Back to Journals » ImmunoTargets and Therapy » Volume 10
Priming Leukemia with 5-Azacytidine Enhances CAR T Cell Therapy
Authors Xu N, Tse B, Yang L, Tang TCY ![]() , Haber M
, Haber M ![]() , Micklethwaite K, Dolnikov A
, Micklethwaite K, Dolnikov A
Received 5 January 2021
Accepted for publication 24 February 2021
Published 28 April 2021 Volume 2021:10 Pages 123—140
DOI https://doi.org/10.2147/ITT.S296161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Ning Xu,1,2 Benjamin Tse,1,2 Lu Yang,1,2 Tiffany CY Tang,1,2 Michelle Haber,1,2 Kenneth Micklethwaite,3– 6 Alla Dolnikov1,2
1Children’s Cancer Institute, University of New South Wales, Sydney, NSW, Australia; 2School of Women’s and Children’s Health, University of New South Wales, Sydney, NSW, Australia; 3Blood Transplant and Cell Therapies Program, Department of Hematology, Westmead Hospital, Sydney, NSW, Australia; 4Sydney Cellular Therapies Laboratory, NSW Health Pathology, Sydney, NSW, Australia; 5Westmead Institute for Medical Research, Sydney, NSW, Australia; 6Sydney Medical School, The University of Sydney, Sydney, NSW, Australia
Correspondence: Alla Dolnikov
Children’s Cancer Institute, University of New South Wales, PO Box 81, Randwick, NSW, 2031, Australia
Tel +61 449 904 174
Email [email protected]
Purpose: Despite the success of chimeric antigen receptor (CAR) T cells in clinical studies, a significant proportion of responding patients eventually relapsed, with the latter correlating with low CAR T cell expansion and persistence.
Methods and Results: Using patient-derived xenograft (PDX) mouse models of CD19+ B cell acute lymphoblastic leukemia (B-ALL), we show that priming leukemia-bearing mice with 5-azacytidine (AZA) enhances CAR T cell therapy. AZA given 1 day prior to CAR T cell infusion delayed leukemia growth and promoted CAR T cell expansion and effector function. Priming leukemia cells with AZA increased CAR T cell/target cell conjugation and target cell killing, promoted CAR T cell divisions and expanded IFNγ+ effector T cells in co-cultures with CD19+ leukemia Nalm-6 and Raji cells. Transcriptome analysis revealed activation of diverse immune pathways in leukemia cells isolated from mice treated with AZA. We propose that epigenetic priming with AZA induces transcriptional changes that sensitize tumor cells to subsequent CAR T cell treatment. Among the candidate genes up-regulated by AZA is TNFSF4 which encodes OX40L, one of the strongest T cell co-stimulatory ligands. OX40L binds OX40, the TNF receptor superfamily member highly specific for activated T cells. TNFSF4 is heterogeneously expressed in a panel of pediatric PDXs, and high TNFSF4 expression correlated with increased CAR T cell numbers identified in co-cultures with individual PDXs. High OX40L expression in Nalm-6 cells increased their susceptibility to CAR T cell killing while OX40L blockade reduced leukemia cell killing.
Conclusion: We propose that treatment with AZA activates OX40L/OX40 co-stimulatory signaling in CAR T cells. Our data suggest that the clinical use of AZA before CAR T cells could be considered.
Keywords: CAR T cells, leukemia, AZA, patient-derived xenografts, gene expression
Introduction
Adoptive therapy with patient T cells modified to express chimeric antigen receptors (CAR) has demonstrated great success in clinical trials.1–3 CAR redirects T cells to recognize and eliminate tumor cells.4 Three CD19-targeting CAR T cell products have been approved for the treatment of relapsed or refractory large-cell lymphomas and B cell acute lymphoblastic leukemia (B-ALL) in children and young adults (Gilead’s axicabtagene ciloleucel and Novartis’s tisagenlecleucel) and mantle cell lymphoma (MCL) in adults (Gilead’s brexucabtagene autoleucel). High response rates ranging from 70% to 94% have been achieved using both CAR T cell products. However, a significant proportion of responding patients eventually relapsed with either CD19– or CD19+ disease.5 Short duration of remission in patients treated with CAR T cells correlated with limited CAR T cell expansion and lack of persistence.6 The in vivo expansion and persistence of adoptively transferred CAR T cells are potentially modulated by premature exhaustion of T cells in patients heavily pre-treated with chemotherapy, T cell exhaustion induced by extensive CAR T cell expansion during manufacture, and the immune-suppressive tumor microenvironment (TME).7–9
DNA methyltransferase (DNMT) inhibitors such as 5-azacytidine (azacitidine, AZA) and 5-aza-2ʹ-deoxycytidine (decitabine, DEC) have been shown to promote immune responses in multiple tumor types.10 DNMT inhibitors up-regulate the expression of tumor-associated antigens (TAA), co-stimulatory ligands and human leukocyte antigen (HLA) molecules important for tumor antigen presentation and activate type I interferon (IFN) signaling in some cancers through the viral defense pathway.11–13 The combination of DNMT inhibitors with checkpoint inhibitors and adoptive T cell therapies has shown efficacy in pre-clinical models of cancer and in the clinic.11,13–18
Here, we show that AZA given prior to infusing CD19-targeting CAR T cells delayed leukemia growth and promoted CAR T cell expansion, increasing CAR T cell effector function in patient-derived xenograft (PDX) mouse models of chemoresistant pediatric CD19+ B-ALL and in co-cultures with CD19+ Nalm-6 and Raji cells. Mechanistically, AZA up-regulated multiple immune regulatory pathways in leukemia cells, particularly TNFSF4 that encodes the T cell co-stimulatory ligand OX40L.19,20 We have shown that high expression of TNFSF4 in PDXs is associated with higher CAR T cell numbers identified in CAR T cell/PDX co-cultures. High OX40L expression was associated with increased CAR T cell killing, and OX40L blockade reduced leukemia cell killing. We propose that priming leukemia with AZA activates OX40L/OX40 co-stimulatory signaling in CAR T cells, promoting their function.
Materials and Methods
All experiments with genetically modified organisms were conducted and complied with institutional biosafety committee guidelines according to Gene Technology Act 2000 (Commonwealth). Ethics approval was obtained from the Sydney Children’s Hospital Network Committee for experiments using human blood to manufacture CAR T cells and B-ALL PDXs. All animal studies were approved by the Animal Care and Ethics Committee of the University of New South Wales and were performed in strict accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes.
