [95% CI 6.3–9.3] months, P=0.0142) in the panitumumab arm. These results are consistent with those seen in the FIRE-3 trial. Investigators in the FIRE trial hypothesized that the depth of response may explain benefit in OS in the absence of benefit in PFS.58
The results of expanded RAS testing from the CRYSTAL (Cetuximab combined with Irinotecan in First-Line Therapy for Metastatic Colorectal Cancer) trial, which studied FOLFIRI with/without cetuximab as first-line therapy, showed that the incidence of expanded RAS mutations was similar to the PRIME study.
In the CRYSTAL study, expanded RAS testing was performed in samples available from 64% of enrolled patients. Of all exon 2 wild-type KRAS patients, 15% had RAS mutations.59 In contrast to the PRIME study, there was no negative effect seen in the CRYSTAL trial when cetuximab was added to FOLFIRI in RAS-positive patients. The reasons for these conflicting results are unclear. It is possible that the use of a different chemotherapy backbone may have influenced these findings. However, given the potential for harm if patients with RAS-positive disease are exposed to anti-EGFR therapy, expanded RAS testing is strongly recommended by ASCO prior to start any patient on these therapies.60
PI3KCA and PTEN
PI3KCA is constitutively activated in 15% of CRCs.61 In CRC cell lines, constitutive activation of PIK3CA leads to increased AKT1 phosphorylation.62 This results in decreased apoptosis and increased cell migration and invasion in vitro. Similarly, PTEN, a negative regulator of PI3K, is inactivated in 20% of CRC.63 PTEN loss leads to PI3K pathway activation.
A retrospective analysis of patients with mCRC treated with cetuximab across multiple centers in Europe suggested that PIK3CA mutations in exon 20 (catalytic domain), assessed by automated sequencing, and PTEN loss assessed by IHC, were associated with resistance to anti-EGFR therapy in mCRC.64 However, the evidence in this arena is conflicting. A different retrospective analysis from the CO.17 study (a randomized Phase III study comparing cetuximab vs BSC) arrived at different conclusions after analyzing the predictive role of PIK3CA mutations and PTEN loss.65 In this analysis, 407 of 572 (71%) samples were tested for PIK3CA using a nested PCR procedure; 205 patients (35%) were assessed for PTEN loss by IHC using a rabbit monoclonal antibody. Neither PIK3CA mutations nor PTEN loss were prognostic of survival in these patients. In addition, they were not predictive of benefit from cetuximab. The lack of standardized methods used to evaluate some of these biomarkers (ie, PTEN) may have contributed to these conflicting results.
MET amplifications
MET is an oncogene that encodes a tyrosine kinase receptor for hepatocyte growth factor. MET may become constitutively activated as a result of copy number variation, overexpression, or activating mutations.66 Genetic aberrations in MET were found in 6 (3%) out of 212 tumor specimens from CRC patients for whom both sequencing and copy number variation analyses were available according to TCGA data from cBioPortal for Cancer Genomics (www.cbioportal.org, data accessed on March 30, 2015). The most common aberrations were missense mutations found in four patients. One patient each had amplification (0.5%) and a homozygous deletion. In a recent work, MET amplifications were identified, at the time of progression to anti-EGFR therapy, in a small subset of patients with mCRC who had initially responded to EGFR inhibition.67 Next-generation sequencing in posttreatment biopsies collected at the time of progression identified MET amplifications in three out of seven patients. MET amplifications were then confirmed by FISH (fluorescence in situ hybridization) analysis (MET/CEP7 ratio >3). Interestingly, in samples from patient-derived xenografts (PDXs) MET amplifications were mutually exclusive with mutations in KRAS, BRAF, NRAS, and PIK3CA and with HER2 amplification. This work suggests that MET testing could help refine the subset of patients most likely to benefit from anti-EGFR therapy. However, given the low incidence of this aberration in mCRC, large pooled analysis will be needed to confirm this hypothesis. In addition, more data are needed to define the optimal cut-off MET amplification that predicts resistance to therapy.
