")
Back to Journals » International Journal of Nanomedicine » Volume 13
Prior anti-CAFs break down the CAFs barrier and improve accumulation of docetaxel micelles in tumor
Authors Pang N, Li J, Sun A , Yang Z, Cheng S, Qi XR
Received 15 April 2018
Accepted for publication 16 July 2018
Published 4 October 2018 Volume 2018:13 Pages 5971—5990
DOI https://doi.org/10.2147/IJN.S171224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Ning Pang, Ji Li, Aning Sun, Zhenzhen Yang, Shixuan Cheng, Xian-Rong Qi
Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery System, Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
Background: Abnormal expression of stromal cells and extracellular matrix in tumor stroma creates a tight barrier, leading to insufficient extravasation and penetration of therapeutic agents. Cancer-associated fibroblasts (CAFs) take on pivotal roles encouraging tumor progression.
Method: To surmount the refractoriness of stroma, we constructed a multi-targeting combined scenario of anti-CAFs agent tranilast and antitumor agent docetaxel micelles (DTX-Ms). Tranilast cut down crosstalk between tumor cells and stromal cells, ameliorated the tumor microenvironment, and enhanced the antiproliferation efficacy of DTX-Ms on cancer cells.
Results: Diverse experiments demonstrated that tranilast enhanced DTX-Ms’ antitumor effect in a two-stage pattern by CAFs ablation, tumor cell migration blocking, and metastasis inhibition. Along with activated CAFs decreasing in vivo, the two-stage therapy succeeded in reducing interstitial fluid pressure, normalizing microvessels, improving micelles penetration and retention, and inhibiting tumor growth and metastasis. Interestingly, tranilast alone failed to inhibit tumor growth in vivo, and it could only be used as an adjuvant medicine together with an antitumor agent.
Conclusion: Our proposed two-stage therapy offers a promising strategy to enhance antitumor effects by breaking down CAFs barrier and increasing micellar delivery efficiency.
Keywords: two-stage therapy, tumor microenvironment normalization, cancer-associated fibroblasts, tranilast, stromal ablation
Plain language summary
Studies reveal that crosstalk between tumor cells and fibroblasts induces refractory tumors. Considering this obstacle, we propose a two-stage combined therapy. The two-stage combined therapy consists of CAFs (cancer-associated fibroblasts) inhibitor tranilast and anti-tumor agent docetaxel micelles. We found that tranilast could be an adjuvant medicine to tackle CAFs (main component of stromal barriers). Docetaxel micelles were administrated after tumor vascular system restoration and more micelles were trapped in tumor to kill cancer cells. Ablation of CAFs by prior anti-CAFs normalized tumor environment and made way for micellar delivery in tumors. Furthermore, stromal ablation did not increase tumor metastasis but inhibited migration. Results reveals that CAFs promote tumor progression and the ablation of stroma contributes to pave the way for anti-tumor therapy. Considering an effectual remedy, tumor cell is not the single target, stromal cell like CAF needs more attention. Our proposed two-stage therapy is a meaningful trial in integrated tumor treatment.
Introduction
Studies on tumor pathology emphasize the existence of tumor microenvironment (TME) apart from a homogeneous collection of neoplastic cells. TME describes the extracellular matrix (ECM), stromal cells, surrounding blood vessels, numerous signaling molecules, and proteolytic enzymes in tumor.1,2 Stromal cells in TME interact with tumor cells by affecting chemoresistance, ECM remodeling, abnormal angiogenesis, epithelial–mesenchymal transition, neoplasm metastasis, and recurrence.3,4 Among these stromal cells, abundant cancer-associated fibroblasts (CAFs) are characterized with contractile feature.5 They are recruited and activated during whole tumor progression. During the transition to premalignant dysplasia, fibroblasts become activated. When it progresses to carcinoma, fibroblasts differentiate into myofibroblasts (CAFs) with expression of growth factors, matrix components, and degrading proteases promoting tumor growth.6 They protect tumor cells from chemotherapy,7–9 promote tumor invasion by remodeling the ECM structure,10,11 and affect angiogenesis etc.
In the case of most solid tumors, CAFs not only interact with tumors cells to communicate signals for tumor progression but also erect tough “walls” that contain enhanced collagen deposition and fibrosis in ECM to hamper drug delivery.12,13 The interstitial fibrosis and a contraction of interstitial space mediated by CAFs probably result in high interstitial fluid pressure (IFP) in the tumor,14,15 which aggravates the drug diffusion dilemma. More than that, abnormal neo-vasculature related to imbalanced angiogenic signals secreted by CAFs serves a bad role to block drug delivery.16–18 Because of these obstacles in the TME, passive targeting nanoparticles may fail to achieve good penetration into the inner part of tumors. Therefore, the targeting at CAFs may help to overcome TME barriers, leading to enhanced nanoparticle delivery efficiency in tumors.19–21
In this study, we focus on a combination therapy of tranilast for TME normalization and docetaxel (DTX) for killing tumor cells. Tranilast is a clinical anti-fibrotic agent.22,23 It can inhibit fibrosis in cancer and reduce angiogenic factors like vascular endothelial growth factor (VEGF), stromal cell-derived factor-1, and transforming growth factor (TGF)-β1 overexpressed in tumor.24–26 Scheme 1 illustrates our proposed two-stage combination therapy (TC therapy); as shown, the solid tumor is well organized with crowded neoplastic cells and stromal cells, surrounded by a large number of active CAFs and aberrantly tortuous blood vessels. Prior administration of tranilast is carried out to downregulate CAFs’ activity, cut off CAFs’ intercommunication with tumor cells, and induce vascular normalization as well. Then, DTX micelles arrive at the tumor tissue via nearby veins. Micelles are passively detained at the tumor as a result of the enhanced permeability and retention (EPR) effect and further diffuse into the inner part to kill tumor cells.
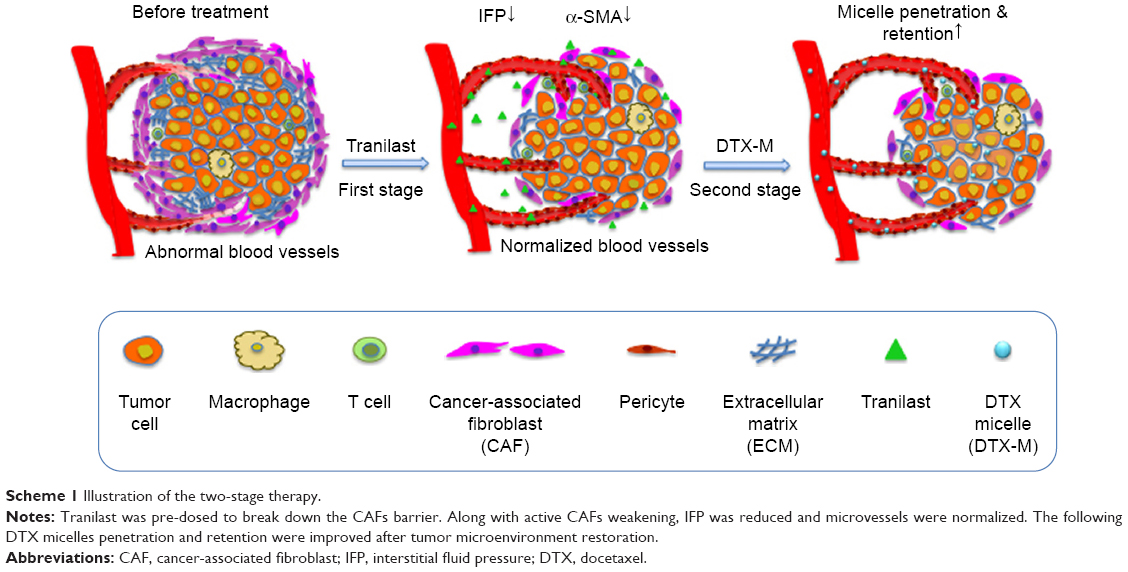 | Scheme 1 Illustration of the two-stage therapy. |
Our work demonstrated that tranilast-assisted therapy inhibited intractable 4T1/3T3 mixed cells, detached CAFs (d-CAFs), and three-dimensional (3D) mixed tumor spheres growth and tumor cell migration in vitro. After the two-stage therapy in vivo, reduced IFP, normalized microvessels, and decreased active CAFs helped to enhance docetaxel micelles (DTX-Ms) retention in tumor. Subsequently, the antitumor efficiency increased significantly. As far as we know, there are no comprehensive researches combining tranilast for TME remediation with chemotherapeutic agents for tumor suppression in the two-stage way. This strategy of breaking nanoparticle delivery barriers of CAFs paves the way for antitumor therapy.
Materials and methods
Reagents and animals
N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) was generously donated by China Pharmaceutical University Pharmaceutical Co, Ltd (Nanjing, Jiangsu, China) and was dissolved in DMSO. DTX was obtained from Jiangsu Hengrui Medicine Co, Ltd (Lianyungang, Jiangsu, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from VWR International (Radnor, PA, USA). Poly(ethylene glycol)-poly(caprolactone) (PEG-PCL) was synthesized in our laboratory previously with a molecular weight of 3,900 at a two-block ratio of 1:1.
Platelet-derived growth factor receptor-β (PDGFR-β) and α-smooth muscle actin (α-SMA) were purchased from Santa Cruz Biotechnology Inc (Dallas, TX, USA). Alexa Fluor® 647 was obtained from Beyotime Biotechnology (Shanghai, China). 3,3′-Dioctadecyloxacarbocyanine perchlorate (DiO) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) were obtained from KeyGen (Jiangsu, China). Hoechst 33258 (1 mg/mL) was from Beyotime Biotechnology. FITC-lectin was purchased from Vector Laboratories Inc (Burlingame, CA, USA).
BALB/C female mice weighing 16–18 g (8 weeks) were obtained from Peking University Health Science Center (license no SCXK [Jing] 2006–0041 and SCXK [Jing] 2006–0010). All animal experiments adhered to the principles of care and use of laboratory animals by the Committee of Peking University Health Science Center and were approved by the Institutional Animal Care and Use Committee of Peking University.
Cell culture
Breast cancer 4T1 cells and fibroblast 3T3 cells were obtained from American Type Culture Collection, Manassas, VA, USA and were cultured in DMEM or RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), and 100 U/mL penicillin and streptomycin.
As for the co-culture system of 4T1/3T3 mixed cells, 4T1 and 3T3 cells were harvested and mixed at a ratio of 1/3. Mixed cells were cultured in 1640/DMEM (mixed ratio were 1/3) complete culture medium.
d-CAF cells from 4T1 tumors in mice
d-CAF cells were derived from 4T1 tumor bearing mouse according to a method described previously.27 In brief, 4T1 cells were injected into the mouse armpit to harvest tumor tissues (>2,000 mm3). Tissues were cut into small pieces (<1 mm3) with sterilized surgical instruments and incubated with DMEM medium containing 0.1% type I collagenase, 5% FBS, and 2% penicillin and streptomycin for 3 hours at 37°C. Then, the cell suspension was filtrated, centrifuged, and finally resuspended in a culture flask. Cells adhering to the flask bottom within 20–30 minutes were thought to be d-CAFs, while non-adherent cells were abandoned. The culture process was repeated five times to achieve nearly pure d-CAFs. The obtained cells were used within 10 passages. The certification of d-CAFs is described in Supplementary materials.