Generation of CAR T Cells
Peripheral blood (PB) from healthy donors was purchased from Australian Red Cross. CAR T cells were generated using peripheral blood mononuclear cells (PBMNC) infected with the SFG.CAR.CD19-28ζ retrovirus (courtesy of Prof. Gianpietro Dotti).21 Briefly, viral supernatant was produced by transfecting Phoenix-AMPHO (ATCC® CRL-3213™, Manassas, VA) with SFG.CAR.CD19-28ζ (5 μg). PBMNCs were stimulated with anti-CD3/anti-CD28 beads and IL-2 (20 U/mL) for 2 days and then incubated for 2 days on virus-coated RetroNectin® (Takara Bio USA, Inc.) covered plates in the presence of IL-2 (20 U/mL). The infected T cells were further expanded for another 5–7 days with human IL-2 (20 ng/mL), IL-7 (10 ng/mL) and IL-15 (10 ng/mL) (Miltenyi Biotec Australia) in T cell medium (TCM), comprised of AIM V medium (Life Technologies, New York, NY) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin/L-glutamine (PSG). CAR expression in the transduced T cells was determined by staining with the polyclonal F(ab’)2 fragment goat anti-human IgG Fc (Jackson ImmunoResearch Laboratories, West Grove, PA). CAR T cell phenotype was determined by flow cytometry using anti-CAR APC, anti-hCD3 FITC, anti-hCD4 PerCP-Cy5.5, anti-hCD8 APC-Cy7, anti-hCD62L PE, and anti-hCD45RO APC (BD Biosciences, San Diego, CA, USA). Data (105 events) were acquired using the BD FACSCanto™ II and analyzed with BD FACSDiva™ software (Becton Dickinson, San Jose, CA). Viable cells were gated based on forward versus side scatter (FSC/SSC).
Cell Lines, PDXs and Culture Media
Human CD19+ B-ALL Nalm-6 cells and Burkitt lymphoma Raji cells were purchased from ATCC® (Manassas, VA) and PDXs were provided by Prof. Richard Lock (Children’s Cancer Institute Australia).22 Leukemia cells were routinely cultured in complete RPMI-1640 (Life Technologies, New York, NY), 10% FCS, HEPES (25 mmol/L), 2-mercaptoethanol (50 nmol/L), and L-glutamine (2 mmol/L). AZA (Sigma-Aldrich, #A2385) was used at doses indicated in figure legends. 5 x 105 target cells per well were co-cultured with CAR T cells at ratios indicated in figure legends. All assays had controls with isolated CAR T cells and target cells. Human IL-2 was added at concentrations of 20 ng/mL. Leukemia cells were co-cultured with CAR T cells (24 hr) at different effector to target ratios (E:T) indicated in figure legends. Each condition was performed in triplicate co-cultures unless indicated otherwise. At the end of the culture, bulk co-cultures were harvested and CAR T cell and target cell numbers were quantified using CountBright™ counting beads (Becton Dickinson, San Jose, CA). 104 events per condition were acquired using the BD FACSCanto™ II and analyzed with BD FACSDiva™ software. CD3+ T cells and CD10+ leukemia cells were analyzed in viable cell gate based on FSC/SSC. CD10 was used to identify leukemia cells due to significant reduction in CD19 expression in leukemia cells following co-culture with CAR T cells.23 Heterologous CAR T and target cell conjugates were identified as CD3+ CD10+ events, and unconjugated effector and target cells were identified as single positive CD3+ and CD10+ events, respectively. Total target cell numbers and the numbers of 7-aminoactinomycin D (7-AAD)+ apoptotic target cells were measured in CD10+ CD3– gate at the end of co-culture.
Cytolytic function of CAR T cells was assessed using degranulation assays.23 Briefly, 105 target cells and CAR T cells were plated in TCM (200 mL) with saturating concentrations of phycoerythrin (PE)-conjugated anti-human CD107a or IgG isotype control and the protein transport inhibitor monensin. Flow cytometry was performed to determine CD107 expression in gated CD3+ T cells.
Carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution assays were performed to monitor CAR T cell divisions. CAR T cells were labeled with CFSE (Invitrogen, Victoria, Australia) and co-cultured with Nalm-6 cells for 48 hrs. CFSE analysis was performed in bulk cultures in CD3+ gate using flow cytometry.23 Gating for divided and undivided CAR T cells was established using CFSE-stained CAR T cells at day 0 (undivided cells). Analysis was performed in duplicate. Two independent experiments were performed.
FSChigh cell numbers, expression of PD-1, CD62L and CD45RO were analyzed in the CD3+ T cell gate to determine T cell activation, exhaustion and differentiation status, respectively. Cells were washed with PBS, incubated with permeabilization solution (10 min, RT) and then with anti-INFγ or anti-CD107a antibodies for flow cytometry analysis in the CD3+ cell gate. All reagents were purchased from Becton Dickinson, San Jose, CA. For each condition, measurements were done in duplicate or triplicate in two independent experiments.
OX40L and OX40 expression in leukemia cells and CAR T cells, respectively, were analyzed using anti-OX40L (CD252) PE (clone 11C3.1, BioLegend, #326,307) and anti-OX40 PerCP (clone ACT35, Becton Dickinson). Anti-OX40L neutralizing antibody (R&D Systems, MAB10541-25) was used for functional blockade of OX40L.
CD19+ chemoresistant pediatric ALL cells derived from individual patient PDXs were co-cultured with CAR T cells in RPMI at E:T 1:1 (48 hr) as per published protocols.22,24 Association between TNFSF4 expression in PDXs and CAR T cell survival was performed using univariate regression analysis with the least squares method in GraphPad Prism 7. R2 was estimated for T cell parameter combinations as the ratio of regression sum of squares to total sum of squares.
