Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 10
Prioritizing single-nucleotide polymorphisms and variants associated with clinical mastitis
Authors Suravajhala P ![]() , Benso A
, Benso A
Received 30 September 2016
Accepted for publication 29 December 2016
Published 12 June 2017 Volume 2017:10 Pages 57—64
DOI https://doi.org/10.2147/AABC.S123604
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Juan Fernandez-Recio
Video abstract presented by Prashanth Suravajhala.
Views: 862
Prashanth Suravajhala,1 Alfredo Benso2
1Department of Molecular Biology and Genetics, Center for Quantitative Genetics and Genomics, Aarhus University, Aarhus, Denmark; 2Department of Control and Computer Engineering, Politecnico di Torino, Torino, Italy
Abstract: Next-generation sequencing technology has provided resources to easily explore and identify candidate single-nucleotide polymorphisms (SNPs) and variants. However, there remains a challenge in identifying and inferring the causal SNPs from sequence data. A problem with different methods that predict the effect of mutations is that they produce false positives. In this hypothesis, we provide an overview of methods known for identifying causal variants and discuss the challenges, fallacies, and prospects in discerning candidate SNPs. We then propose a three-point classification strategy, which could be an additional annotation method in identifying causalities.
Keywords: clinical mastitis, single-nucleotide polymorphisms, variants, associations, diseases, linkage disequilibrium, GWAS
Introduction
In the face of effective treatment strategies, identifying causal single-nucleotide polymorphisms (SNPs) plays an important role in prioritizing biomarkers. The methodologies for understanding and determining which genes are linked to a certain disease are aimed at detecting quantitative trait loci (QTLs) associated with the phenotypes. While there have been broad approaches established in identifying causal genes, polymorphisms, and variants affecting a range of diseases including inflammatory diseases,1 it would be remarkable to predict whether the SNPs function as the actual causal variants to diseases. Recent advances using bioinformatics and systems biology approaches seem to be amenable in functionally mapping genes and variants associated with the diseases.2 The most commonly used methods are pathway analyses,3 functional mapping/association methods,4 structural variants and single-nucleotide variant calling,5 a relationship between genotypes and expressed phenotypes,6,7 incorporated workflows, and computational frameworks.8 A detailed review on the promises and challenges of genome-wide association studies (GWAS) for studying complex traits is beyond the scope of this article; nevertheless, apart from the methods discussed above, we point to reviews.9–11 Although these mapping strategies are aimed to discover causal SNPs, integrated bioinformatics and systems biology methods are not thoroughly evaluated. Furthermore, multiple nucleotide variants, insertions–deletions, copy number variations, translocations, and mobile elements could also play an important role for the fact that these variant types are more difficult to detect from short read data. The SNP markers are identified by improving the integrated data from association studies and novel gene/functional mapping strategies. In addition, pathway fractional analysis serves to predict these SNP markers, which can be further validated in vitro for susceptibility to diseases or for linking changes in gene expression to phenotypic variations. The genomic variation can be specially associated with noncoding/introns, and intergenic and intragenic–intronic sequences, each with a small effect, further suggesting that several regulatory sequences might be involved in causing the diseases. As significant fractions of these noncoding sequences are transcriptionally regulated, the impact of such variations associated with diseases/traits – pleiotropic effect – cannot be undervalued. With the effort in finding the causal mutation for quantitative/complex traits, many associated variants are reported from GWAS across species, but only a few cases had led to the discovery of real causal gene/variant.12,13 For example, a significant number of candidate SNPs/variants between the genes vitamin D-binding protein precursor (group-specific component) and neuropeptide FF receptor 2 genes on chromosome 6 in cow are known to be putative candidates for bovine clinical mastitis.14 More recently, imputed sequence variants have been rigorously used for association studies, and udder conformation traits including mastitis were identified in noncoding regions of the genome.15 This region underlying the peaks of associations with bovine clinical mastitis has certain traits specific for vitamin D components across all eutherians including humans.16 Conversely, strong linkage disequilibrium (LD), especially in these regions, affects the subregions underlying the peaks of associations with the disease. Thus, there remains a challenge to identify bona fide candidate SNPs for such regions using integrated bioinformatics and systems biology approaches by choosing a region among denser peaks of interest to determine whether the approaches such as transcription factor binding sites (TFBSs), enhancer elements, and methylation play a role in the identification of causal mutations or genes associated with diseases, thereby serving as the major determinants of variation specific to diseases.