Preparation and characterization of DTX-Ms
The micelles were prepared via film hydration method.28,29 DTX and PEG-PCL were dissolved in acetonitrile and the dissolvent was removed through rotary evaporation to form a thin film; then, physiological saline was added for vortex and ultrasonic process. They were filtrated through a 0.22-μM filter membrane to obtain DTX-Ms. Using the same method, DiR-loaded micelles (DiR-Ms) were prepared.
A dynamic light scattering system (Malvern Zetasizer 3000HS; Malvern Instruments, Malvern, UK) was used to measure the diameter and zeta potential of DTX-Ms. Micelles morphological features were observed via a transmission electron microscope (TEM, JEM-2100F, 200 kV; JEOL, Tokyo, Japan).
Construction of mixed tumor spheres
The two-cell-type mixed tumor spheres comprised 4T1 and 3T3 cells. Briefly, 2% (w/v) agarose dissolved in serum-free DMEM formed a gel layer at the bottom of 48-well plates and each well was filled with the culture medium. 4T1 and 3T3 cells were collected to obtain a mixed cell suspension at different ratios (3/1, 1/1, 1/3, and 1/5, respectively). Twenty microliters of cell suspension formed dew-like liquid drops on the plate lid for 48 hours incubation. Cells aggregated into cellular congeries on plate lid and then they were transferred into plate chambers for another 24 hours. To certify mixed tumor spheres, 3T3 cells were pre-stained with cell fluorescent probe DiO (green fluorescence). Mixed tumor spheres were photographed with a confocal laser scanning microscope (CLSM).
Combination therapy regimens illustration
The following experiments were conducted according to alternative combination therapy regimens. One was a co-dose combination therapy (CC therapy) that meant tranilast and DTX-Ms were administrated simultaneously. The other was TC therapy, indicating that tranilast was pre-dosed with proper time interval (TI) before DTX-Ms.
Cytotoxicity test
4T1, 3T3, 4T1/3T3 (1/3) mixed cells, and d-CAFs were initially seeded in 96-well plates (5,000–8,000 cells per well). After overnight incubation, a series of tranilast, DTX-Ms, or drug combinations was added. In order to reduce the vehicle’s (DMSO) effect on cells, the final concentration of DMSO was controlled below 0.2% (v/v). MTT (5 mg/mL) was added after 48 hours incubation. The plates were placed in the dark for 4 hours at 37°C. After emptying the plates, 200 μL DMSO was pipetted into each well. MTT formazan was dissolved entirely for 10 minutes in the dark, and the absorbance at 570 nm was measured to calculate the inhibition rate.
Cell scratch wound assay
4T1 or 3T3 cells (1×106/mL) were incubated overnight in 6-well plates to grow to confluence, then a thin wound was introduced by scratching with a pipette tip. The system was transferred into low FBS (2%) culture medium to reduce the effect of cell proliferation on experimental results without destroying cell viability. Tranilast (200 μM), DTX-Ms (10 μM), or a drug combination was added. Images were captured at 10 and 20 hours and interval lengths were semi-quantitatively analyzed with the software Image J 2.1.4.7 (National Institutes of Health, Bethesda, MD, USA).
Antitumor efficiency in mixed tumor spheres
Mixed tumor spheres (4T1 and 3T3 mixed at 1/3) were processed with PBS, tranilast, DTX-Ms, CC therapy, and TC therapy (TI =24 hours). The final concentrations of tranilast and DTX-Ms were 2,000 and 100 μM, respectively. Tumor spheres width (W) and length (L) were monitored every day. The sphere volumes were calculated according to the formula: volume =1/2×W2×L. The volume ratio (Vd/V0) was calculated, in which Vd represented the daily recorded volume and V0 the initial volume at day 0.
Animal model and antitumor therapies in vivo
BALB/C female mice were subjected to 4×106 4T1 cells injection subcutaneously at the right armpit. After tumor volume increased to 150–200 mm3, mice were divided into five groups (n=9) randomly and underwent various treatments. Tranilast in 0.5% sodium carboxyl methyl cellulose was administrated by gavage at 100 mg/kg per time, except for CC therapy (tranilast was administrated at 300 mg/kg once to keep the total dose constant). DTX-Ms were dosed via tail vein injection at 10 mg/kg per time. Tumor volume were recorded: volume =1/2S2L. S means short diameter of tumor and L means long diameter of tumor. Growth inhibitory effects (GIE) were calculated by comparing the mean tumor volume of each treatment (VT) with the volume of control (VC), GIE = (1−VT/VC)×100%.
At the end of the last treatment (day 21), the tumor and organs were detached to obtain tumor and organ coefficients according to the formula: coefficient = tumor or organ weight/body weight ×100%.
IFP and micellar distribution in tumor
In IFP assay, mice (n=3) were anesthetized with chloral hydrate on day 19. IFP was measured with the aid of a wick-in-needle IFP measuring instrument.30
On day 20, mice (n=3) were injected with DiR-Ms intravenously to conduct an in vivo imaging test through noninvasive in vivo imaging systems (IVIS) spectrum. Mice were photographed at 2, 6, and 24 hours. The radiant efficiencies at the tumor site were analyzed with IVIS spectrum system software (Living Image 4.3.1, PerkinElmer Inc., Waltham, MA, USA).
Vascular perfusion, immunofluorescence, and H&E assays
FITC-lectin was injected intravenously on day 21 to indicate the tumor vascular system. Tumors (n=2) of DTX-Ms or TC therapy were removed 1 hour after injection to prepare frozen sections, and the sections were stained with Hoechst 33258 (10 μg/mL) to direct nuclei.
For the immunofluorescence assay, the sections were permeated in Triton-X 100 following PBS washing, methanol (containing 10% H2O and 10% H2O2), and 2% BSA solution. Sections were treated with the primary antibody for overnight incubation and secondary antibody for 1 hour incubation. Cell nuclei were stained with Hoechst 33258 (10 μg/mL). All results were monitored via a CLSM.
For the H&E assay, the tissues were embedded into paraffin and 5-μM thick sections were prepared. Before staining, the sections were dewaxed in xylene and rehydrated by decreasing concentrations of ethanol. Then, the slides were washed in PBS, stained with H&E, and observed with an inverted fluorescence microscope (Olympus IX73; Olympus Corporation, Tokyo, Japan).
Statistical analysis
All data obtained were analyzed with IBM SPSS Statistics 19.0 (IBM Corporation, Armonk, NY, USA) and presented as mean ± standard deviation. One-way or two-way analysis of variance was performed according to statistical methods. A P-value <0.05 was considered significant (*), while P-values <0.01 were regarded as highly significant (**).
Results
Preparation and characterization of DTX-loaded micelles
DTX has poor solubility, short elimination half-life, and low permeability, belonging to class IV of the Biopharmaceutical Classification System.31,32 Biodegradable and biocompatible PEG-PCL micelles were employed as DTX vector because of their capability of i) increasing drug solubility and stability; ii) maintaining longer residence time in vivo; iii) penetrating tumor vessels and targeting tumors via the EPR effect; and iv) reducing nonspecific organ toxicity.33–35
Blank or drug-loaded micelles, 25–30 nm in diameter, demonstrated narrow distribution (polydispersity index ~0.17) and neutral zeta potential (Table 1). DTX loading capacity was up to 80% at the drug-lipid proportion of 1:20 (Table 1). TEM images indicated that the micelles were spheroids approximately (Figure 1).
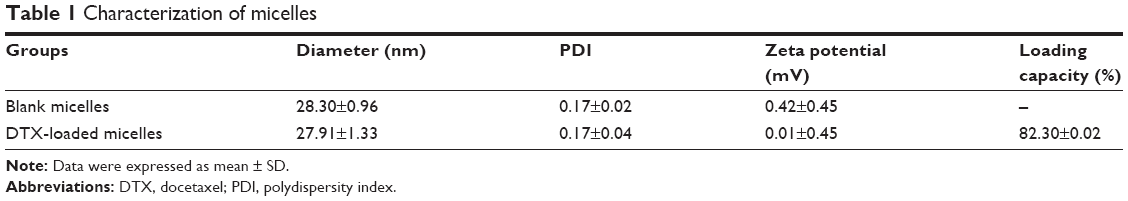 | Table 1 Characterization of micelles |
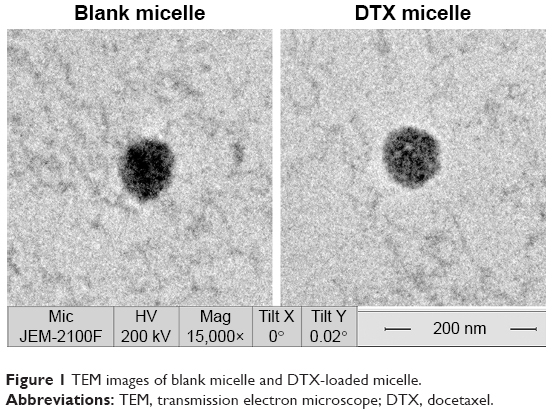 | Figure 1 TEM images of blank micelle and DTX-loaded micelle. |
Combination therapy enhances antitumor effects in intractable 4T1/3T3 mixed cells and d-CAFs
DTX aims at tubulin to cause a stabilization of microtubules and thereby results in cell-cycle arrest, mitosis damage, and apoptosis.36 4T1 were treated with free DTX and DTX-Ms for 48 hours in the presence or absence of tranilast (Figure 2A). DTX and DTX-Ms resulted in proliferative inhibition on 4T1 in a dose-dependent manner, and DTX-Ms revealed priority over free DTX in the range of 1.25–10 μM. Tranilast inhibits cell proliferation through cell cycle arrest beyond G1/S phase to interrupt protein synthesis at high concentrations.37,38 As DTX and tranilast acted on differed targets, theoretically, their actions on the tumor were relatively independent. The combination therapy ratio of DTX and tranilast was set at 1:20 (mol/mol) according to previous researches.39,40 The addition of tranilast in a 20-fold mole ratio of free DTX or DTX-Ms (1.25–20 μM) stimulated greater cytotoxicity (Figure 2A), revealing tranilast’s synergistic effect on antitumor cell proliferation.