Animal Modeling
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred in the animal facility at Children’s Cancer Institute. Eight to ten-week-old mice were infused intravenously (IV) with chemoresistant pediatric CD19+ B-ALL cells (ALL-2, 3 x 106 cells/mouse).22–24 Mice were randomized and allocated to groups. Numbers of mice per group are indicated in figure legends. Single doses of AZA (2.5 mg/kg) were given IV at 1 or 4 weeks post ALL-2 infusion in Experiments 1 and 2, respectively. CAR T or untransduced T cells (5 x 106 bulk T cells/mouse) were injected intraperitoneally (IP) 24 hrs post-AZA injection. Leukemia cell engraftment was monitored by flow cytometry. CD19+ leukemia cell engraftment in PB, bone marrow (BM) and spleen (SPL) was analyzed in the mouse (m) CD45– human (h) CD45+ gate. Anti-mCD45 PerCP and PerCP-Cy5.5 (clone 30-F11), anti-hCD45 APC-H7 and APC-Cy7 (clone 2D1) and anti-hCD19 PE and FITC (clone HIB19) were used, respectively, in these experiments. Anti-CD3 FITC and PE (clone UCHT1) and anti-CAR-APC were used to determine CAR T cell engraftment. Expression of CD62L, CD45RO and PD-1 was analyzed in the CD3+ T cell gate to determine T cell differentiation and exhaustion status, respectively. Anti-PD-L1 PE (clone MH1) and PD-L2 APC (clone MIH18) were used to determine PD-L1 and PD-L2 expression in CD19+ leukemia cells. Phenotypic CAR T cell analysis was performed using anti-CD3, CD4, CD8, CD62L PE (clone DREG-56) and CD45RO PerCP-Cy5.5 (clone UCHL1) prior to infusion. All antibodies were purchased from Becton Dickinson, San Jose, CA.
To discriminate the effect of AZA on alloreactivity of CAR T cells from its effect on antigen-specific activation, CD19– CD10+ early T cell precursor leukemia (ETP-1) cells were used as target cells.22 CD10+ leukemia cell engraftment was analyzed in the mCD45– hCD45+ gate. Mice were monitored daily for signs of graft versus host disease (GVHD): weight loss, hair loss, altered posture, or reduced mobility.25 Mice that showed clinical signs of GVHD were sacrificed and their blood was analyzed for the presence of human leukemia and T cells.
Gene Expression Analysis
NSG mice were injected with ALL-2 cells (5 x 106 cells/mouse). At week 8, when hCD45+ cell engraftment in PB reached 50–65%, mice were either injected with AZA (n = 2) or PBS (n = 2). It is relevant that 10 mg/kg rather than 2.5 mg/kg AZA dose was used in mice with high leukemia burden since 2.5 mg/kg AZA had minimal effect in the setting of advanced disease. Total RNA was isolated from splenocytes using the RNeasy Mini-Kit (Qiagen, Melbourne, Australia) 24 hrs after AZA or PBS injection. Biotinylated complementary RNA was prepared using the Illumina TotalPrep RNA Amplification Kit Q10 (Ambion, TX, USA). Complementary RNA was hybridized to Sentrix® HumanRef-8 BeadChips (Illumina, version 2.0) containing 24,526 human genes. All procedures for hybridization, signal detection, and analysis were performed according to BeadStation 500X system protocols. Raw data were normalized to the background with detection P ≤ 0.01 and fold-change ratio ≥1-fold. Annotation clustering was applied to the significant genes using standard correlation analysis with DAVID software (DAVID Bioinformatics Resources, National Institute of Allergy and Infectious Diseases, National Institute of Health). Genes with >1 log fold-change were filtered for further analysis. Gene set enrichment analysis (GSEA) of the differentially expressed genes was performed as described previously and using standard parameters.
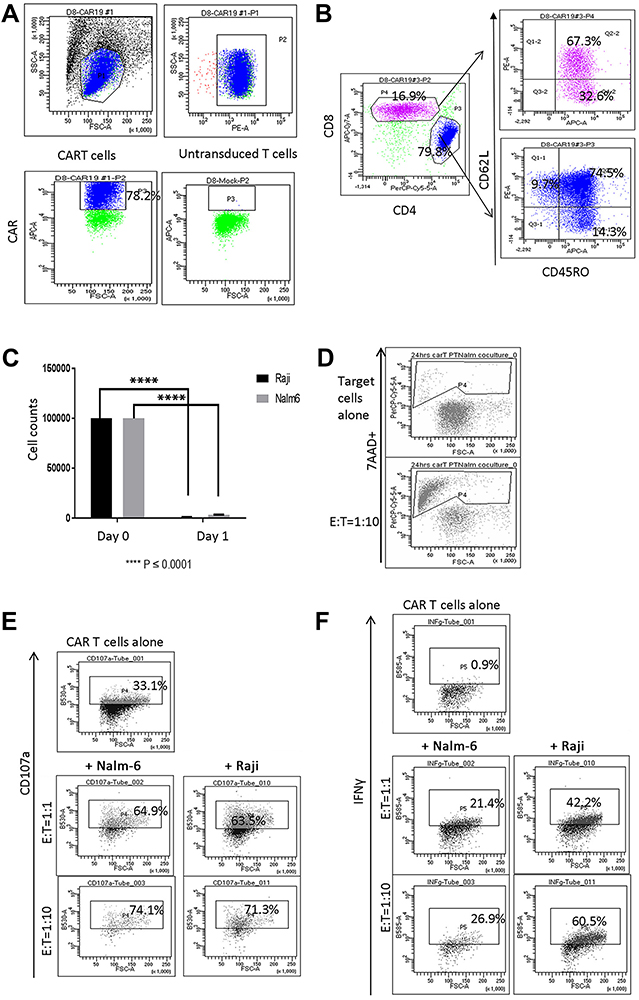 |
Figure 1 Characterization of CAR T cells following ex vivo expansion. (A) Live lymphocytes were gated based on SSC/FSC fluorescence (left top panel). CAR expression (P3 gate, left bottom panel) was analyzed in the CD3+ P2 gate (right top panel). CAR expression in untransduced T cells is shown in right bottom panel in P3 gate. (B) CAR T cells were composed of CD4+ and CD8+ cells. Both CD4+ and CD8+ CAR T cells were represented by CD62L+ CD45RO+ Tcm, CD62L– CD45RO+ Tem and CD62L+ CD45RO– naïve T (Tn) cells. (C) Reduced numbers of CD19+ Raji and Nalm-6 cells were observed in 24-hour co-cultures with CAR T cells (E:T = 1:1, ****P < 0.0001). (D) Co-culture with Nalm-6 cells at 1:10 E:T ratio increased the proportion of 7AAD+ apoptotic target cells identified in CD10+ gates. (E) CAR T cells co-cultured with Nalm-6 and Raji cells exhibited increased degranulation, measured using anti-CD107a in CD3+ gates. (F) IFNγ expression in CAR T cells co-cultured with target cells. IFNγ expression was analyzed in the CD3+ gate. Flow cytometry analysis of one representative experiment is shown in (D–F). |
Differential expression analysis of TNFSF4 in the panel of 7 chemoresistant pediatric B-ALL PDXs was performed using gene expression data from www.ncbi.nlm.nih.gov/geo (accession no. GSE52991). Microarray gene expression data were imported from GEO using R Studio’s xlsx package.