Three-point classification strategy to discern candidate SNPs
A three-point classification approach, based on functional annotation, regulatory regions, and constrained elements, is proposed to identify causal variants and further validate as SNP as a cause (Figure 1). The three-point classification and its associated parametric annotations are described with illustrative examples.
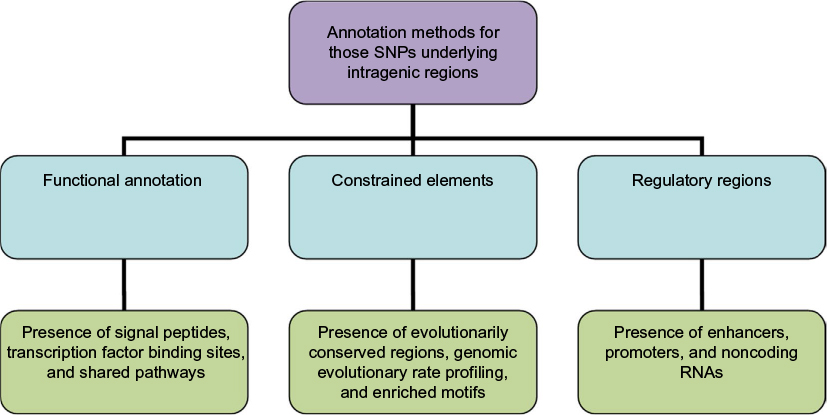 | Figure 1 Approaches in identifying the candidate SNPs: the SNPs are annotated using three annotation features in the form of classifiers (light blue) and the candidates are confirmed from those that match all these features. However, for those candidate SNPs that are highly enriched, only the regulatory regions can be used for confirmation. Abbreviations: SNPs, single-nucleotide polymorphisms; RNAs, ribonucleic acids. |
Improved functional annotation
The interaction between genes and transcription factors is important for understanding gene regulation and the origin of protein complexes components. For example, identifying TFBS regions and signal peptides (SPs) nearing the protein is useful to understand the details of the regulatory networks and pathways associated with the gene. To show this, we have selected a highly enriched LD region in cow that is associated with various phenotypic traits, viz. calf size, carcass weight, and somatic cell count/score. If these regions contained TFBS or signal peptides, it would be straightforward to assume that the underlying SNPs would be very good candidates for being associated with the disease (Figure 2). While the somatic cell count is a cell count of somatic cells in the milk indicating the (trait) quality of milk, the carcass weight is considered as a production trait to determine the yield grade of the animal.