EGFR S492R
At the present time, the two approved anti-EGFR monoclonal antibodies are considered largely equivalent in terms of efficacy. This was studied in ASPECCT (A Study of Panitumumab Efficacy and Safety Compared to Cetuximab in Patients With KRAS Wild-Type Metastatic Colorectal Cancer), a noninferiority trial with 1,010 patients with wt-KRAS mCRC refractory to chemotherapy who were randomized to receive either panitumumab or cetuximab. OS was the same in both arms (10.4 months with panitumumab vs 10.0 months with cetuximab) and AEs were similar with slightly greater skin toxicity in the panitumumab arm although less infusion reactions (one patient vs nine in the cetuximab arm).68 As described above, the decreased incidence of infusion reactions is likely secondary to panitumumab being a fully human monoclonal antibody. A small Phase II trial showed no responses to panitumumab in 20 patients with mCRC who had previously progressed on cetuximab.69 However, acquired mutations in the extracellular domain of EGFR (including S492R, S464L, G465R, and I491M) which may predict resistance to cetuximab but not panitumumab, were recently identified.70,71 Retrospective analysis of samples from ASPECCT revealed that 16% of patients in the cetuximab arm but only 1% of those in the panitumumab arm developed EGFR S492R mutations. Those with EGFR S492R mutations in the cetuximab arm were also shown to have worse OS (11.9 vs 13.8 months, HR =1.75, 95% CI: 1.23–2.50).72 The role of these mutations in assisting therapeutic decisions involving the selection of the most appropriate monoclonal antibody still needs to undergo prospective validation before it can be implemented in the clinic.
p53
p53 is a tumor suppressor gene with a critical role in checkpoint activation during cell cycle progression.73 Mutations in p53 are present in 50% of patients with mCRC.63 The role of p53 as a biomarker of response to anti-EGFR therapies is still unclear. A retrospective analysis in 64 patients with chemorefractory mCRC treated with cetuximab plus chemotherapy showed a longer time to progression in those whose tumor specimens were p53 mutated vs wild-type.74 However, mass parallel sequencing analysis from tumor specimens collected during a Phase III trial that compared panitumumab plus BSC vs BSC alone, failed to show that p53 status was predictive of benefit from panitumumab.75
HER2
Activation of HER2 signaling through HER2 amplifications or activating mutations in the kinase domain of HER2 is present in 7% of patients with advanced CRC.63 HER2 activation predicts resistance to EGFR inhibitor in CRC PDX. In PDX models representative of this molecular subset, dual HER2 inhibition induced tumor regression.76 A recent study showed promising RR (35%) in patients with HER2-positive CRC patients treated with lapatinib plus trastuzumab. HER2 was considered positive if tumor was IHC 3+ or 2+ and FISH positive (HER2:CEP17 >2).
FGFR1 amplifications and PDGFRA mutations
Whole exome sequencing of KRAS wild-type CRC PDX recently identified FGFR1 amplifications in 3% of PDX resistant to EGFR inhibition.77 In addition, mutations near the catalytic domain of PDGFRA were also found in 3% of tumorgrafts resistant to EGFR. Of note, single-agent pathway inhibition with BGJ398 (FGFR inhibitor) or imatinib (PDGFR inhibitor) failed to show activity in these models despite them harboring actionable aberrations. However, tumor regressions were seen when these agents were combined with cetuximab indicating that perhaps combination therapy will be needed for these molecular subsets.
Toxicity profile from first-line randomized trials
In the PRIME study, the addition of panitumumab increased the incidence of AE. This had no impact on patient-reported quality of life; however, up to 20% of the patients discontinued treatment due to AEs in the panitumumab arm. The distribution of grade 3–4 AE was similar regardless of KRAS status. The most common AE with panitumumab was acneiform rash (37% and 31% for KRAS wild-type and KRAS mutant, respectively). The incidence of skin toxicity in the control arm was less than 2%. Rash was typically located in face/upper trunk and appeared early during treatment, typically during the 1st month. In the KRAS wild-type group, patients who developed grade 2–4 skin rash had improved clinical outcomes measured by RR, PFS, and OS compared to those with grade 0–1 skin rash. Thirty percent of patients had a delayed onset of rash with similarly improved clinical outcomes as did those with early-onset rash. Data from the STEPP (Skin Toxicity Evaluation Protocol with Panitumumab) clinical trial shows that preemptive antibiotic treatment with doxycycline decreases the incidence of grade ≥2 rash secondary to panitumumab from 62% to 29%.78 Similarly, the addition of panitumumab increased the incidence of grade 3–4 diarrhea (18% vs 9%), hypokalemia (10% vs 5%), and fatigue (10% vs 3%).
In the PEAK study, patients in the panitumumab arm experienced AEs of any grade more frequently than those in the bevacizumab arm (91% vs 83%); however, the percentage of patients who discontinued treatment due to AEs, one-third, was similar. Again, the most common grade 3–4 AE was skin rash, 32% vs 2% in the panitumumab vs bevacizumab group. Additional grade 3–4 AEs that were increased in the panitumumab arm included hypokalemia (11% vs 5%) and hypomagnesemia (7% vs 0%).