Statistical Analysis
ANOVA one-way analysis was performed to test for systematic within-subjects differences. GraphPad Prism 7 (GraphPad Software, La Jolla, CA) was used for statistical analysis for both in vitro and in vivo experiments. The number of mice per condition is indicated in figure legends corresponding to each experiment. Data was presented as mean ± standard deviation (SD). The significance level P < 0.05 was used in all experiments.
Results
Priming with AZA Delays Leukemia Growth and Promotes CAR T Cell Expansion in PDX Mouse Models of B-ALL
Retroviral gene transfer was used to manufacture CAR T cells with high levels of CAR (78%). These were enriched with CD4+ T cells, and predominantly CD62L+ CD45RO+ central memory (Tcm) and CD62L– CD45RO+ effector memory (Tem) cells (Figure 1A and B). Reduced CD19+ target Nalm-6 and Raji cell numbers and increased proportion of apoptotic 7AAD+ target cells were observed in 24 hr co-cultures with CAR T cells (Figure 1C and D). Additionally, co-culture with target cells promoted CAR T cell degranulation and increased IFNγ expression (Figure 1E and F).
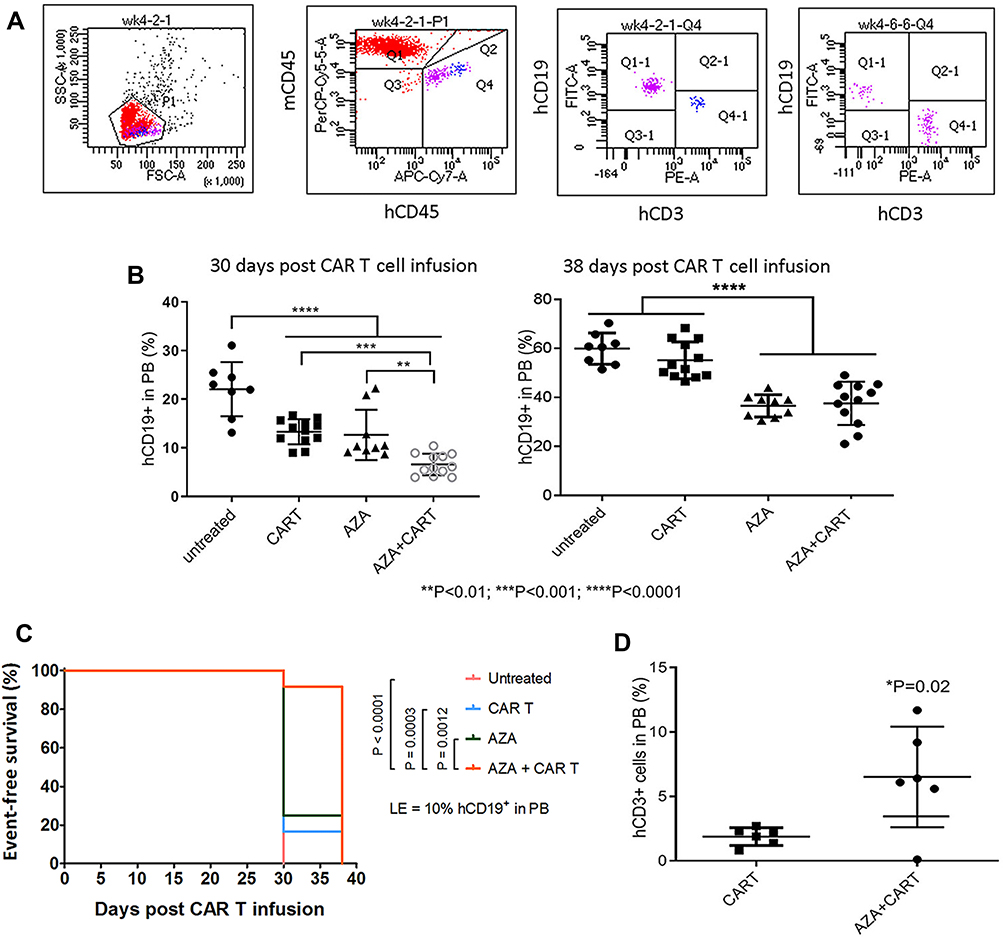 |
Figure 2 Host priming with AZA reduced leukemia cell growth and promoted CAR T cell expansion in low leukemia burden model. Mice were transplanted with CD19+ ALL-2 cells and randomized in 4 groups: untreated (n = 8), mice treated with CAR T cells alone on day 8 (CAR T, n = 12), mice treated with AZA (2.5 mg/kg) on day 7 (AZA, n = 9), mice treated with AZA on day 7 and then infused with CAR T cells on day 8 (AZA + CAR T, n = 12). (A) Flow cytometry analysis of CD19+ leukemia and CD3+ CAR T cells from PB of mice. Live cells were gated based on FSC/SSC fluorescence (P1 gate, left panel). Mouse CD45+ (mCD45+) and human CD45+ (hCD45+) cells were analyzed in the P1 gate (second left panel). Human (hCD19+) and human CD3+ (hCD3+) cells were identified in the hCD45+ Q4 gate (second and first right panels). One representative dot plot is shown. (B) hCD19+ leukemia cell engraftment in PB measured at days 30 and 38 post CAR T cell infusion (left and right panels, respectively). (C) Kaplan-Meier survival analysis showing significant differences in event-free survival (EFS) between untreated and CAR T and/or AZA-treated groups. (D) CD3+ T cells identified in PB of 6/12 mice from CAR T and AZA + CAR T groups at day 30, with each dot plot representing the analysis of individual mice from CAR T and AZA + CAR T groups. |
Administration of CAR T cells to NSG mice reduced hCD19+ ALL-2 cell engraftment in PB analyzed 30 days post T cell infusion (Figure 2A and B). Single doses of AZA given alone reduced leukemia cell engraftment, and further reduced hCD19+ cell engraftment in mice when combined with CAR T cells (Figure 2B). Event-free survival (EFS) analysis was performed using leukemia event (LE) defined as 10% of hCD19+ blasts in PB (Figure 2C). Priming with AZA significantly improved EFS in mice infused with CAR T cells (Figure 2C).