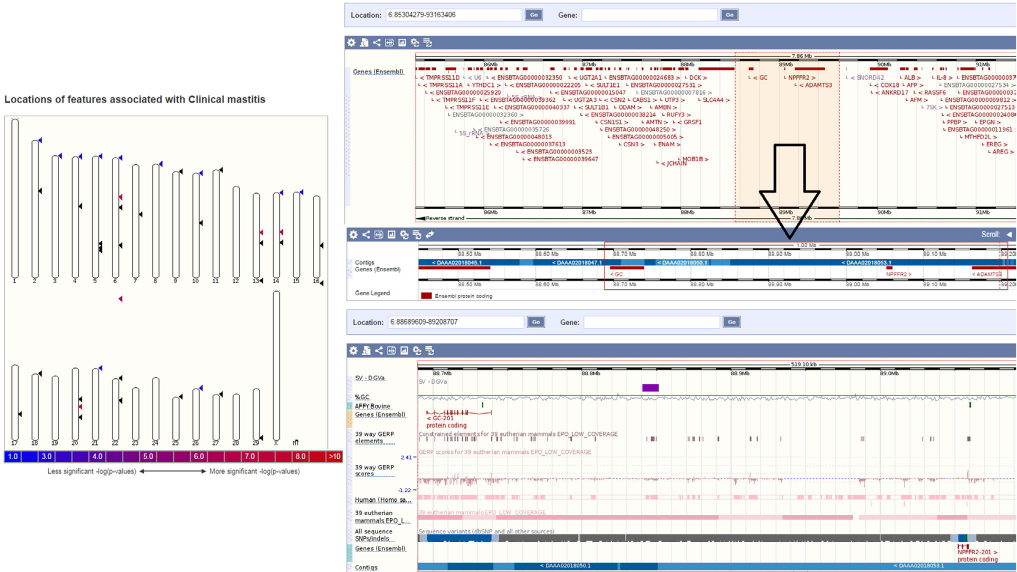 | Figure 2 (Left) Location of features with clinical mastitis in cow (http://www.ensembl.org).40 (Right) Select regions in chromosome 6 of cow (chromosome 6: 88689609–89208707) with high LD demarcated in selection with arrow. The panel below shows the zoomed in region containing the genes, SNPs, GERP, phenotypic traits conjoining the regions, and other information. Inferring the candidate SNPs from these approaches would essentially allow us to find novel biomarkers for genotyping. Abbreviations: GERP, genomic evolutionary rate profiling; LD, linkage disequilibrium; SNPs, single-nucleotide polymorphisms. |
Signal peptide
The sorting signal present in the protein is usually at the N-terminal region. A cleavage site is also associated with each SP. A strategy for prioritizing SNPs occurring within these regions needs a great deal of functional understanding of the cell processes implicated in the diseases.17 Tools such as SignalP18 can be used to predict the presence of SPs and their cleavage sites.
Transcription factor-binding affinity
As SNPs presence peaks significantly in the intronic/non-coding regions, searching the TFBS that span across 5′ untranslated regions (UTRs), intronic, and 3′ UTRs could be very helpful. Common TFBS associated with diseased genes such as myc, jun, and zinc finger are searched for alternative targets with TFBS prediction tools such as PhysBinder,19 and experimental prediction tools such as Qiagen’s ChIP qPCR search (http://www.sabiosciences.com/chipqpcrsearch.php) can be used as validation tool if the SNPs lie in those regions. Furthermore, computing the percentage of identified true positives as a weighted average of the precision and recall for the TFBS regions would allow to better understand the role of enriched motifs that are essentially conserved sequences. For instance, SP1, a well-known transcription factor associated with immune diseases, has a selection for an enriched motif. The enriched motifs, when checked for exon/intron specificity, help us to identify the level of conservation in the TFBS and can be visualized through sequence logos20 (Figure 3). Bickhart and Liu21 have detected TFBS in cattle genome using phylogenetic footprinting tools. However, the challenge would be to validate them with different prediction scores.
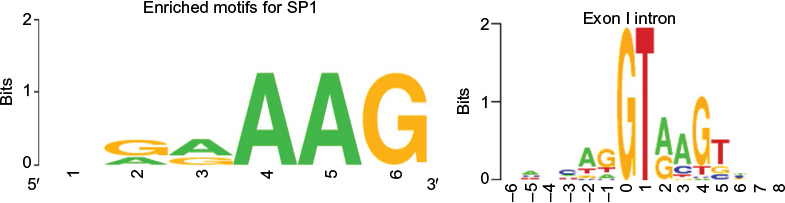 | Figure 3 Enriched motifs seen for SP1 (GAAAG) and the panel next to it shows the exon/intron boundary where the consensus motif (GTA(G)AG) can essentially be seen in eukaryotes. Note: Sequence logos: http://weblogo.berkeley.edu.41 Abbreviation: SP, signal peptide. |
Shared pathways
Previous efforts helped in identifying relevant gene networks using Ingenuity Pathway Analysis in milk-yielding traits22 and in understanding pathways of the mammary gland involved in the pathogenesis of bovine Escherichia coli mastitis.23 The disease-specific phenotypic traits/data share similar genetic variation, and so the phenotypic variation may be associated with it. With complementary approaches existing,24 possibly the shared phenotypic traits might be connected with shared pathways25 and so the genes and pathways with the related phenotypes might be collectively associated with similar outcomes, thus influencing the heterogeneity of a disease.