Conclusion
The addition of panitumumab to FOLFOX in first-line treatment resulted in improved RR, PFS, and OS for patients with KRAS exon 2 wild-type mCRC. Unfortunately, the identification of predictive biomarkers of response to targeted therapy in mCRC is lagging behind compared to other malignancies. KRAS exon 2 mutations were identified as a negative predictor of response to anti-EGFR therapy in patients with mCRC nearly a decade ago.22 However, a large percentage of patients with KRAS exon 2 wild-type mCRC still derive no benefit from the addition of anti-EGFR therapies. It is clear that additional biomarkers that can be used for patient stratification are needed in this setting.
Efforts to identify new biomarkers in mCRC have included testing archival tissue from different studies with anti-EGFR therapies to search for additional mutations in 1) different exons within KRAS, 2) different members of the RAS family, 3) additional genes that may activate pathways downstream of KRAS and therefore drive resistance to therapy (HER2, PIK3CA, PTEN), and 4) genes involved in alternative signaling pathways (MET, FGFR1).77 In randomized trials with panitumumab in the first line, 17%–23% of KRAS exon 2 wild-type patients harbored additional mutations within KRAS (exon 3 and 4) or different members of the RAS family (NRAS).37,49 In both trials, expanded RAS analysis was performed using dideoxy DNA Sanger sequencing (detection threshold of 5% copies). It is important to note that expanded RAS analysis across trials testing cetuximab has been done using different methods. Pyrosequencing was performed in samples from the FIRE-3 study.8 BEAMing (Bed Emulsion Amplification and Magnetics) was used in the OPUS and CRYSTAL studies (5% sensitivity cut-off selected for the analysis).59,79 The prevalence of RAS mutations in KRAS exon 2 wild-type specimens from CRYSTAL, FIRE, and OPUS trials were 14.7%, 16%, and 31%, respectively.
Several key questions will need to be addressed in the future. First, is a dynamic definition of expanded RAS mutations needed or even acceptable? A recent meta-analysis showed a correlation higher than 92% between KRAS mutations in primary and matched metastases in CRC.80 However, most of those studies used detection methods with thresholds greater than 5% per allele copies. Available technologies like BEAMing have detection thresholds as low as 0.01%. It is plausible that if the detection threshold is lowered, additional RAS mutations could be identified. Second, do all RAS isoforms signify a negative predictive role? Retrospective analysis suggests this is true, at least when detection thresholds >5% were used.49 However, it will be challenging to evaluate the predictive role of RAS mutations present only in rare clones or even the individual role of some of these mutations with low prevalence. Indeed, although some early data suggested that the subgroup of patients with KRAS G13D mutations may receive some benefit from cetuximab, the results from ICECREAM study reported recently showed no benefit from single agent cetuximab in patients with KRAS G13D-positive disease.48 These patients should be spared from anti-EGFR therapies similar to other patients with RAS mutations.
Third, should BRAF be included in initial testing when data supporting the use of anti-EGFR therapy in this subset of patients in the first line are conflicting? Fourth, the role of additional biomarkers beyond RAS will need to be better defined. For instance, the negative predictive value of PIK3CA mutations or PTEN loss gleaned in a retrospective analysis across different European centers could not be confirmed in samples from a randomized study (C0.17).65 Finally, will the new molecular subtype classifications of CRC, largely based on gene expression analysis, allow better patient stratification and treatment selection?81 Results from ongoing prospective clinical trials are awaited to answer this question.