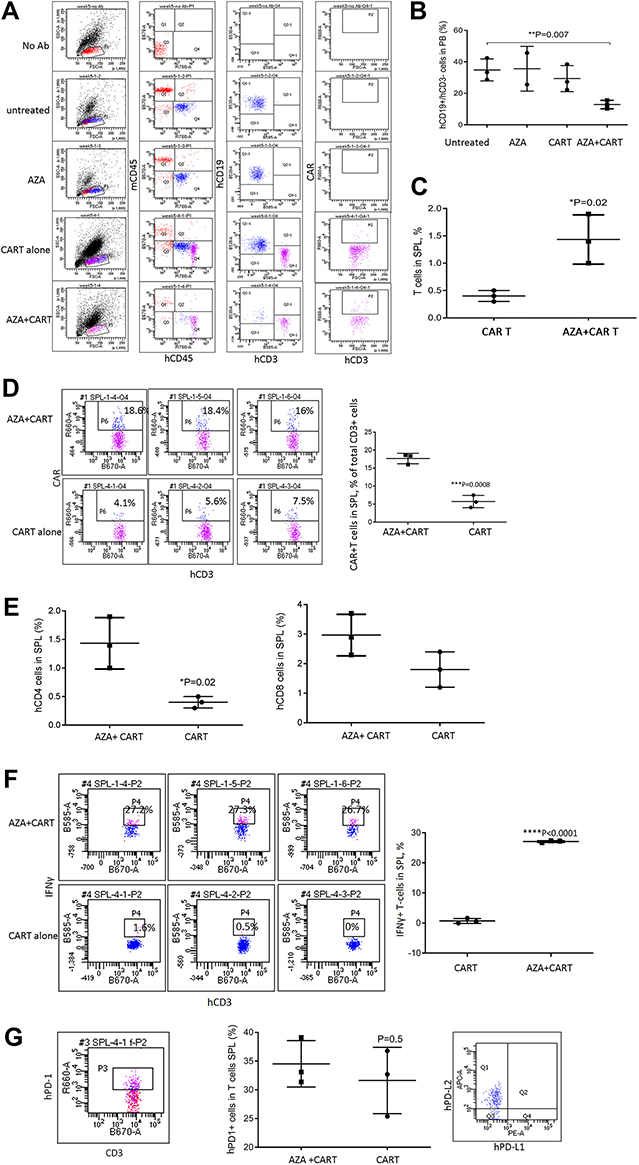 |
Figure 3 Host priming with AZA delays leukemia cell growth and promotes CAR T cell expansion in high leukemia burden models. Mice transplanted with ALL-2 cells, were treated with AZA and CAR T cells 4 weeks post ALL-2 injection, with AZA injected 24 hrs prior to CAR T cells. (A) Expression of mCD45, hCD45, hCD19 and hCD3 is shown in one representative flow cytometry dot plot for each condition. (B and C) hCD19+ leukemia and hCD3+ T cell engraftment in PB at 1 week post CAR T cell infusion, respectively. Each group except AZA alone (n = 2) contained 3 mice. (D) Host priming with AZA increased CAR T cell numbers in SPL analyzed post-mortem at week 5. Each dot plot represents analysis of individual mice from CAR T or AZA + CAR T groups. (E) Host priming with AZA increased %CD4+ and %CD8+ (not significant). (F) Host priming with AZA increased the proportion of IFNγ+ T cells in the SPL of mice infused with CAR T cells. (G) PD-1 expression in hCD3+ T cells (left and middle panels) and PD-L1/PD-L2 expression in hCD19+ cells isolated from the SPL of mice infused with CAR T cells (right panel). |
CD3+ T cells were identified in PB of 6/12 mice infused with CAR T cells alone or AZA + CAR T cells. However, mice that were given AZA prior to CAR T cells exhibited an increased proportion of CD3+ T cells at day 30 post CAR T cell infusion, suggesting that reduced leukemia cell burden in this group was associated with higher CAR T cell expansion (Figure 2D). It is unlikely that AZA or its metabolites, with half-life of 41 min and 4 hrs respectively may mediate the direct effect on CAR T cells, suggesting that CAR T cell expansion in AZA-primed mice rather results from the modulation of immune capacity of target cells to stimulate or inhibit T cell function.
The effect of priming with AZA was further examined in advanced leukemia settings when AZA and CAR T cells were injected at 4 weeks post-ALL-2 injection and analyzed 1 week later (Week 5). Both CAR T cells and AZA given alone had minimal effect in the setting of advanced disease. However, priming with AZA upfront of CAR T cell infusion significantly reduced leukemia engraftment in the PB and increased proportions of CD3+, CAR+ and CD4+ T cells in the spleen (Figure 3A–E). Importantly, priming with AZA increased the numbers of IFNγ+ T cells (Figure 3F). T cells from CAR T alone and AZA + CAR T groups expressed similar levels of PD-1 (Figure 3G). Activation of the inhibitory PD-1 checkpoint in T cells is controlled by the availability of specific PD-1 ligands, PD-L1 and PD-L2.26 Flow cytometry analysis revealed PD-L2 but not PD-L1 expression in ALL-2 cells (Figure 3G), and host priming with AZA did not modulate PD-L2 expression (data not shown).
AZA Potentiates CAR T Cells in Co-Cultures with CD19+ Leukemia Cells
CAR T cells were co-cultured with CD19+ B-ALL Nalm-6 cells pre-treated with AZA (0.4 μM, 24 hrs) or untreated cells. Multiparameter flow cytometry analysis was performed to determine the effect of leukemia priming with AZA on the formation of effector:target (E:T) CD3+ CD10+ cell conjugates, target CD10+ cell cytolysis, and proliferative and inflammatory functions of CAR T cells.23 Pre-treatment with AZA increased the proportion of CD10+ leukemia cells forming CD3+ CD10+ conjugates (Q2/Q1 + Q2) and reduced the numbers of single CD10+ target cells (Q1) at E:T = 10 and E:T = 20 (Figure 4A–C). Additionally, priming with AZA increased the proportion of 7-AAD+ apoptotic cells identified in CD10+ gate (Figure 4D, P4 gate).