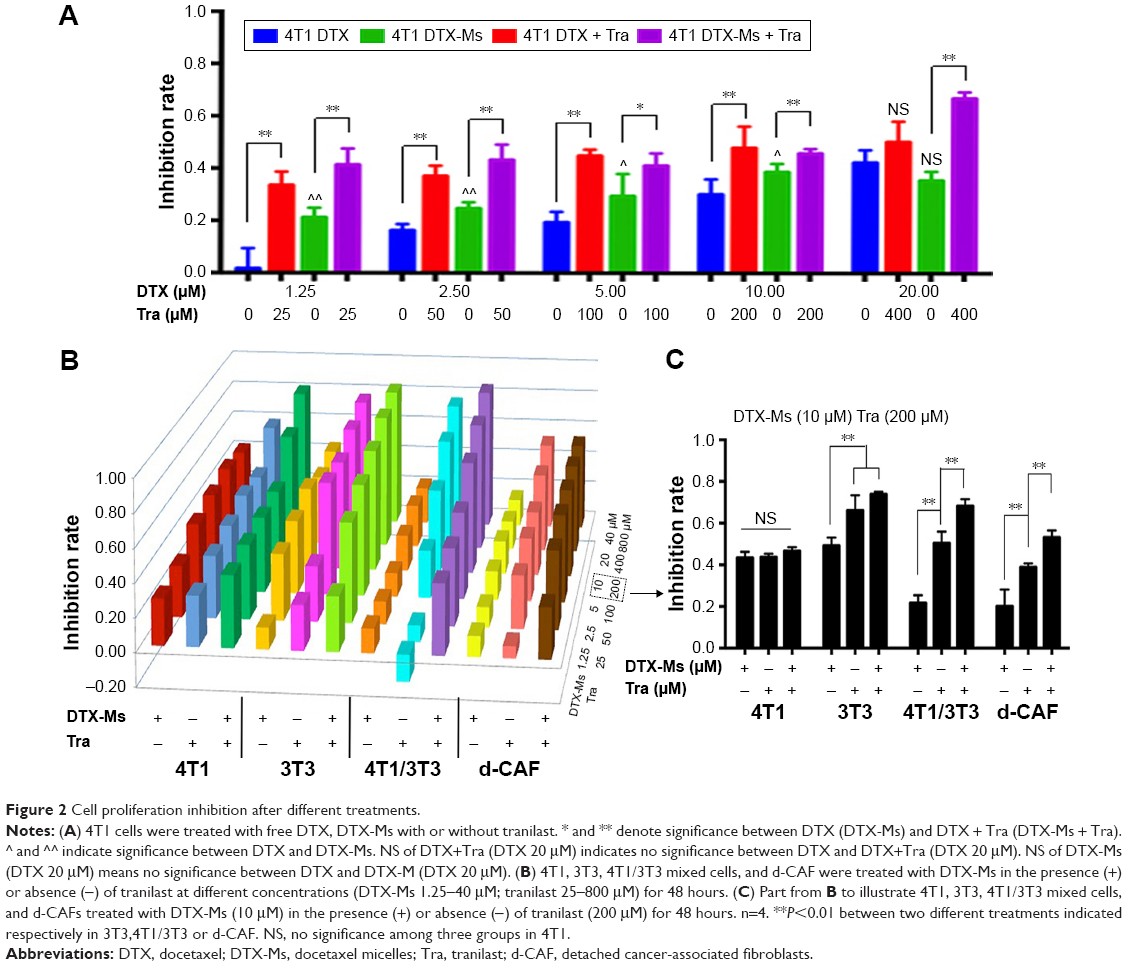 | Figure 2 Cell proliferation inhibition after different treatments. |
CAFs detached from 4T1 bearing mice (d-CAFs) have proved their fibrous feature of elevated α-SMA and PDGFR-β protein expression (Figure S1). A series of DTX-Ms (1.25–40 μM) and/or tranilast (25–800 μM) were added into 4T1, 3T3, 4T1/3T3 mixed cells, and d-CAFs to study the feasibility of combination therapy. Data are displayed in Figures 2B and S2. Obviously, DTX-Ms, tranilast, and the combination therapy demonstrated good antiproliferation effects on 4T1 and 3T3 cells. However, for 4T1/3T3 mixed cells and d-CAFs, DTX-Ms showed a much weaker antiproliferation effect. This may imply that crosstalk between tumor cells and fibroblasts induces 3T3 to become CAF-like and 4T1 to become chemoresistant.41 The off-target of DTX-Ms on d-CAFs might be due to lower differentiation and proliferation of fibroblasts. Combined administration exhibited dose-dependent inhibition on intractable 4T1/3T3 and d-CAFs at the listed concentrations. With DTX-Ms 10 μM and tranilast 200 μM for example in 4T1/3T3 and d-CAFs (Figure 2C), 3.14-fold and 2.64-fold inhibition rate was found for the combination therapy compared with DTX-Ms alone, 1.35-fold and 1.37-fold compared with tranilast alone. We obtained a combination index (shown as q-value) (Supplementary materials) to evaluate the combination efficacy. For 4T1 and 3T3, DTX-Ms and tranilast manifested an additive effect at most concentrations. Specially, addition or even synergism was shown at all listed concentrations on 4T1/3T3 mixed cells and d-CAF (Table S1).
Combination therapy decreases cell migration
In malignant cancer, metastasis is a tricky problem due to its abundant stromal components.42 Tumor cell mobility is a symbolic character of metastasis. After scratching, cells on the edge of a wound are inclined to migrate toward the vacant site, making the scratch heal (Figure 3). 4T1 cells have an aggressive trait43 while 3T3 cells are weakly migratory. The scratch healing of 3T3 was completely inhibited by tranilast, DTX-Ms, and the combination therapy. The treatments of tranilast, DTX-Ms, and especially the combined administration weakened the migration property of 4T1. MTT assays certified that DTX-Ms and tranilast resulted in cytotoxicity on cells at 48 hours with DTX-Ms 10 μM or tranilast 200 μM (Figure 2C), which might cause some interference on cell migration inhibition. However, the photographs show that cell viability was still relatively good at 10 and 20 hours. This meant that the drugs mainly caused the migration inhibition without destroying cell health.
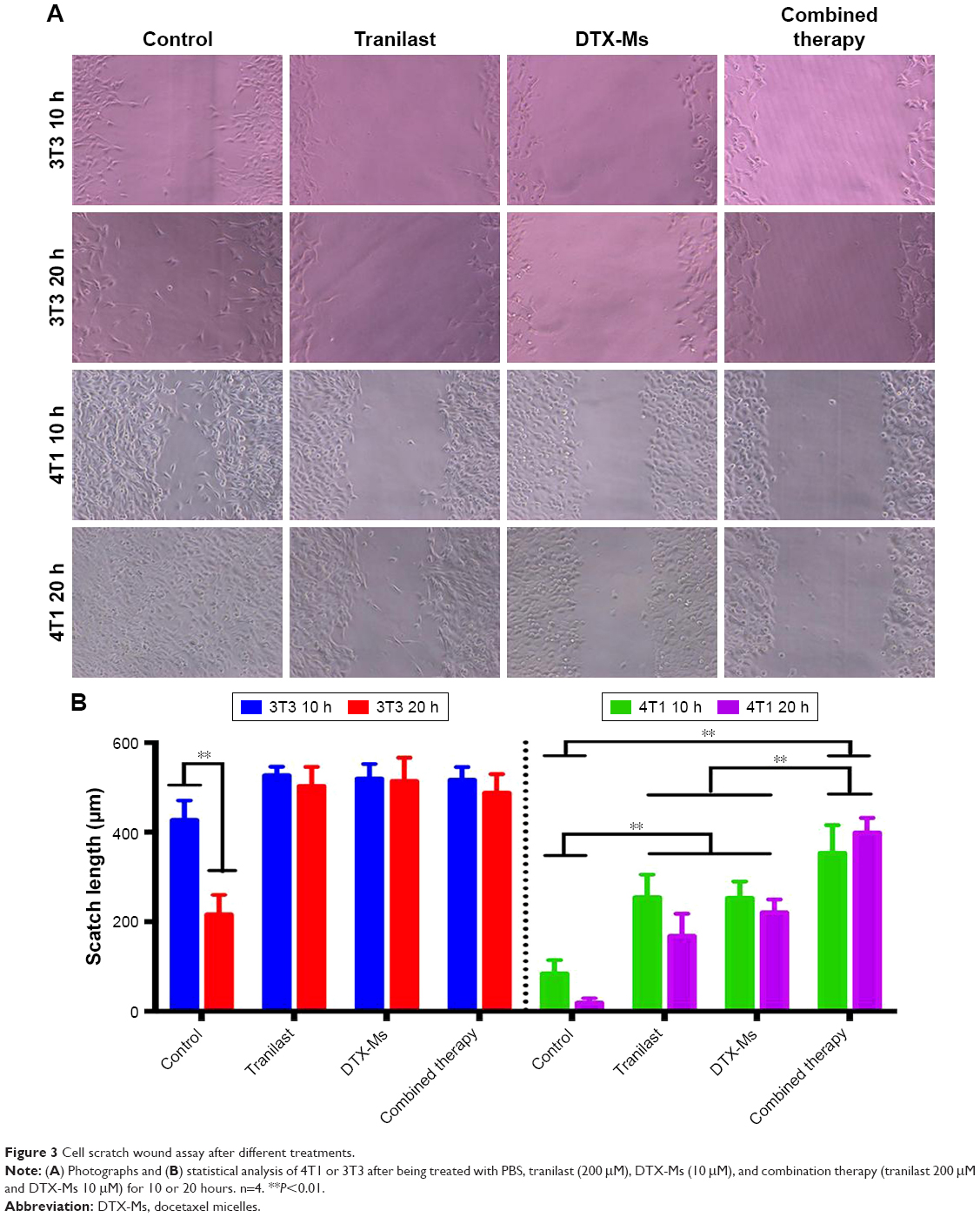 | Figure 3 Cell scratch wound assay after different treatments. |
Combination therapies inhibit 3T3/4T1 mixed tumor spheres growth
Tumor cells cultured in 3D model feature cell polarity, cell differentiation, and various kinds of signaling cascades.44,45 Mixed tumor spheres of diverse ratios were fabricated through the pendant drop method as described earlier.46 As the ratio of 3T3 increased, tumor spheroids became compactible with sharp edges (Figure S3A). Pre-stained 3T3 by DiO showed uniform dispersion of heterogeneous cells in spheres (Figures 4A and S3B). We chose 1/3 as the ratio of 4T1/3T3 mixed tumor spheres for the following assays. At this ratio, the tumor spheres took on compactible balls with a proper amount of 3T3 scattered evenly.