In summary, RAS mutations have been identified as a negative predictor of response to EGFR inhibitors, including panitumumab, in patients with mCRC. A number of additional biomarkers are currently investigated for which there is conflicting evidence. As only a subset of RAS-wild type patients benefit from anti-EGFR therapies, additional work in this arena is urgently needed to refine patient stratification.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. | ||
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. | ||
Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. | ||
Nishihara R, Wu K, Lochhead P, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med. 2013;369:1095–1105. | ||
De Angelis R, Sant M, Coleman MP, et al. Cancer survival in Europe 1999–2007 by country and age: results of EUROCARE-5 – a population-based study. Lancet Oncol. 2014;15:23–34. | ||
Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: evidence in terms of response rate. Advanced Colorectal Cancer Meta-Analysis Project. J Clin Oncol. 1992;10:896–903. | ||
Koopman M, Antonini NF, Douma J, et al. Sequential versus combination chemotherapy with capecitabine, irinotecan, and oxaliplatin in advanced colorectal cancer (CAIRO): a phase III randomised controlled trial. Lancet. 2007;370:135–142. | ||
Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–1075. | ||
Venook AP, Niedzwiecki D, Lenz HJ. CALGB/SWOG 80405: phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC) [abstr LBA3]. J Clin Oncol. 2014;32(Suppl):5s. | ||
Roberts RB, Min L, Washington MK, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:1521–1526. | ||
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. | ||
Guy PM, Platko JV, Cantley LC, et al. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc Natl Acad Sci U S A. 1994;91:8132–8136. | ||
Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. | ||
Cohen S, Ushiro H, Stoscheck C, et al. A native 170,000 epidermal growth factor receptor-kinase complex from shed plasma membrane vesicles. J Biol Chem. 1982;257:1523–1531. | ||
Li S, Schmitz KR, Jeffrey PD, et al. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. | ||
Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. | ||
Luetteke NC, Qiu TH, Fenton SE, et al. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development. 1999;126:2739–2750. | ||
Luetteke NC, Qiu TH, Peiffer RL, et al. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73:263–278. | ||
Iwamoto R, Yamazaki S, Asakura M, et al. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci U S A. 2003;100:3221–3226. | ||
Spano JP, Fagard R, Soria JC, et al. Epidermal growth factor receptor signaling in colorectal cancer: preclinical data and therapeutic perspectives. Ann Oncol. 2005;16:189–194. | ||
Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803–1810. | ||
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. | ||
Shia J, Klimstra DS, Li AR, et al. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: an immunohistochemical and chromogenic in situ hybridization study. Mod Pathol. 2005;18:1350–1356. | ||
Personeni N, Fieuws S, Piessevaux H, et al. Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin Cancer Res. 2008;14:5869–5876. | ||
Cappuzzo F, Finocchiaro G, Rossi E, et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol. 2008;19:717–723. | ||
Sartore-Bianchi A, Moroni M, Veronese S, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25:3238–3245. | ||
Baselga J, Pfister D, Cooper MR, et al. Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J Clin Oncol. 2000;18:904–914. | ||
Cetuximab FDA label, 2015. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory#labelinfo. Accessed October 28, 2015. | ||
Yang XD, Jia XC, Corvalan JR, et al. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol. 2001;38:17–23. | ||
Yang XD, Jia XC, Corvalan JR, et al. Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res. 1999;59:1236–1243. | ||
Panitumumab FDA label, 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125147s200lbl.pdf. Accessed October 28, 2015. | ||
Foon KA, Yang XD, Weiner LM, et al. Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int J Radiat Oncol Biol Phys. 2004;58:984–990. | ||
Rowinsky EK, Schwartz GH, Gollob JA, et al. Safety, pharmacokinetics, and activity of ABX-EGF, a fully human anti-epidermal growth factor receptor monoclonal antibody in patients with metastatic renal cell cancer. J Clin Oncol. 2004;22:3003–3015. | ||
Weiner LM, Belldegrun AS, Crawford J, et al. Dose and schedule study of panitumumab monotherapy in patients with advanced solid malignancies. Clin Cancer Res. 2008;14:502–508. | ||
Stephenson JJ, Gregory C, Burris H, et al. An open-label clinical trial evaluating safety and pharmacokinetics of two dosing schedules of panitumumab in patients with solid tumors. Clin Colorectal Cancer. 2009;8:29–37. | ||
Douillard JY, Siena S, Cassidy J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25:1346–1355. | ||
Schwartzberg LS, Rivera F, Karthaus M, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32:2240–2247. | ||
Van Cutsem E, Siena S, Humblet Y, et al. An open-label, single-arm study assessing safety and efficacy of panitumumab in patients with metastatic colorectal cancer refractory to standard chemotherapy. Ann Oncol. 2008;19(1):92–98. | ||
Kohne CH, Hofheinz R, Mineur L, et al. First-line panitumumab plus irinotecan/5-fluorouracil/leucovorin treatment in patients with metastatic colorectal cancer. J Cancer Res Clin Oncol. 2012;138:65–72. | ||
Saltz LB, Lenz HJ, Kindler HL, et al. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J Clin Oncol. 2007;25:4557–4561. | ||
Hecht JR, Mitchell E, Chidiac T, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27:672–680. | ||
Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–572. | ||
de Gramont A, Watson S, Ellis LM, et al. Pragmatic issues in biomarker evaluation for targeted therapies in cancer. Nat Rev Clin Oncol. 2015;12:197–212. | ||
Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–2048. | ||
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. | ||
De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–1820. | ||
Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. | ||
Segelov E, Thavaneswaran S, Waring P, et al. Abstract 32LBA. The AGITG ICECREAM Study: the Irinotecan Cetuximab Evaluation and Cetuximab Response Evaluation Amongst Patients with a G13D Mutation – analysis of outcomes in patients with refractory metastatic colorectal cancer harbouring the KRAS G13D mutation. Paper presented at: European Cancer Congress, 2015, Vienna, Austria. | ||
Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. | ||
Maughan TS, Adams RA, Smith CG, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–2114. | ||
Pietrantonio F, Petrelli F, Coinu A, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51:587–594. | ||
Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014;53:852–864. | ||
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–113. | ||
Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16:1306–1315. | ||
Southwest Oncology Group. S1406 Phase II study of irinotecan and cetuximab with or without vemurafenib in BRAF mutant metastatic colorectal cancer. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02164916. NLM identifier: NCT02164916. Accessed October 28, 2015. | ||
Novartis Pharmaceuticals. Study of LGX818 and cetuximab or LGX818, BYL719, and cetuximab in BRAF mutant metastatic colorectal cancer. Available from: https://www.clinicaltrials.gov/ct2/show/NCT01719380. NLM identifier: NCT01719380. Accessed October 28, 2015. | ||
Schellens JH, Van Geel R, Bendell JC, et al. Final biomarker analysis of the phase I study of the selective BRAF V600 inhibitor encorafenib (LGX818) combined with cetuximab with or without the α-specific PI3K inhibitor alpelisib (BYL719) in patients with advanced BRAF-mutant colorectal cancer. Paper presented at: AACR 106th Annual Meeting, April 18–22, 2015, Philadelphia, PA [Abst #CT136]. | ||
Rivera F, Karthaus M, Hecht JR. First-line treatment with modified FOLFOX6 (mFOLFOX6) + panitumumab (pmab) or bevacizumab (bev) in wild-type (WT) RAS metastatic colorectal carcinoma (mCRC): tumor response outcomes beyond RECIST [abstr 660]. J Clin Oncol. 2015;33(Suppl 3). | ||
Van Cutsem E, Lenz HJ, Kohne CH, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015;33:692–700. | ||
Allegra CJ, Rumble RB, Hamilton SR, et al. Extended RAS gene mutation testing in metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol. Epub October 5, 2015. | ||
Garrido-Laguna I, Hong DS, Janku F, et al. KRASness and PIK3CAness in patients with advanced colorectal cancer: outcome after treatment with early-phase trials with targeted pathway inhibitors. PLoS One. 2012;7:e38033. | ||
Samuels Y, Diaz LA Jr, Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. | ||
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. | ||
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. | ||
Karapetis CS, Jonker D, Daneshmand M, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer – results from NCIC CTG/AGITG CO.17. Clin Cancer Res. 2014;20:744–753. | ||
Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. | ||
Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. | ||
Price TJ, Peeters M, Kim TW, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014;15:569–579. | ||
Wadlow RC, Hezel AF, Abrams TA, et al. Panitumumab in patients with KRAS wild-type colorectal cancer after progression on cetuximab. Oncologist. 2012;17:14. | ||
Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–223. | ||
Arena S, Bellosillo B, Siravegna G, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015;21(9):2157–2166. | ||
Price TJ, Newhall K, Peeters M, et al. Prevalence and outcomes of patients (pts) with EGFR S492R ectodomain mutations in ASPECCT: panitumumab (pmab) vs. cetuximab (cmab) in pts with chemorefractory wild-type KRAS exon 2 metastatic colorectal cancer (mCRC). J Clin Oncol. 2015;33(Suppl): abstr e14623. ASCO Annual Meeting Proceedings. | ||
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. | ||
Oden-Gangloff A, Di Fiore F, Bibeau F, et al. TP53 mutations predict disease control in metastatic colorectal cancer treated with cetuximab-based chemotherapy. Br J Cancer. 2009;100:1330–1335. | ||
Peeters M, Oliner KS, Parker A, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19:1902–1912. | ||
Kavuri SM, Jain N, Galimi F, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832–841. | ||
Bertotti A, Papp E, Jones S, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526:263–267. | ||
Lacouture ME, Mitchell EP, Piperdi B, et al. Skin toxicity evaluation protocol with panitumumab (STEPP), a phase II, open-label, randomized trial evaluating the impact of a pre-Emptive Skin treatment regimen on skin toxicities and quality of life in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:1351–1357. | ||
Tejpar S, Lenz HJ, Kohne CH. Effect of KRAS and NRAS mutations on treatment outcomes in patients with metastatic colorectal cancer (mCRC) treated first-line with cetuximab plus FOLFOX4: new results from the OPUS study [abstr LBA444]. J Clin Oncol. 2014;32(Suppl 3). | ||
Mao C, Wu XY, Yang ZY, et al. Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci Rep. 2015;5:8065. | ||
Isella C, Terrasi A, Bellomo SE, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–319. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.