Skimming the regulatory regions
To identify SNPs underlying the regulatory regions, it may be possible to look for the functional effect of SNPs. For example, the presence of promoters, enhancers, or silencers accompanied by noncoding RNA sequences would facilitate a strong correlation of genes interacting with them. These, in turn, could serve as biomarkers for disease diagnosis and therapy allowing us to understand the varied phenotypic traits linked to a disease, for example, from the GWAS. Regions could be skimmed by checking the regions for structural variants/regulatory elements using the variant effect predictor from Ensembl.org. Inferring noncoding RNAs within the genome would mean that the upstream or downstream regions harboring the SNPs could play a regulatory role. Recent efforts on the exploration of genetic variants using regulatory genomics approaches in complex diseases have provided insights into easy detection of causal variants.26 Finding the syntenic regions to nearest taxa, such as dogs and chimps, for the presence of any long noncoding RNAs (lncRNA) would be an added verification.27 A blast search with the well-reported human lncRNAs from databases such as Noncode (www.noncode.org) and the highly significant regions that meet the e-value (expectant value) threshold of <0 are considered. The reason why lncRNAs and not small noncoding RNAs like miRNAs could serve as important candidates is that we believe that miRNAs, being highly conserved with 22–23 mers (when compared with >200 bp lncRNAs), may not harbor mutations specific to a disease. To understand the transcripts that are single and multiple exonic, Koufariotis et al28 have indeed looked for lncRNA in various tissues. As an example, we have analyzed the lipopolysaccharide-induced mastitis-specific RNA-Seq gene expression data sets to see whether they have any ncRNAs spanning these regions.29 From our annotation, we perceive that they indeed are a part of multi-exonic regions and we found ~45 lncRNAs and 2 miRNAs associated with the differentially expressed gene data sets (Figure 4). On the contrary, we found them not to be harbored near the intronic regions, where SNPs specific to mastitis are seen. The location of the SNP, such as intergenic, intragenic–intronic, downstream, or upstream regions, serves as a run-up to the evolutionary conserved regions (ECRs). To show this, we proposed a third classifier in the form of constrained elements.
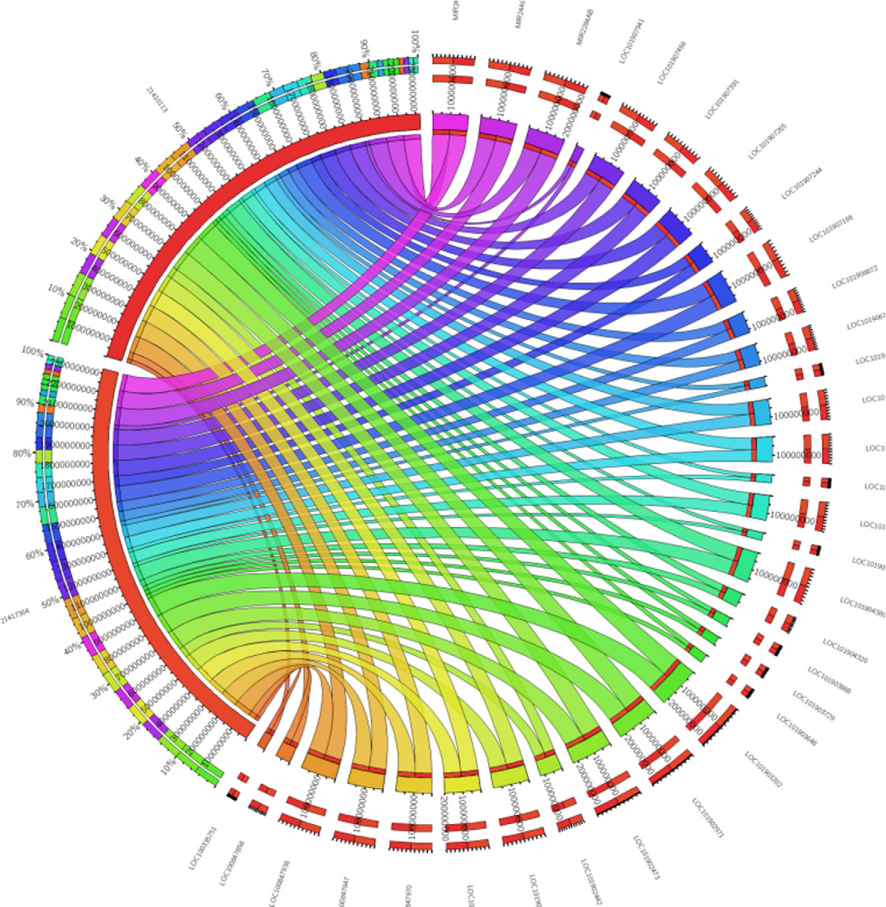 | Figure 4 Forty-five lncRNAs and two miRNAs shown in the form of circos figure associated with the differentially expressed gene data sets. |
Constrained/enriched elements