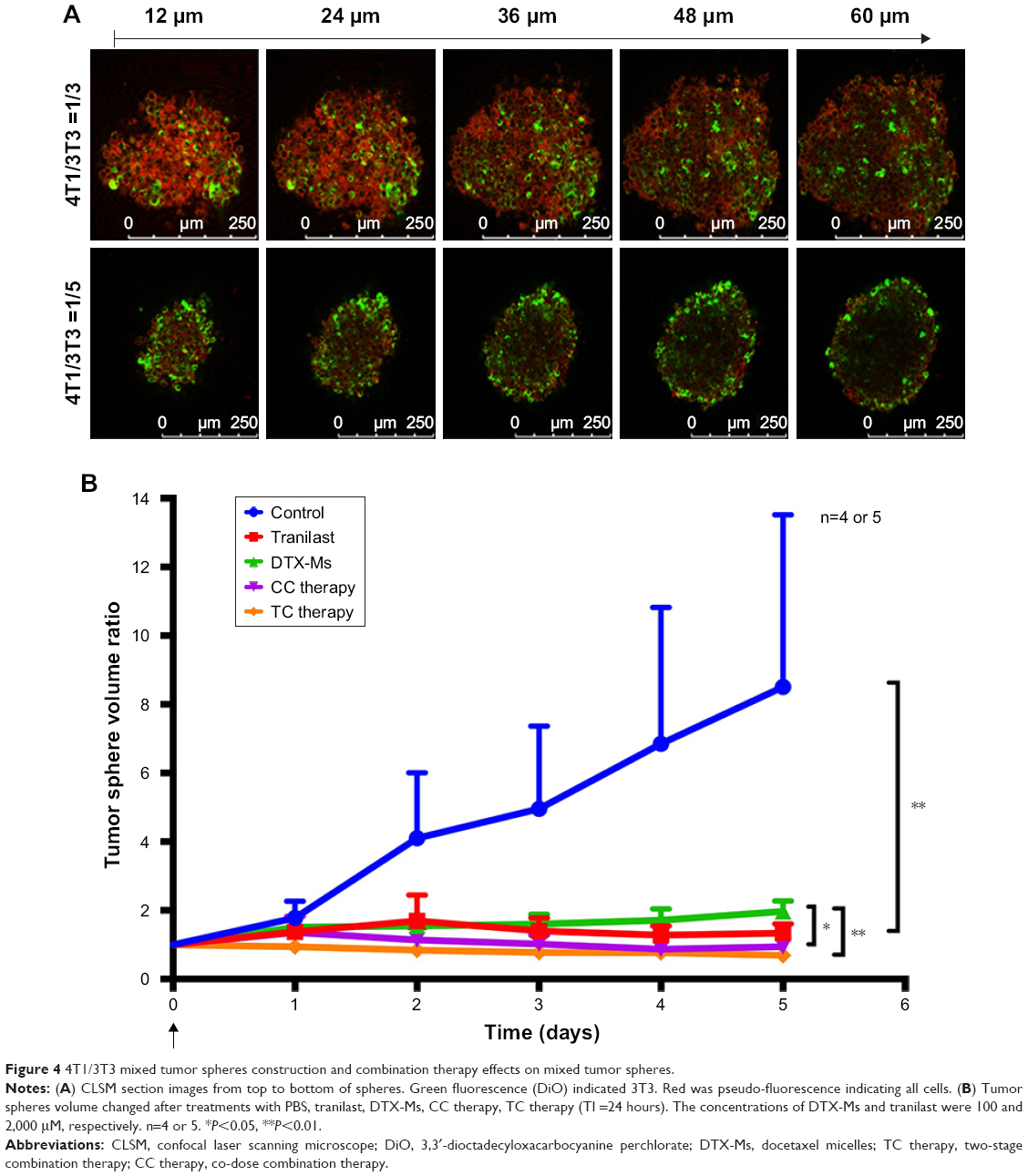 | Figure 4 4T1/3T3 mixed tumor spheres construction and combination therapy effects on mixed tumor spheres. |
The constructed 4T1/3T3 mixed tumor spheres were divided into five groups randomly and dosed with corresponding agents at day 0, and the sphere volume was monitored for 5 days (Figure 4B). We evaluated spheres growth by comparing the mean volume daily (Vd) with the volume at day 0 (V0) of each group and the volume ratio (Vd/V0) was then calculated. At day 5, results of control, tranilast, DTX-Ms, CC therapy, and TC therapy were 8.51, 1.33, 1.97, 0.94, and 0.69, respectively. Combination therapies (CC therapy and TC therapy) had remarkable suppression on mixed tumor spheres growth.
TC therapy decreases IFP, normalizes microvessels, and destroys active CAFs in tumor
As shown in Figure 5A, five treatments were conducted on 4T1 tumor bearing mice until TME formation.47 At day 19, mice (n=3) were anesthetized to test IFP. The IFP of control was extraordinarily high, while the other four contributed to decrease IFP dramatically (P<0.05) (Figure 5B). Particularly, TC therapy caused the strongest inhibition on IFP (decreased 51.7% to control).
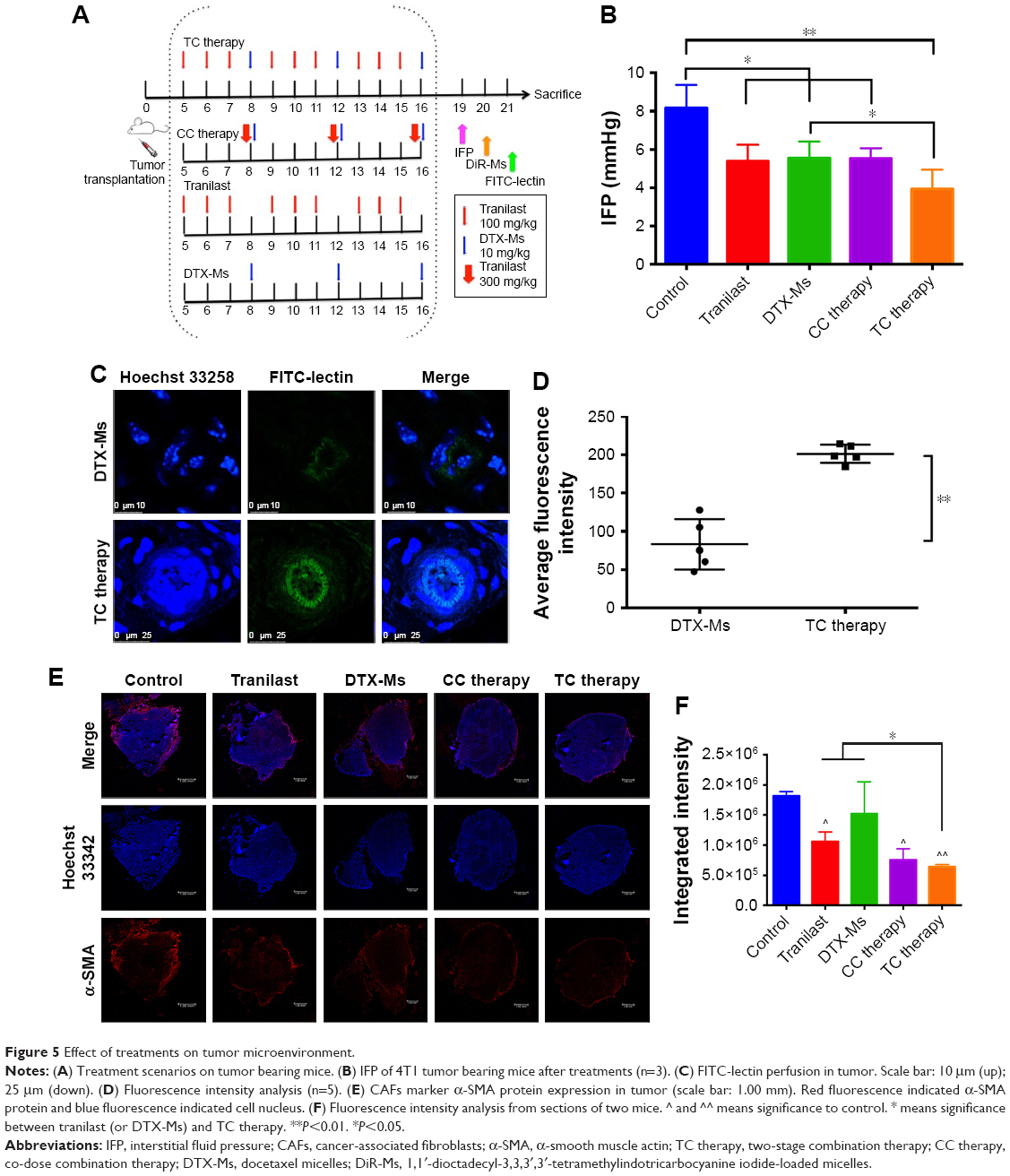 | Figure 5 Effect of treatments on tumor microenvironment. |
FITC-lectin was intravenously injected via the tail vein to visualize the microvascular system. Only microvessels with valid blood perfusion could be marked with green fluorescence.48,49 After DTX-Ms treatment, microvessels at the tumor periphery showed abnormal capillaries structure with thinned endothelial cells and lack of pericyte support (Figures 5C and S4), which implied weak perfusion ability.50 Nevertheless, in TC therapy, tumor vessels showed a well-organized vascular structure with valid blood perfusion. Detailed results are presented in Figure S4. The fluorescence intensity of the TC therapy perfusion level was 2.42 times higher than that of DTX-Ms (Figure 5D).
In order to find out the relationship between TME alteration (means IFP reduction and vascular normalization herein) and CAFs, we chose CAFs protein marker α-SMA to study CAFs accumulation in tumor. Although CAFs are composed of multi-resource subtypes, α-SMA-positive fibroblasts can be considered as the dominating CAFs.27 As shown in Figure 5E, CAFs displayed at the tumor periphery mainly act as a tough barrier for the tumor. Red fluorescence indicated α-SMA protein decrease after tranilast and DTX-Ms treatments. Particularly, TC therapy demonstrated minimum α-SMA expression. The fluorescence intensity of DTX-Ms, tranilast, CC therapy, and TC therapy decreased 16.3%, 41.5%, 58.3%, and 64.8%, respectively, compared with the control (Figure 5F).
TC therapy improves micelles retention in tumor
The superiority of micelles delivery lies in their passive targeting at the tumor site; yet, dense stroma and elevated IFP hamper micelles from penetrating into the tumor.51 After TC therapy, microvessels and IFP were normalized, and the penetration and retention of micelles in tumor should have been increased. In order to verify this hypothesis, mice (n=3) were injected with simulative micelles DiR-Ms to observe micelle retention in tumor. Mice were photographed via IVIS at 2, 6, and 24 hours after injection (Figures 6A and S5 show more details). DiR-Ms were passively targeted at the tumor due to EPR effect. The radiant efficiency in control, tranilast, and TC therapy was greater than that in DTX-Ms and CC therapy (Figure 6B).
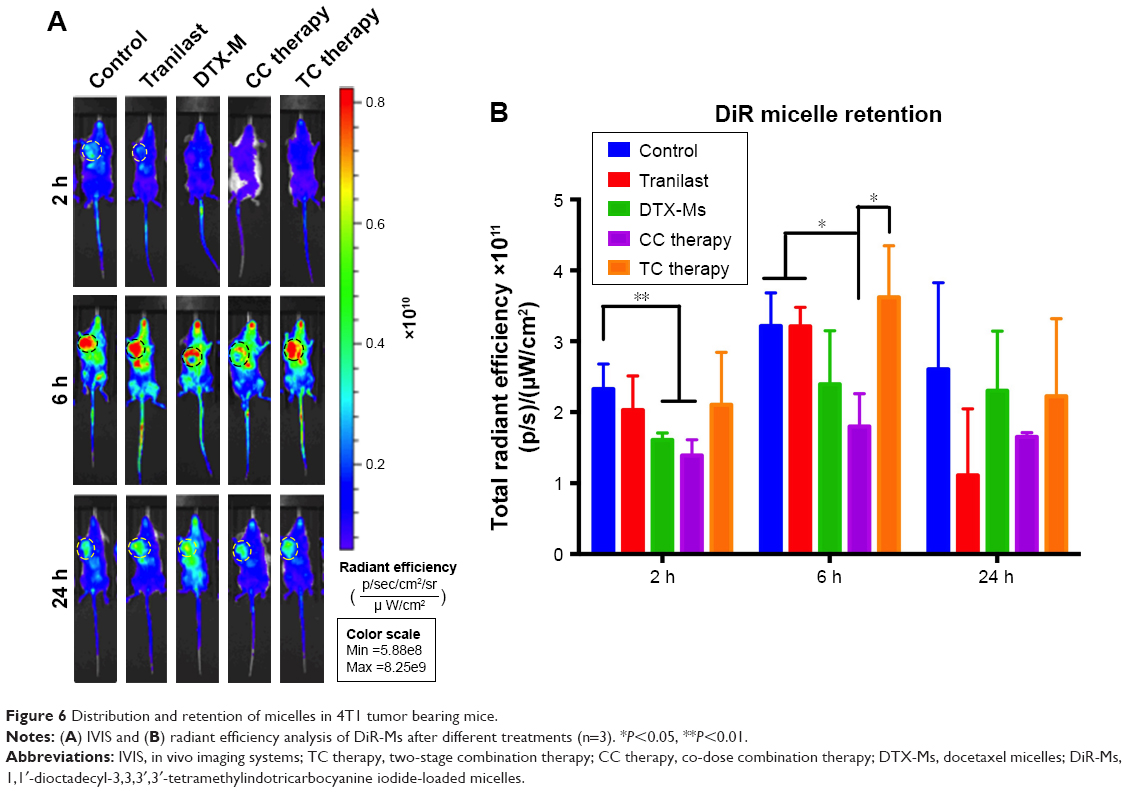 | Figure 6 Distribution and retention of micelles in 4T1 tumor bearing mice. |
Combination therapies inhibit tumor growth and metastases in vivo
In this malignant case, tranilast showed little tumor growth inhibition (Figure 7A) compared with control. For TC therapy, tumor volume growth inhibition became noteworthy (Figure 7A) and the GIE of TC therapy was 32.0% at day 20, higher than DTX-Ms (15.3%) and CC therapy (17.5%). The consequence of tumor coefficients (Figure 7B) was in agreement with tumor volume alteration. Five days after all treatments were completed, the tumor weight coefficient of TC therapy decreased 58.0% compared with control, higher than DTX-Ms (47.9%) and CC therapy (27.2%).