 |
Figure 4 Pre-treatment of leukemia cells with AZA potentiates CAR T cells in co-cultures. (A and B) Nalm-6 cells were primed with AZA (0.4 μM, 24 hrs) or remained untreated. Equal numbers of viable cells were then co-cultured with CAR T cells for another 24 hrs at (E) T = 1:1, 1:10, 1:20. Proportions of CD3+ CD10+ T cell/leukemia-cell conjugates were determined as number of events in Q2/Q1 + Q2. (C) single CD10+ target cell numbers were determined in Q1. (D) Proportions of apoptotic 7AAD+ target cells (P4) were analyzed in CD3+ gate. (E) CFSE-labeled CAR T cells cultured alone (in the presence of IL-2 only) or co-cultured with Nalm-6 cells for 48 hrs at (E) T=1:1 exhibit dye dilution during 48 hr culture with each peak representing one division. It is relevant that all CFSE+ T cells analyzed prior to culture were located in P6 (data not shown). Rapidly dividing CFSEdim T cells that underwent 4 divisions were determined in P10 gate. Each condition was done in duplicate, with 1 representative histogram for each condition shown. (F) CD3+ T cell numbers per culture. (G) PD-1 expression in CAR T cells before and after 24-hour co-culture with Nalm-6 cells analyzed in CD3+ T cell gates. One representative flow cytometry analysis is shown for each condition. (H) hPD-L1/hPD-L2 expression in Nalm-6 cells. |
Antigen stimulation in CAR T and Nalm-6 co-cultures increased the proportion of dividing CFSEdim T cells compared to CAR T alone culture (Figure 4E, top and middle panels). However, CD3+ T cell numbers were reduced in co-cultures presumably due to activated T cell death (ATCD).23 Importantly, priming with AZA increased the proportion of rapidly dividing CFSEdim T cells identified in P10 gate and CD3+ T cell numbers identified at the end of co-culture (Figure 4E and F respectively). It is relevant that the proportion of apoptotic 7AAD+ T cells was not modulated by pre-treatment of Nalm-6 cells with AZA (data not shown).
Unlike CAR T cells cultured with only IL-2 (CAR T only), CAR T cells co-cultured with Nalm-6 cells rapidly up-regulated PD-1 (Figure 4G). Pre-treatment of Nalm-6 cells with AZA did not significantly modulate PD-1 in CAR T cells (Figure 4G). Similar to ALL-2 cells derived from PDXs, Nalm-6 cells did not express PD-L1 but expressed PD-L2, an alternative PD-1-specific ligand that activates the PD-1 checkpoint in T cells (Figure 4H). Pre-treatment with AZA did not affect PD-L1 or PD-L2 expression in Nalm-6 cells (Figure 4H), suggesting that AZA does not modulate the PD-1 checkpoint.
Pre-treatment of target cells with AZA increased target cell killing, expanded CAR T cells in co-cultures with CD19+ Burkitt lymphoma Raji cells (Figure 5A and B), and increased the numbers of CD62L– CD45RO+ Tem and effector IFNγ+ CD3+ T cells (Figure 5C and D). Pre-treatment with AZA did not modulate PD-L1 or PD-L2 expression in Raji cells, and PD-1 expression in CAR T cells was not affected by priming leukemia cells with AZA (Figure 5E and F respectively). Collectively, our in vitro data suggest that priming target cells with AZA increases functional capacity and improves the anti-tumor potency of CAR T cells.
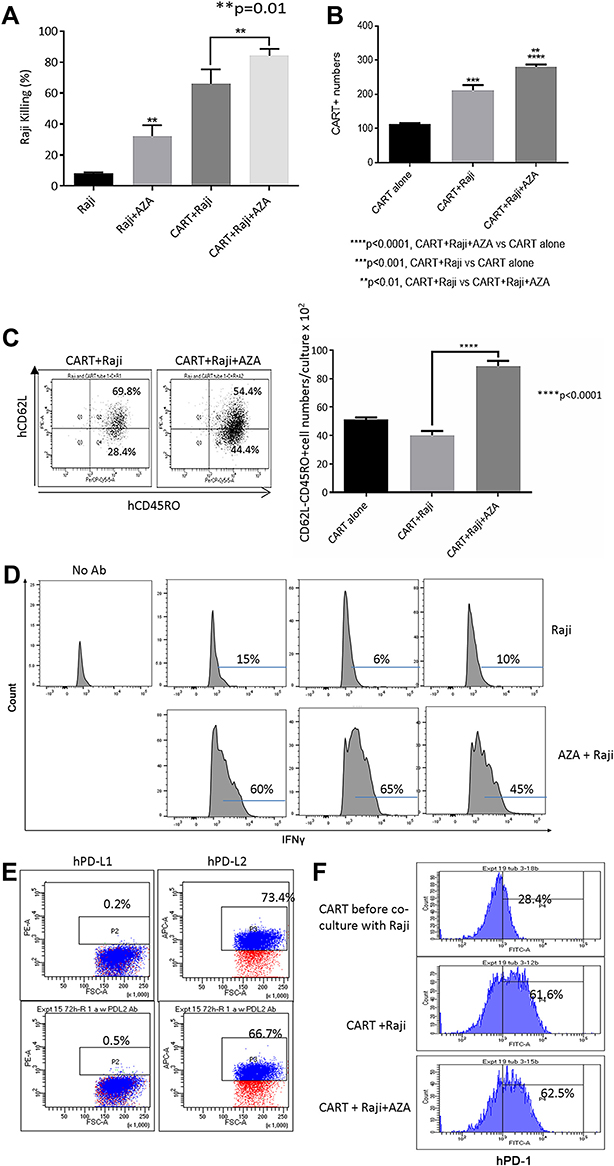 |
Figure 5 Priming Raji cells with AZA modulates CAR T cell function in co-cultures. Pre-treatment with AZA promoted Raji cell killing (A), increased CAR T and hCD62L– CD45RO+ Tem numbers (B and C) and increased proportion of IFNγ+ CAR T cells co-cultured with Raji cells, with IFNγ expression in CD3+ T cells measured in triplicate (D). Flow cytometry measurements of PD-L1/PD-L2 expression in Raji cells (E) and PD-1 expression in CAR T cells at the end of co-culture (F). |
Gene Expression in Leukemia Cells Isolated from Mice Primed with AZA
To determine the mechanisms triggered by AZA priming, 4 mice were injected with ALL-2 cells, and 2 of those mice were treated with AZA (ALL-2 + AZA mice) at week 8 when leukemia engraftment reached 56.75% in average in PB (data not shown) or remained untreated (ALL-2 mice). RNA was isolated from splenocytes 24 hrs post-AZA treatment. Gene expression analysis revealed 1682 genes modulated by AZA, with 828 genes up-regulated and 854 down-regulated. GSEA revealed the up-regulation of numerous immune pathways identified in leukemia cells isolated from AZA-treated mice (Supplementary Table S1). AZA up-regulated genes encoding cancer testis antigens, integrins and HLA-I (HLA-B, C, F and H) and HLA-II (HLA-DRB1, 3, 4, 5 and HLA-A29.1, HLA-DQA1 and 2, HLA-DPA1) that are key for antigen presentation and processing. Genes encoding immune stimulatory ligands (LITAF, TNFAIP8L2, TNFSF4, IL-32) and chemokines CXCL16 and CCL25 promoting T cell migration, and epigenetically deregulated in pediatric B-ALL-26,27 were up-regulated following treatment with AZA (Figure 6). In addition, reduced expression of genes encoding IL-10 and TNFSF14 that negatively regulate T cell function was identified in leukemia cells isolated from AZA-treated mice. Remarkably, AZA also down-regulated genes that encode CD24, which is over-expressed in many cancers.28 GSEA also highlighted pathways with previously underappreciated roles in regulating the responses of leukemia cells to cytotoxic T lymphocyte (CTL)-mediated killing (Supplementary Table S1). Pre-treatment with AZA did not modulate target CD19 gene expression or the expression of genes encoding PD-L1 or PD-L2, which is consistent with flow cytometry data. Up-regulation of genes encoding pro-apoptotic proteins Bax, CASP9, FASL, FADD and down-regulation of genes encoding anti-apoptotic Bcl2L1 were registered in leukemia cells derived from AZA-treated hosts (Figure 6), suggesting that pre-treatment with AZA activates pro-apoptotic programs in leukemia cells. Up-regulation of CDKN1A in leukemia cells derived from AZA-treated mice suggests that AZA suppresses leukemia cell cycling in addition to apoptosis.