Conservation across the genome by checking its syntenic blocks may validate the presence of conserved SNPs. In the light of finding ECR, blasting the regions (paralogons) that are conserved against the organism of interest would be a useful resource to add. Also, genomic evolutionary rate profiling (GERP) may be considered, to a certain degree, to find whether the conservation is specifically called as a constrained element.30 While these conservation regions are estimated across a wide range of organisms, the genome-wide consistency check would restrict information on their conservation wherein the SNP may be considered as a candidate if detected to be lying in both ECR and GERP. The syntenic regions are made assuming that the sequence blocks are in synteny, and the alignments are grouped as blocks apart in the genome browser. In those regions, the enriched motifs can be approximated based on the TFBS and enhancers found using a database enhancer region such as cap analysis gene expression tags from the FANTOM5 project.31
Keeping in view of the fact that introns harbor important functional elements which we might miss from the annotation strategy as discussed above, the top significant SNPs from the regions of interest are checked if they form any sequence patterns. A false discovery rate adjusted –log10 P-value cutoff for the regions would denote the most significant peak associations for causal detection. As an example illustrated earlier for high-density SNPs in clinical mastitis-specific region, considering 50 SNPs from those regions with significant associations and 20 top SNPs each with effective P-value scores for those set of chromosomes would determine a scale of how much genetic variation is seen. To identify functional elements in these regions, and to tag them as candidates based on the effective GERP score and ECR, would mean establishing the position of SNP regions corresponding to known constrained elements. The latter part of functional analysis is helpful in detecting pre-mRNA splicing variants, 5′ UTR regions, which show less conservation but a high level of genetic variation. The prioritized SNPs flanking the GERPs and those SNPs underlying the enhancers and constrained elements assume that these patterns are significantly associated with genetic variation. In discriminating these candidates, we are then able to identify causative SNPs that could possibly explain their role in phenotypic associations. The “Genomic Repeat Element Analyzer for Mammals” validates how many genes form a part of the repeat elements and family members, and whether they are conserved or specific to these organisms.32 However, GeneMANIA predictions33 by Cytoscape, as shown in Figure 5, would serve as a confirmatory tool to check whether associations or pathway mappings exist among the genes. In each instance, this will allow us to mark the queried genes with the corresponding annotation and check if any of these genes form a peer interaction network.