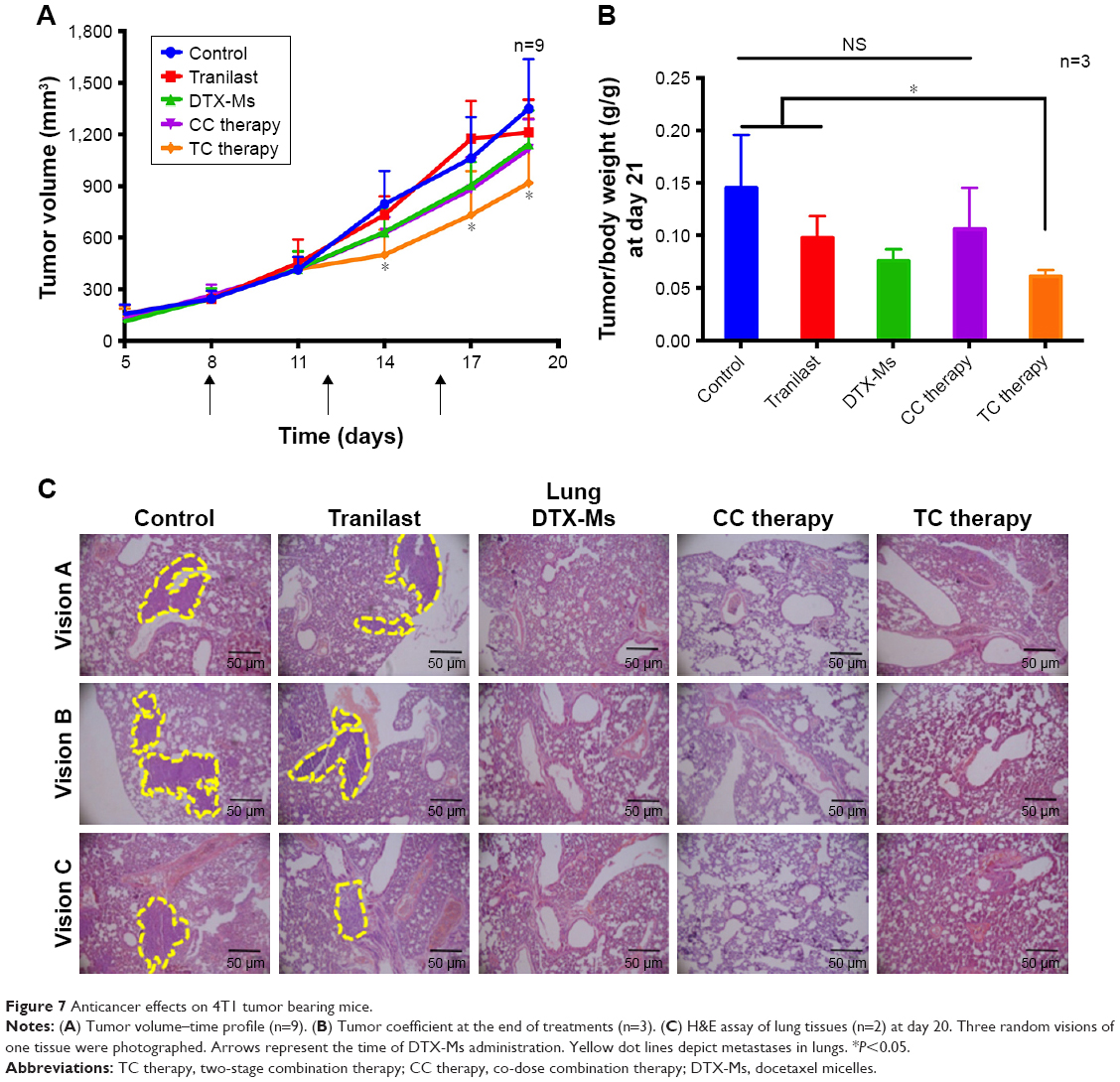 | Figure 7 Anticancer effects on 4T1 tumor bearing mice. |
Breast cancer cells may metastasize and relapse even after removal of the primary tumor.52 4T1 tumor model metastasizes to the lung or bone via the hematogenous route.53,54 After DTX-Ms, CC therapy, and TC therapy treatments, no obvious metastases were found (Figure 7C). However, there were legible metastasis nodules in control and tranilast regimens.
Combination therapies without severe toxic action in vivo
The body weight of every mouse was recorded. Except that the weight of control increased for tumor growth, the body weight in the other four treatments fluctuated within the range of 16–18 g (Figure 8A). Organ coefficients did not show significant difference among these groups (Figure 8B). Taken together, TC therapy did not stimulate severe toxic reaction.
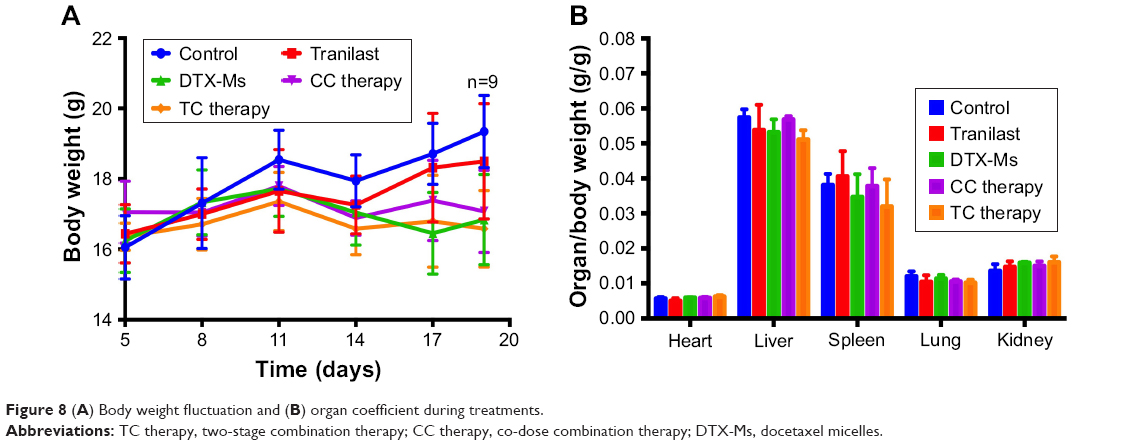 | Figure 8 (A) Body weight fluctuation and (B) organ coefficient during treatments. |
Discussion
Breast cancer is characterized by a rich tumor environment.55 CAFs surround tumor nests to form a tough barrier protecting cells from traditional chemotherapy.56,57 This research focuses on a promising scenario of combination therapy, consisting of CAFs inhibitor tranilast and antitumor agent DTX delivered in micelles. Tranilast aims at targeting CAFs to dismantle stromal barrier as well as intercommunication between CAFs and tumor cells, normalizes TME, and thus makes way for the following DTX-Ms.
DTX is delivered in 30-nm micelles for the reason that large sizes hamper nanoparticles from penetrating into the tumor core, and very small sizes (<8 nm) cannot avoid renal filtration.58,59 Besides, proper small sizes are necessary to verify microvessels normalization effect on drug delivery improvement.60 Neutral zeta potential is favored because nanoparticles with a zeta potential ranging from −20 to +10 mV distribute homogenously in the tumor for ECM presenting an electrostatic bandpass to suppress diffusion of positively and negatively charged particles.61
As shown in Figure 2, DTX-Ms presented low inhibition ability on 4T1/3T3 and d-CAFs, but the antiproliferation effect could be strengthened by the combination therapy. Besides, the combination therapy weakened the high migration property of 4T1 cells (Figure 3), which showed potential in decreasing tumor metastasis. Combination therapy stood out in antitumor potential in vitro especially when tumor cells and CAFs coexisted (Figure 4).
Various treatments decreased tumor IFP (Figure 5B); however, the mechanisms behind this were differed. Tumor sizes of control and tranilast groups were similar, but their IFPs were different. Tumor size is not the only reason affecting IFP; dense stroma (ECM deposition), microvascular density, and abnormal angiogenesis work in a comprehensive way.18,62,63 CAFs have been proved to promote ECM production to increase interstitial and mechanical pressure to elevate IFP.9,64,65 Disruption of active CAFs (Figure 5E and F) may account for IFP reduction via ECM reduction.
CAFs are thought to highly express angiogenic stimulators like VEGFs, TGF-β, platelet-derived growth factors etc.66–68 In tumors, imbalanced action of highly expressed angiogenic stimulators exceeds the action of angiogenic inhibitors. The imbalance results in functionally and structurally abnormal tumor microvessels that are unable to deliver drug efficiently.68 Studies propose that the application of antiangiogenic agents can rebuild the balance and normalize tumor vasculature, increasing the penetration of drugs throughout the tumor to enhance chemotherapy outcome.68 In our study, the downregulation of CAFs by tranilast potentially reduced angiogenic stimulators to rebuild the balance and restored normalized vasculature. Through TC therapy, FITC-lectin perfusion (Figure 5C and D) presented an integrated microvascular structure and enhanced blood perfusion compared with DTX-Ms. Thus, targeting at CAFs (Figure 5E and F) would help to decrease IFP and restore well-organized capillaries (Figure 5C and D).
Previous studies has proved that angiogenesis can be weakened by inhibiting CAF-secreted cytokines and microvessels in the tumor can be normalized to increase micelles penetration.69,70 On this basis, highly enhanced micelle retention was observed in TC therapy (Figure 6), which was thought to correspond to IFP reduction and microvessels normalization. The control and tranilast groups also showed high radiant efficiency for remarkable EPR effect in breast cancer as a result of high vascular density.71,72 However, the radiant efficiency in CC therapy was low. We tested the simultaneous administration of DTX-Ms and tranilast on EA.hy926 (Supplementary materials) and proved the remarkable toxicity of combined compounds on endothelial cells (Figure S6), which might destroy vessels function.73–75 The EPR effect in tumors is an extremely complex process with many factors and cannot be fully explained by this experiment.