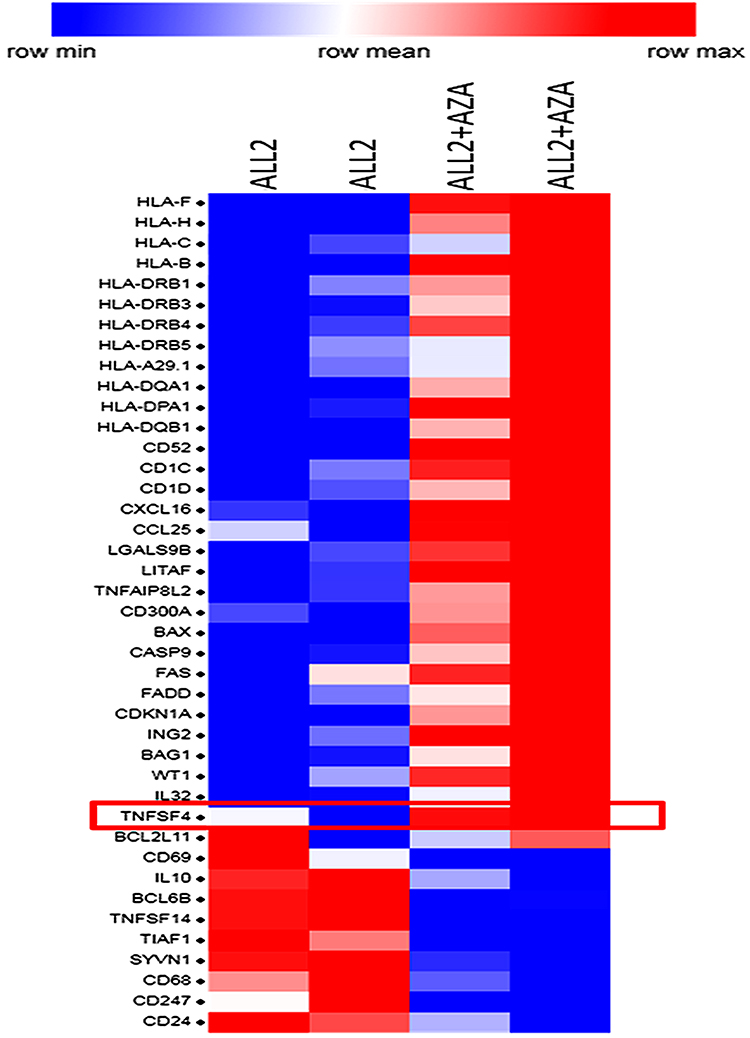 |
Figure 6 Priming leukemia-bearing mice with AZA modulates gene expression in leukemia cells. Selected differentially expressed genes in hCD19+ leukemia cells isolated from the spleen of 2 AZA-treated mice (ALL-2 + AZA) and 2 untreated ALL-2 mice. Genes up-regulated by AZA are shown in red, and down-regulated in blue. |
Host Priming with AZA Promotes Allo- and Xenoreactivity of Human Untransduced T Cells but Only Weakly Modulates CAR19T Cell Allo/Xeno-Responses
Priming mice with AZA promoted graft versus leukemia (GVL) effects induced by untransduced (mock) T cells, although without statistical significance (Figure 7A–C). Increased T cell expansion and differentiation to more cytotoxic CD62L– CD45RO+ Tem in the expense of less cytotoxic Tcm was observed in mice primed with AZA before mock T cell infusion (Figures 7D and 8A). Additionally, mice infused with mock T cells following AZA priming were apathetic, appeared ruffled, had rapid breath, and exhibited weight loss over the last week (Supplementary Figure S1), a surrogate marker for the severity of xenogeneic GVH (xGVH) reactivity.25 xGVH was confirmed in post-mortem analysis (data not shown). Increased allo- and xenoreactivity of mock T cells isolated from AZA-primed mice is consistent with the up-regulation of genes encoding HLAs critical for allo- and xeno-antigen presentation (Figure 6).