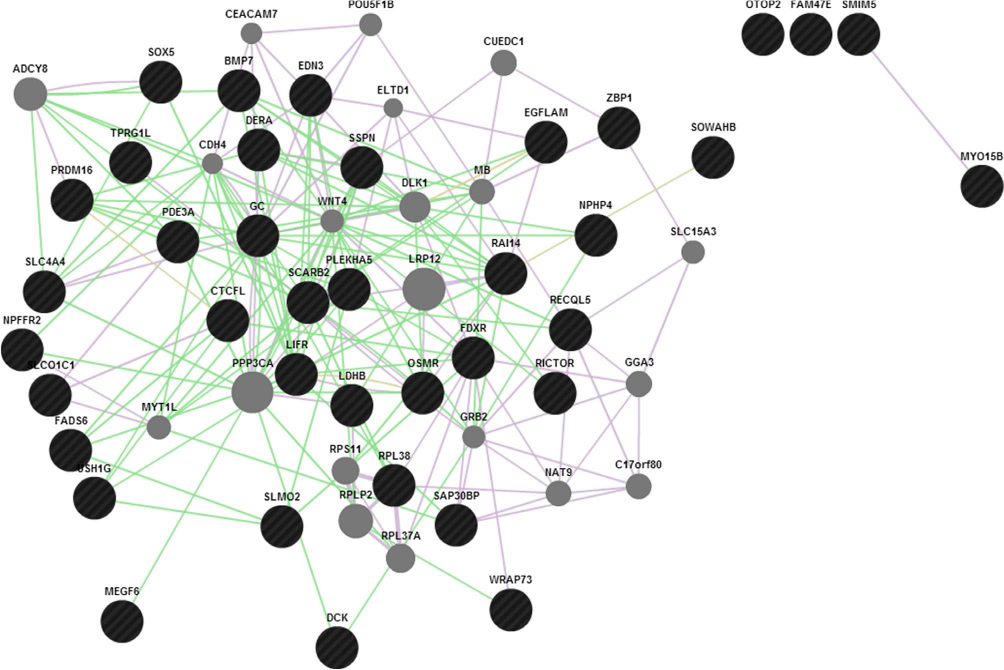 | Figure 5 GeneMANIA predictions for the candidate genes associated with traits lying in the regions (GC, NPFFR2, and SLC4A4). Abbreviations: GC, group-specific component; NPFFR2, neuropeptide FF receptor 2. |
Current challenges and promises in prioritizing the SNPs
Prioritizing SNPs requires different methods for identifying causal relationship between genes. There is a growing number of challenges and promises in this next generation sequencing (NGS) era to understand the available knowledge that can be used to predict the bona fide SNPs. Identifying DNase I hypersensitive sites (DHSs) can perhaps discern a catalog of variants that are regulatory and transcriptionally active. Williams et al34 have identified such regions comparing livestock genomes, and substantial efforts on sidelines have been in place. Given the fact that there are good annotated references for these genomes, identifying DHS would prove vital. Furthermore, as the DHS profiles are documented in the human ENCODE project based on the cell type, the complexity of finding the SNPs regulating the genetic expression would measure the same catalog of such DHS from the human ENCODE project.35 Wherever there are open chromatin elements, the presence of DHS and their enrichment does not necessarily mean that there are enhancer elements. A careful integration of these annotation methods could designate an SNP as a causal. So we ask “Are the CpG islands (CGI) predisposed by the presence of SNPs”? The epigenetics of CpG-rich regions attribute to understanding the polymorphisms related to SNPs (cgSNPs). However, with our current goal of identifying bona fide SNPs, we may not validate the potential effects such as methylation. Loss of CGI sites that are significantly enriched in these QTL regions along with histone marks, H3k4Me signals, and methylation events can distinguish prospects for high-throughput identification of putative enhancers to complement experimental approaches.36
Recently, Functional Annotation of Animal Genomes, an international project consortium,37 has set up a committee on various data generated from eutherian mammalian resources, including bovine genome project. In addition, the 1000 bull genomes38 project has provided the bovine research community a set of regulatory data including a noncoding repertoire that is periodically published. Apart from the predicted coding and noncoding elements, the primary resources that supply information for annotations from actual empirical observations of TFBS, DHSs, CTCF motifs, expression QTLs, and so on would be of great value. Furthermore, tools such as InnateDB would be of great resource to identify candidate genes and pathways specific to innate pathway.39
Conclusion
We have discussed how the three classification features are helpful in detecting causative variants associated with a disease such as clinical mastitis. In prioritizing a set of variants based on bioinformatics annotation, there is indeed a technical, perhaps also an economical advantage in going for a complete targeted sequencing of the LD segment underlying the association peak. Unfortunately, it is still early to reach consensus on statistical and functional evidence, especially when the data are imperfect, which may lead to wrong conclusions. As the new and new methods pop up, we hope the next generation of SNP/genetic variants annotation would definitely bring a complex and yet noticeable resource of information with features/standards of annotation records from heterogeneous data sets, including functional annotation, enhancer elements, methylation and regulatory events, pathways, associations and interactions, spectrum of noncoding SNPs, and so on, and discern SNP prioritization using an accurate and computable confidence scores. While considering such a wide array of highly sensitive, if not less-stringent, classifiers/features, we might devalue the scale of causal SNP prediction. In this process, a thorough definition of “causal SNPs” should be constructed as “all causal variants may be a part of candidate or bona fide SNPs.”