Antitumor efficacy in the tumor model showed that TC therapy caused the strongest inhibition on tumor volume growth (Figure 7A) without noticeable lung metastasis (Figure 7C) and severe systemic toxicity (Figure 8). Antitumor effect of TC therapy lasted 5 days even after the treatments were completed (Figure 7B). The difference between tranilast and tranilast-assisted treatments indicated that tranilast alone was not enough in antitumor trials, but it could strengthen DTX-Ms’ antitumor effect. Previous researches demonstrate that the mechanism of tranilast involves TGF-β signaling and TGF-β-Smad2/3 signaling in TME stands out to mediate CAFs transition and properties acquirements.40,76,77 So, tranilast should be utilized as an adjuvant medicine together with an antitumor agent in prior administration to achieve better effects.
Taken together, the TC therapy offers new scope in antitumor treatment. Studies also suggest that the two-stage therapy deserves further analyses on appropriate dose, effective treatment window, therapeutic TI, as well as the molecular mechanisms on stroma and tumor.
Acknowledgment
We would like to acknowledge the assistance of the National Natural Science Foundation of China (no 81673365, no 81473156) for funding this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Crotti S, Piccoli M, Rizzolio F, Giordano A, Nitti D, Agostini M. Extracellular Matrix and Colorectal Cancer: How Surrounding Microenvironment Affects Cancer Cell Behavior? J Cell Physiol. 2017;232(5):967–975. | ||
Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015;368(1):7–13. | ||
Fuxe J, Karlsson MC. TGF-β-induced epithelial-mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol. 2012;22(5–6):455–461. | ||
McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov. 2013;12(3):217–228. | ||
Xouri G, Christian S. Origin and function of tumor stroma fibroblasts. Semin Cell Dev Bio. 2010;21(1):40–46. | ||
Mueller MM, Fusenig NE. Friends or foes – bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–849. | ||
Zhang H, Xie C, Yue J, et al. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol Carcinog. 2017;56(3):1150–1163. | ||
Son B, Lee S, Youn H, Kim E, Kim W, Youn B. The role of tumor microenvironment in therapeutic resistance. Oncotarget. 2017;8(3):3933–3945. | ||
Marusyk A, Tabassum DP, Janiszewska M, et al. Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical Outcomes. Cancer Res. 2016;76(22):6495–6506. | ||
Gaggioli C, Hooper S, Hidalgo-Carcedo C. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Bio. 2007;9(12):1392–1400. | ||
Sanz-Moreno V, Gaggioli C, Yeo M, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20(2):229–245. | ||
Pankova D, Chen Y, Terajima M, et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Cancer Res. 2016;76(22):6495–6506. | ||
Sakai S, Iwata C, Tanaka HY, et al. Increased fibrosis and impaired intratumoral accumulation of macromolecules in a murine model of pancreatic cancer co-administered with FGF-2. J Control Release. 2016;230:109–115. | ||
Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. | ||
Sriraman SK, Aryasomayajula B, Torchilin VP. Barriers to drug delivery in solid tumors. Tissue Barriers. 2014;2(3):e29528–e29565. | ||
Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. | ||
Mongiat M, Andreuzzi E, Tarticchio G, Paulitti A. Extracellular matrix: a hard player in angiogenesis. Int J Mol Sci. 2016;17(11):1822–1848. | ||
Mohammadi M, Chen P. Effect of microvascular distribution and its density on interstitial fluid pressure in solid tumors: A computational model. Microvasc Res. 2015;101:26–32. | ||
Provenzano PP, Cuevas C, Chang AE, Goel VK, von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. | ||
Goodman TT, Olive PL, Pun SH. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int J Nanomed. 2007;2(2):265–274. | ||
Ji T, Ding Y, Zhao Y, et al. Peptide assembly integration of fibroblast-targeting and cell-penetration features for enhanced antitumor drug delivery. Adv Mater. 2015;27(11):1865–1873. | ||
Azuma H, Banno K, Yoshimura T. Pharmacological properties of N-(3′,4′-dimethoxycinnamoyl) anthranilic acid (N-5’), a new anti-atopic agent. Br J Pharmacol. 1976;58(4):483–488. | ||
Isaji M, Miyata H, Ajisawa Y. Tranilast: A New Application in the Cardiovascular Field as an antiproliferative drug. Cardiovasc Drug Rev. 1998;16(3):288–299. | ||
Saito H, Fushida S, Harada S, et al. Importance of human peritoneal mesothelial cells in the progression, fibrosis, and control of gastric cancer: inhibition of growth and fibrosis by tranilast. Gastric Cancer. 2018;21(1):1–13. | ||
Ohshio Y, Hanaoka J, Kontani K, Teramoto K. Tranilast inhibits the function of cancer-associated fibroblasts responsible for the induction of immune suppressor cell types. Scand J Immunol. 2014;80(6):408–416. | ||
Chakrabarti R, Subramaniam V, Abdalla S, Jothy S, Prud’homme GJ. Tranilast inhibits the growth and metastasis of mammary carcinoma. Anticancer Drugs. 2009;20(5):334–345. | ||
Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99:186–196. | ||
Wang AT, Liang DS, Liu YJ, Qi XR. Roles of ligand and TPGS of micelles in regulating internalization, penetration and accumulation against sensitive or resistant tumor and therapy for multidrug resistant tumors. Biomaterials. 2015;53:160–172. | ||
Tong S-W, Xiang B, Dong D-W, Qi X-R. Enhanced antitumor efficacy and decreased toxicity by self-associated docetaxel in phospholipid-based micelles. Int J Pharm. 2012;434(1–2):413–419. | ||
Sun L, Liu Y-J, Yang Z-Z, Qi X-R. Tumor specific delivery with redox-triggered mesoporous silica nanoparticles inducing neovascularization suppression and vascular normalization. RSC Adv. 2015;5(68):55566–55578. | ||
Gao K, Sun J, Liu K, Liu X, He Z. Preparation and characterization of a submicron lipid emulsion of docetaxel: submicron lipid emulsion of docetaxel. Drug Dev Ind Pharm. 2008;34(11):1227–1237. | ||
Kuppens IE, Bosch TM, van Maanen MJ, et al. Oral bioavailability of docetaxel in combination with OC144-093 (ONT-093). Cancer Chemother Pharmacol. 2005;55(1):72–78. | ||
Ebrahim Attia AB, Ong ZY, Hedrick JL, et al. Mixed micelles self-assembled from block copolymers for drug delivery. Curr Opin Colloid Interface Sci. 2011;16(3):182–194. | ||
Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab. 2009;10(8):836–841. | ||
Mou Q, Ma Y, Zhu X, Yan D. A small molecule nanodrug consisting of amphiphilic targeting ligand-chemotherapy drug conjugate for targeted cancer therapy. J Control Release. 2016;230:34–44. | ||
Cortes JE, Pazdur R. Docetaxel. J Clin Oncol. 1995;13(10):2643–2655. | ||
Subramaniam V, Chakrabarti R, Prud’homme GJ, Jothy S. Tranilast inhibits cell proliferation and migration and promotes apoptosis in murine breast cancer. Anticancer Drugs. 2010;21(4):351–361. | ||
Chakrabarti R, Subramaniam V, Abdalla S, Jothy S, Prud’homme GJ. Tranilast inhibits the growth and metastasis of mammary carcinoma. Anticancer Drugs. 2009;20(5):334–345. | ||
Li Y, Jin M, Shao S, et al. Small-sized polymeric micelles incorporating docetaxel suppress distant metastases in the clinically-relevant 4T1 mouse breast cancer model. BMC Cancer. 2014;14:329. | ||
Subramaniam V, Chakrabarti R, Prud’homme GJ, Jothy S. Tranilast inhibits cell proliferation and migration and promotes apoptosis in murine breast cancer. Anticancer Drugs. 2010;21(4):351–361. | ||
Zhang J, Miao L, Guo S, et al. Synergistic anti-tumor effects of combined gemcitabine and cisplatin nanoparticles in a stroma-rich bladder carcinoma model. J Control Release. 2014;182(1):90–96. | ||
Kaushik S, Pickup MW, Weaver VM. From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016;35(4):655–667. | ||
Pulaski BA, Terman DS, Khan S, Muller E, Ostrand-Rosenberg S. Cooperativity of Staphylococcal aureus enterotoxin B superantigen, major histocompatibility complex class II, and CD80 for immunotherapy of advanced spontaneous metastases in a clinically relevant postoperative mouse breast cancer model. Cancer Res. 2000;60(10):2710–2715. | ||
Weaver VM, Petersen OW, Wang F, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137(1):231–245. | ||
Cukierman E. Cell migration analyses within fibroblast-derived 3-D matrices. Methods Mol Biol. 2005;294:79–93. | ||
Gao W, Xiang B, Meng TT, Liu F, Qi XR. Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials. 2013;34(16):4137–4149. | ||
Sckell A, Safabakhsh N, Dellian M, Jain RK. Primary tumor size-dependent inhibition of angiogenesis at a secondary site: an intravital microscopic study in mice. Cancer Res. 1998;58(24):5866–5869. | ||
Thurston G, McLean JW, Rizen M, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 1998;101(7):1401–1413. | ||
Debbage PL, Griebel J, Ried M, Gneiting T, Devries A, Hutzler P. Lectin intravital perfusion studies in tumor-bearing mice: micrometer-resolution, wide-area mapping of microvascular labeling, distinguishing efficiently and inefficiently perfused microregions in the tumor. J Histochem Cytochem. 1998;46(5):627–639. | ||
Dvorak HF. Tumor stroma, tumor blood vessels, and antiangiogenesis therapy. Cancer J. 2015;21(4):237–243. | ||
Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res. 1989;37(1):77–104. | ||
Pulaski BA, Clements VK, Pipeling MR, Ostrand-Rosenberg S. Immunotherapy with vaccines combining MHC class II/CD80+ tumor cells with interleukin-12 reduces established metastatic disease and stimulates immune effectors and monokine induced by interferon γ. Cancer Immunol Immunother. 2000;49(1):34–45. | ||
Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res. 2000;2(5):331–334. | ||
Zhang C, Winnard PT Jr, Dasari S, et al. Label-free Raman spectroscopy provides early determination and precise localization of breast cancer-colonized bone alterations. Chem Sci. 2018;9(3):743–753. | ||
Majidinia M, Yousefi B. Breast tumor stroma: A driving force in the development of resistance to therapies. Chem Biol Drug Des. 2017;89(3):309–318. | ||
Ryan MC, Orr DJA, Horgan K. Fibroblast stimulation of breast cancer cell growth in a serum-free system. Br J Cancer. 1993;67(6):1268–1273. | ||
Busch S, Acar A, Magnusson Y, Gregersson P, Rydén L, Landberg G. TGF-beta receptor type-2 expression in cancer-associated fibroblasts regulates breast cancer cell growth and survival and is a prognostic marker in pre-menopausal breast cancer. Oncogene. 2015;34(1):27–38. | ||
Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2(1):14–33. | ||
Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. | ||
Chauhan VP, Stylianopoulos T, Martin JD, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7(6):383–388. | ||
Lieleg O, Baumgärtel RM, Bausch AR. Selective filtering of particles by the extracellular matrix: an electrostatic bandpass. Biophys J. 2009;97(6):1569–1577. | ||
Jain RK, Tong RT, Munn LL. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res. 2007;67(6):2729–2735. | ||
Swartz MA, Fleury ME. Interstitial flow and its effects in soft tissues. Annu Rev Biomed Eng. 2007;9(1):229–256. | ||
Paraiso KH, Smalley KS. Fibroblast-mediated drug resistance in cancer. Biochem Pharmacol. 2013;85(8):1033–1041. | ||
Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure and stromal resistance in pancreas cancer. Br J Cancer. 2013;108(1):1–8. | ||
Liu J, Liao S, Diop-Frimpong B, et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A. 2012;109(41):16618–16623. | ||
Hanahan D, Folkman J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell. 1996;86(3):353–364. | ||
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. | ||
Cavaco A, Rezaei M, Niland S, Eble JA. Collateral Damage Intended-Cancer-Associated Fibroblasts and Vasculature Are Potential Targets in Cancer Therapy. Int J Mol Sci. 2017;18(11):2355–2398. | ||