Acknowledgments
PS thanks Goutam Sahana, Bernt Guldbrandtsen, Mogens Lund, and Peter Soerensen, all from the Centre for Quantitative Genetics and Genomics, Aarhus University, Denmark, for some useful discussions.
Author contributions
PS analyzed the data, proposed the methods, and wrote the initial draft. AB endorsed the methods and helped in fine tuning the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Yamada R, Ymamoto K. Recent findings on genes associated with inflammatory disease. Mutat Res. 2005;573(1–2):136–135. | ||
Shameer K, Denny JC, Ding K, et al. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet. 2014;133(1):95–109. | ||
Lee YH, Kim JH, Song GG. Pathway analysis of a genome-wide association study in schizophrenia. Gene. 2013;525(1):107–115. | ||
Camargo M, Rivera D, Moreno L, et al. GWAS reveals new recessive loci associated with non-syndromic facial clefting. Eur J Med Genet. 2012;55(10):510–514. | ||
Cui H, Dhroso A, Johnson N, Korkin D. The variation game: cracking complex genetic disorders with NGS and omics data. Methods. 2015;79–80:18–31. | ||
Gui LS, Zhang YR, Liu GY, Zan LS. Expression of the SIRT2 gene and its relationship with body size traits in Qinchuan cattle (Bos taurus). Int J Mol Sci. 2015;16(2):2458–2471. | ||
Bigham AW, Julian CG, Wilson MJ, et al. Maternal PRKAA1 and EDNRA genotypes are associated with birth weight, and PRKAA1 with uterine artery diameter and metabolic homeostasis at high altitude. Physiol Genomics. 2014;46(18):687–697. | ||
Liu Y, Maxwell S, Feng T, et al. Gene, pathway and network frameworks to identify epistatic interactions of single nucleotide polymorphisms derived from GWAS data. BMC Syst Biol. 2012;6(Suppl 3):S15. | ||
Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187(2):367–383. | ||
Hou L, Zhao H. A review of post-GWAS prioritization approaches. Front Genet. 2013;4:280. | ||
Price AL, Spencer CC, Donnelly P. Progress and promise in understanding the genetic basis of common diseases. Proc Biol Sci. 2015;282(1821):20151684. | ||
Hall MA, Moore JH, Ritchie MD. Embracing complex associations in common traits: critical considerations for precision medicine. Trends Genet. 2016;32(8):470–484. | ||
Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187(2):367–383. | ||
Sahana G, Guldbrandtsen B, Thomsen B. Genome-wide association study using high-density single nucleotide polymorphism arrays and whole-genome sequences for clinical mastitis traits in dairy cattle. J Dairy Sci. 2014;97(11):7258–7275. | ||
Pausch H, Emmerling R, Schwarzenbacher H, Fries R. A multi-trait meta-analysis with imputed sequence variants reveals twelve QTL for mammary gland morphology in Fleckvieh cattle. Genet Sel Evol. 2016;48(1):14. | ||
Olsen HG, Knutsen TM, Lewandowska-Sabat AM, et al. Fine mapping of a QTL on bovine chromosome 6 using imputed full sequence data suggests a key role for the group-specific component (GC) gene in clinical mastitis and milk production. Genet Sel Evol. 2016; 48(1):79. | ||
Jarjanazi H, Savas S, Pabalan N, Dennis JW, Ozcelik H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins. 2008;70(2):394–403. | ||
Petersen TN, Brunak S, von Heijne G and Nielsen H (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 8(10):785–786. | ||
Broos S, Soete A, Hooghe B, Moran R, van Roy F, De Bleser P. PhysBinder: Improving the prediction of transcription factor binding sites by flexible inclusion of biophysical properties. Nucleic Acids Res. 2013;41(Web Server issue):W531–W534. | ||
Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. | ||
Bickhart DM, Liu GE. Identification of candidate transcription factor binding sites in the cattle genome. Genomics Proteomics Bioinformatics. 2013;11(3):195–198. | ||
Iso-Touru T, Sahana G, Guldbrandtsen B, Lund MS, Vilkki J. Genome-wide association analysis of milk yield traits in Nordic Red Cattle using imputed whole genome sequence variants. BMC Genet. 2016;17:55. | ||
Buitenhuis B, Røntved CM, Edwards SM, Ingvartsen KL, Sørensen P. In depth analysis of genes and pathways of the mammary gland involved in the pathogenesis of bovine Escherichia coli-mastitis. BMC Genomics. 2011;12:130. | ||
Mahurkar S, Moldovan M, Suppiah V, O’Doherty C. Identification of shared genes and pathways: a comparative study of multiple sclerosis susceptibility, severity and response to interferon beta treatment. PLoS One. 2013;8(2):e57655. | ||
Brodie A, Tovia-Brodie O, Ofran Y. Large scale analysis of phenotype-pathway relationships based on GWAS results. PLoS One. 2014; 9(7):e100887. | ||
Liao X, Lan C, Liao D, Tian J, Huang X. Exploration and detection of potential regulatory variants in refractive error GWAS. Sci Rep. 2016; 6:33090. | ||
Lomelin D, Jorgenson E, Risch N. Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 2010;20(3):311–319. | ||
Koufariotis LT, Chen YP, Chamberlain A, Vander Jagt C, Hayes BJ. A catalogue of novel bovine long noncoding RNA across 18 tissues. PLoS One. 2015;10(10):e0141225. | ||
Jiang L, Sørensen P, Røntved C, Vels L, Ingvartsen KL. Gene expression profiling of liver from dairy cows treated intra-mammary with lipopolysaccharide. BMC Genomics. 2008;9:443. | ||
Cooper GM, Stone EA, Asimenos G; NISC Comparative Sequencing Program., Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15(7):901–913. | ||
Andersson R, Gebhard C, Miguel-Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. | ||
Chandrashekar DS, Dey P, Acharya KK. GREAM: a web server to short-list potentially important genomic repeat elements based on over-/under-representation in specific chromosomal locations, such as the gene neighborhoods, within or across 17 mammalian species. PLoS One. 2015;10(7):e0133647. | ||
Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue): W214–W220. | ||
Williams SH, Mouchel N, Harris A. A comparative genomic analysis of the cow, pig, and human CFTR genes identifies potential intronic regulatory elements. Genomics. 2003;81(6):628–639. | ||
Encode project consortium publications issue. Available from: http://genome.cshlp.org/content/17/6.toc. Accessed March 17, 2017. | ||
Kleftogiannis D, Kalnis P, Bajic VB. Progress and challenges in bioinformatics approaches for enhancer identification. Brief Bioinform. 2016;17(6):967–979. | ||
The FAANG Consortium. Available from: http://www.faang.org. Accessed March 17, 2017. | ||
The 1000 bull genomes. Available from: http://www.1000bullgenomes.com. Accessed March 17, 2017. | ||
Breuer K, Foroushani AK, Laird MR, et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 2013;41(Database issue):D1228–D1233. | ||
Aken BL, Ayling S, Barrell D. The Ensembl gene annotation system. Database (Oxford). 2016;2016. | ||
http://weblogo.berkeley.edu/ [homepage on the Internet]. Available from: http://weblogo.berkeley.edu/. Accessed June 1, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.