Johansson-Percival A, He B, Li ZJ, et al. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat Immunol. 2017;18(11):1207–1217. | ||
Taurin S, Nehoff H, van Aswegen T, Greish K. Tumor vasculature, EPR effect, and anticancer nanomedicine: connecting the dots. In: Bae, Han Y, Mrsny, Randall J, Park Kinam, editors. Cancer Targeted Drug Delivery. New York: Springer; 2013:207–239. | ||
Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6. | ||
Xie F, Cai W, Liu Y, et al. Vaccarin attenuates the human EA.hy926 endothelial cell oxidative stress injury through inhibition of Notch signaling. Int J Mol Med. 2015;35(1):135–142. | ||
Li S, Dang YY, Oi Lam Che G, et al. VEGFR tyrosine kinase inhibitor II (VRI) induced vascular insufficiency in zebrafish as a model for studying vascular toxicity and vascular preservation. Toxicol Appl Pharmacol. 2014;280(3):408–420. | ||
Durrant D, Corwin F, Simoni D, et al. cis-3,4′,5-Trimethoxy-3′-aminostilbene disrupts tumor vascular perfusion without damaging normal organ perfusion. Cancer Chemother Pharmacol. 2009;63(2):191–200. | ||
Suzawa H, Kikuchi S, Arai N, Koda A. The mechanism involved in the inhibitory action of tranilast on collagen biosynthesis of keloid fibroblasts. Jpn J Pharmacol. 1992;60(2):91–96. | ||
Kojima Y, Acar A, Eaton EN, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–20014. |
Supplementary materials
Supplementary experimental procedures
Characterization and certification of d-CAFs detached from 4T1 tumors in mice
CAFs were obtained from 4T1 tumor bearing mice named d-CAFs purified for five passages. To identify d-CAFs, the fifth-passage cells were cultured in glass bottom culture dishes for 24 hours, fixed with 4% paraformaldehyde, and permeabilized by 0.2% Triton-X 100. Then, α-SMA and PDGFR-β primary antibodies (1:500 dilution) were added for overnight incubation at 4°C, followed by secondary antibody Alexa Fluor® 647 dye (1:500 dilution) incubation. The cell nuclei were stained with Hoechst 33258 (10 μg/mL) for 30 minutes in room temperature. Normal fibroblast 3T3 cells and tumor 4T1 cells were stained at the same time as positive and negative controls, respectively. All cells were observed using a CLSM (TCS SP8 MP FLIM; Leica Microsystems, Wetzlar, Germany).
Construction and certification of mixed tumor spheres with a series of ratios
Mixed tumor spheres were constructed via the pendant drop method. Mixed cell suspension (totally containing 1,000 cells) of 4T1 and 3T3 cells were collected in ratios of 3/1, 1/1, 1/3, and 1/5 to form cell aggregations on plate lid. After being transferred into wells and incubation for another 24 hours, all the formed spheres were photographed by an inverted fluorescence microscope (Olympus IX73; Olympus Corporation). For observing cell distribution, 3T3 cells were pre-marked with DiO before cell mixing. After spheres formation, they were observed under a confocal laser scanning microscope (TCS SP8 MP FLIM).
Evaluation of combined therapy from cytotoxicity tests
Docetaxel micelles (DTX-Ms) act on tubulin while tranilast works on TGF-β signaling.2 For distinct mechanisms and targets, the combination index proposed by Jin3 is used to discuss combination efficacy. After cytotoxicity tests on 4T1, 3T3, 4T1/3T3, and d-CAF, q-values were calculated to evaluate the combination index according to the following formula:
|
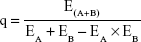
where EA+B is the efficacy (indicates inhibition rate here) of the combined therapy, and EA and EB are the efficacies (indicates inhibition rate here) of DTX-Ms and tranilast individually. The q-value at each concentration implies the combination index, of which q<0.85 indicates antagonism, 0.85≤q<1.15 means addition, and q≥1.15 refers to synergism.
Cytotoxicity of DTX-Ms and tranilast combination on endothelial cells
Co-dose combination therapy of DTX-Ms and tranilast may cause severe cytotoxicity on endothelial cells; EA.hy926 was used as endothelial cells to conduct the cytotoxicity experiment. EA.hy926 cells were collected and seeded (8,000 cells per well) on 96-well plates; after overnight incubation, DTX-Ms with or without tranilast was added into the wells for 48 hours. Then, we used MTT (solution: 5 mg/mL) formazan method as 4T1 and other cells to measure cell vitality.
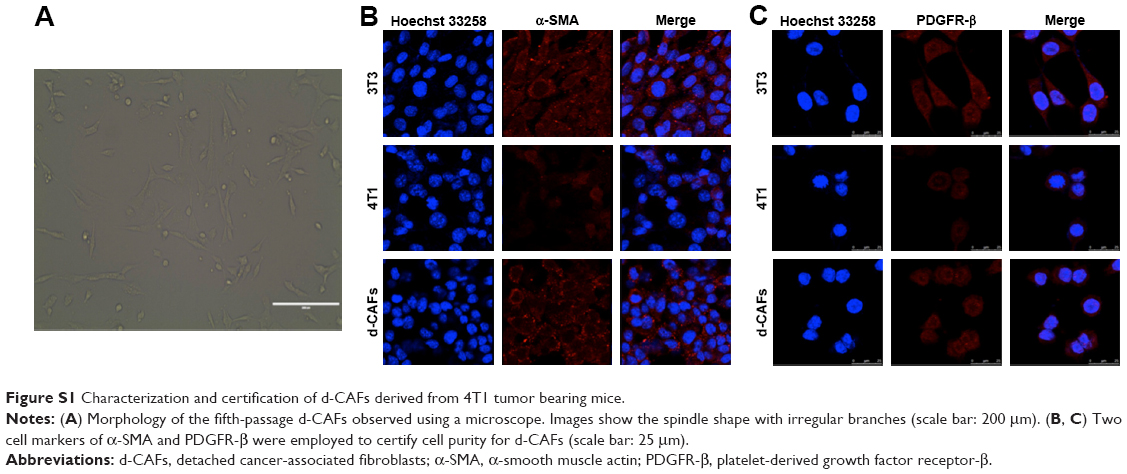 | Figure S1 Characterization and certification of d-CAFs derived from 4T1 tumor bearing mice. |
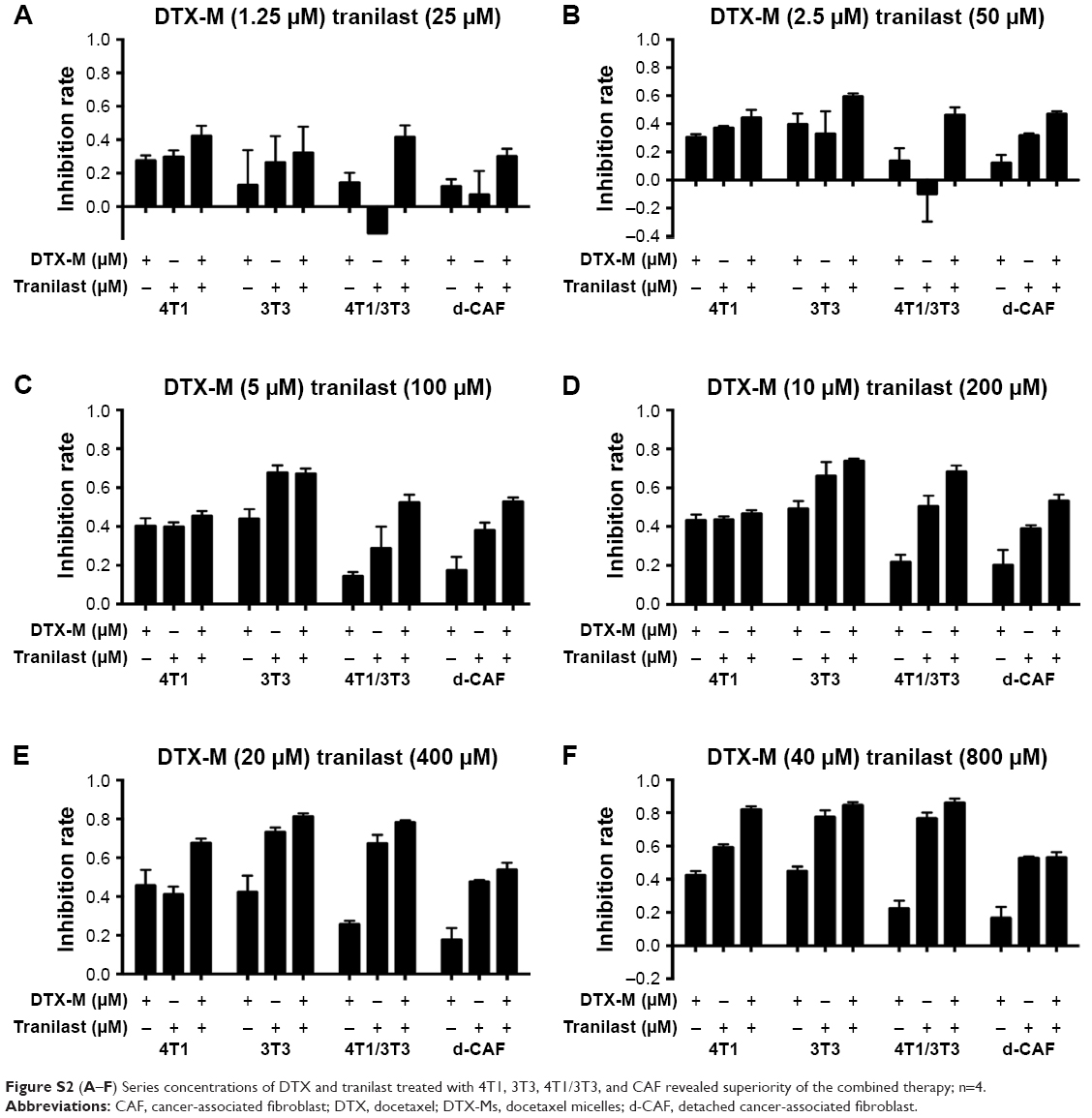 | Figure S2 (A–F) Series concentrations of DTX and tranilast treated with 4T1, 3T3, 4T1/3T3, and CAF revealed superiority of the combined therapy; n=4. |
 | Table S1 Combined evaluation on cells |
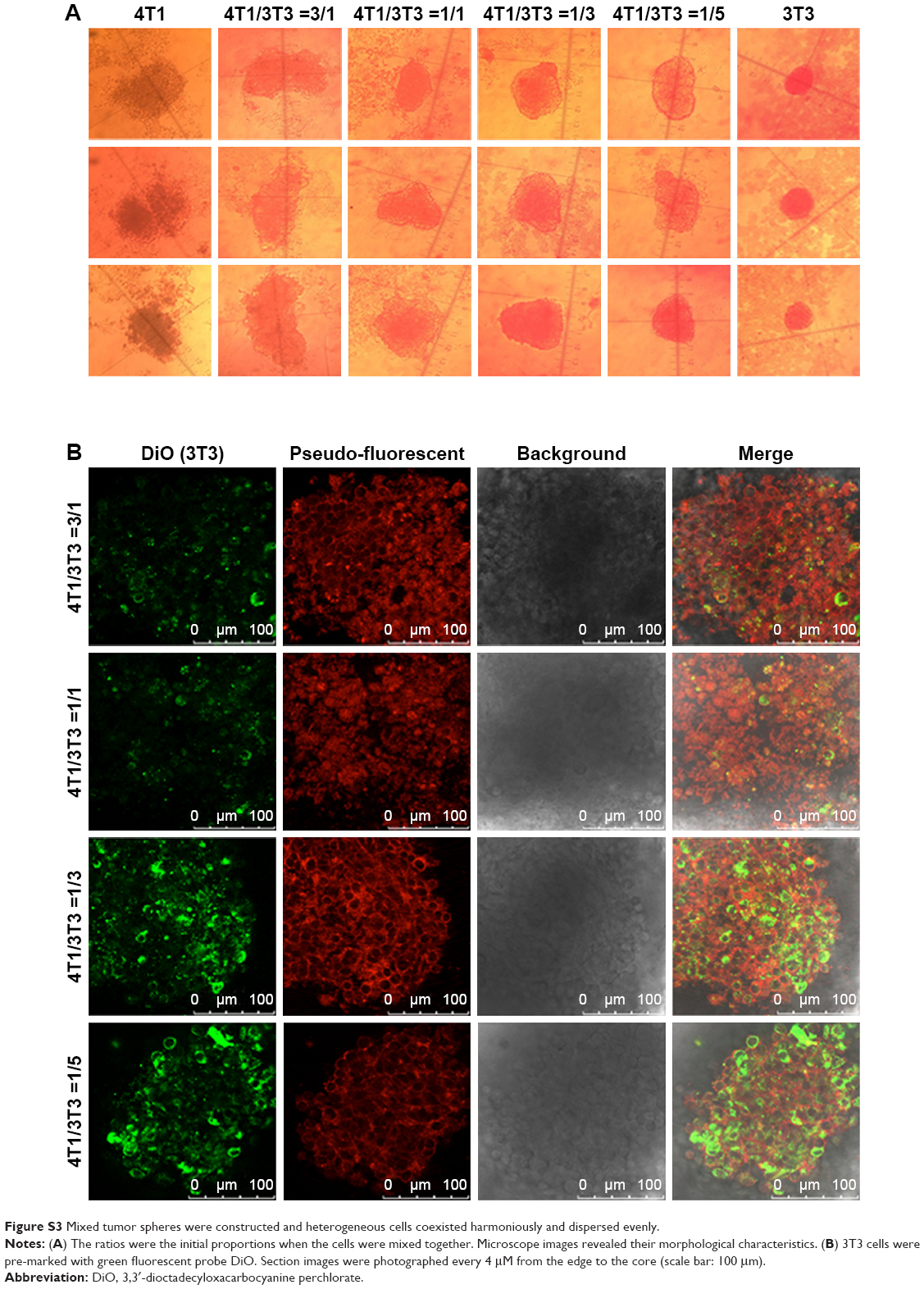 | Figure S3 Mixed tumor spheres were constructed and heterogeneous cells coexisted harmoniously and dispersed evenly. |
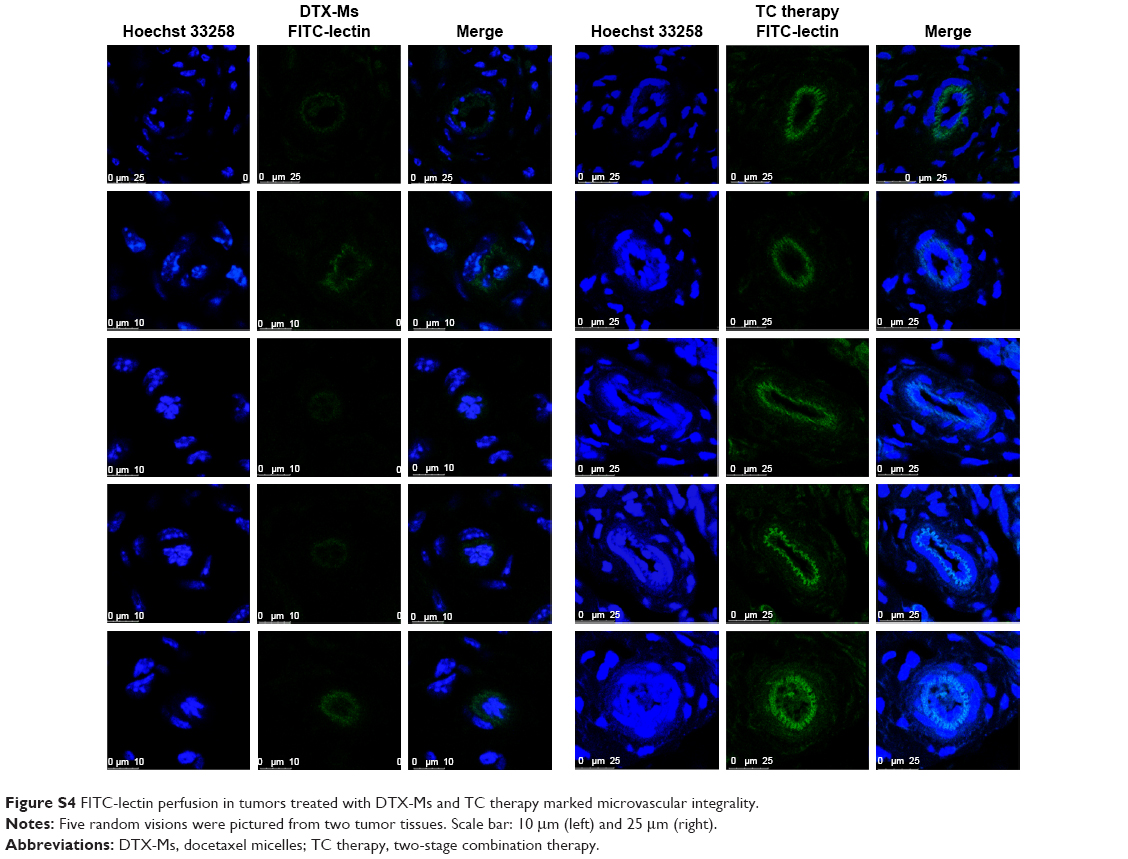 | Figure S4 FITC-lectin perfusion in tumors treated with DTX-Ms and TC therapy marked microvascular integrality. |
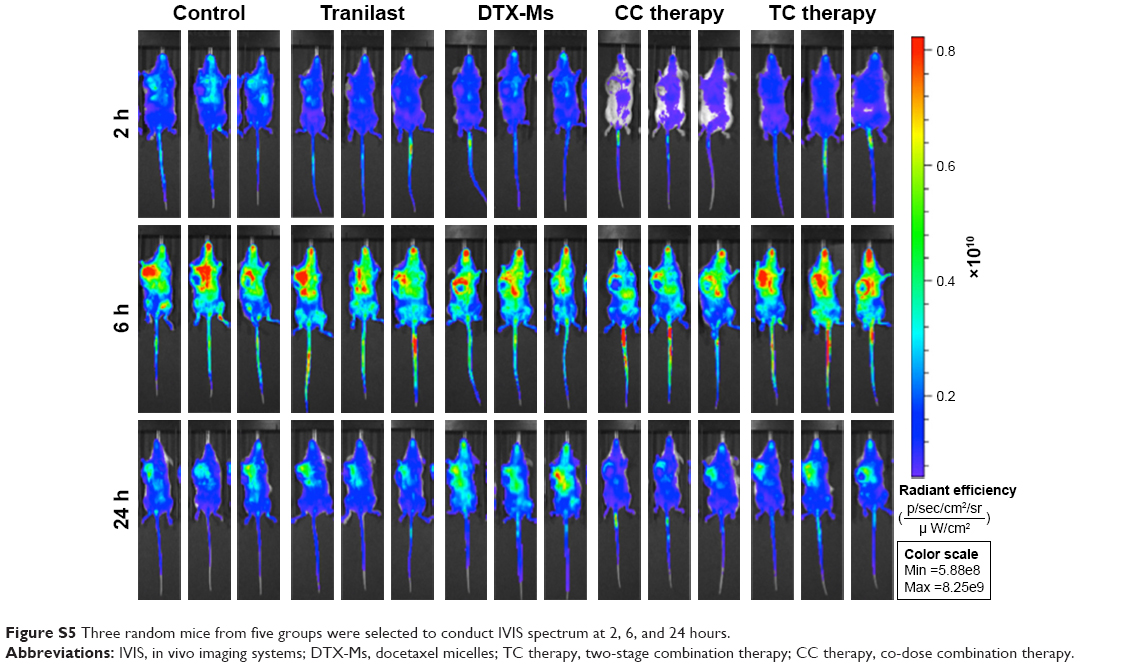 | Figure S5 Three random mice from five groups were selected to conduct IVIS spectrum at 2, 6, and 24 hours. |
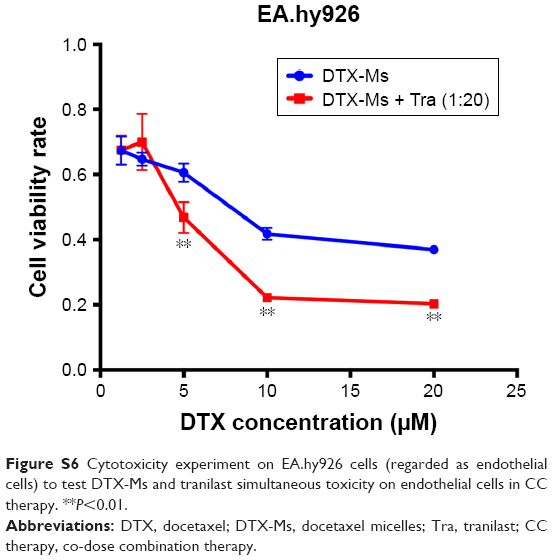 | Figure S6 Cytotoxicity experiment on EA.hy926 cells (regarded as endothelial cells) to test DTX-Ms and tranilast simultaneous toxicity on endothelial cells in CC therapy. **P<0.01. |
References
Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliver Rev. 2016;99(Pt B):186–196. | ||
Rogosnitzky M, Danks R, Kardash E. Therapeutic potential of tranilast, an anti-allergy drug, in proliferative disorders. Anticancer Res. 2012;32(7):2471–2478. | ||
Jin ZJ. Addition in drug combination. Acta Pharmacol Sin. 1980;1(2):70–76. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.