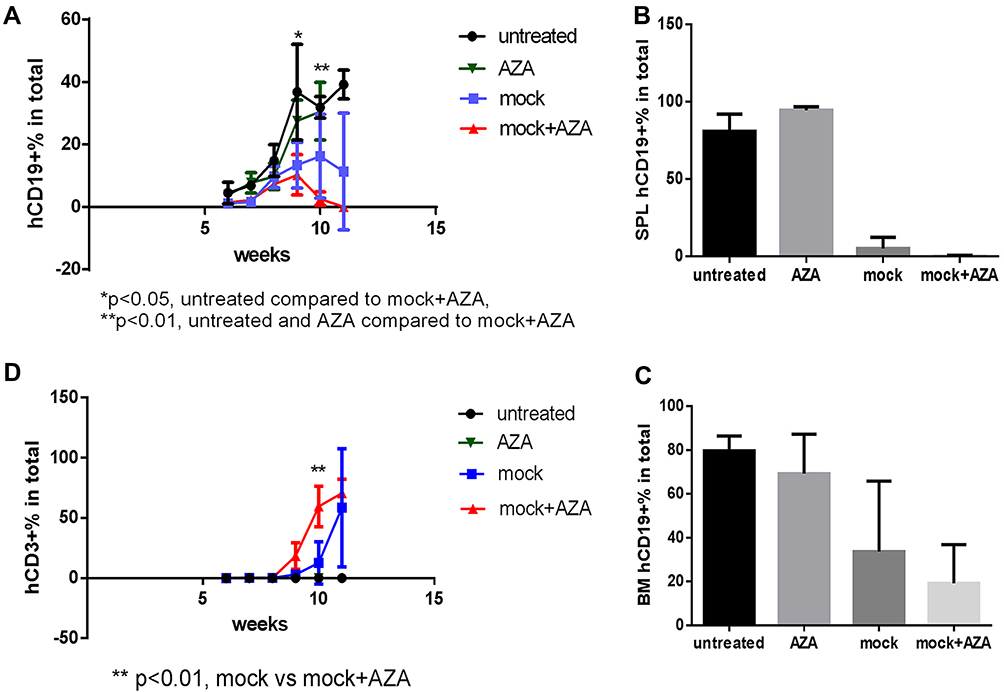 |
Figure 7 Host priming with AZA promoted GVL effect mediated by mock T cells. Untransduced (mock) T cells were injected 1 week post ALL-2 infusion, and AZA was injected 1 day prior to mock T cell infusion as in Figure 3. (A–C) Human CD19+ leukemia cell engraftment in PB, SPL, and bone marrow (BM). (D) hCD3+ T cell engraftment in PB. |
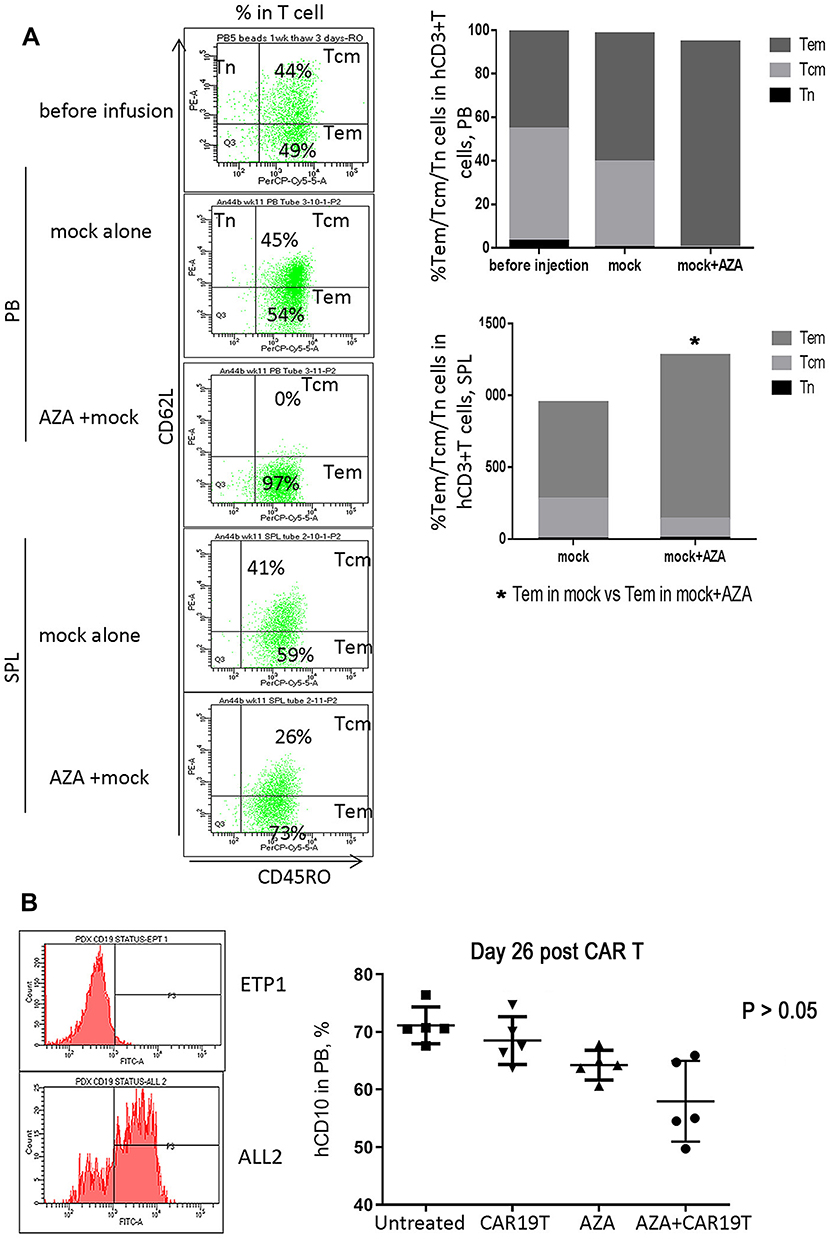 |
Figure 8 Host priming with AZA promoted GVL effect mediated by mock T cells (continued). (A) Host priming with AZA promoted T cell differentiation of infused mock T cells into CD62L– CD45RO+ Tem in expense of CD62L+ CD45RO+ Tcm. Representative dot plots of CD62L and CD45RO expression are shown. (B) hCD19 expression in leukemia ETP-1 and ALL-2 cells. hCD10+ cell engraftment in PB in mice infused with CD19– CD10+ ETP-1 cells and treated with CAR19 T, AZA, or AZA + CAR19 T (n = 5 per group). |
To discriminate alloreactivity from antigen-specific reactivity of CAR19T cells and to determine whether alloreactivity of CAR T cells could potentially contribute to the increased GVL effect in AZA-pre-treated mice in mismatched settings, mice bearing CD19– CD10+ ETP-1 cells were treated with CAR19 T cells (Figure 8B, left panel). CAR19 T cells alone did not reduce CD19– CD10+ ETP-1 cell engraftment in the PB of mice, thus exhibiting low allo-reactivity, while AZA alone slightly reduced ETP-1 cell engraftment, although not significantly (Figure 8B, left panel). Priming with AZA reduced CD10+ cell engraftment in mice infused with CAR19T cells, suggesting increased alloreactivity in CAR19T cells isolated from AZA-treated mice. However, this effect did not reach significance (Figure 8B, right panel). Thus, increased alloreactivity of CAR19T cells in AZA-primed mice appears to only marginally contribute to the increased GVL effect against CD19+ leukemia. Importantly, unlike mock T cells, CAR19 T cells did not induce xGVHD, with the latter correlating with reduced allo- and xenoreactivity of human CAR T cells used following ex vivo expansion.29
TNFSF4/OX40L Up-Regulation in AZA-Primed Leukemia Cells May Play a Role in Potentiating CAR T Cell Function
Gene expression analysis showed significant up-regulation of the TNFSF4 gene in ALL-2 cells isolated from AZA-primed mice (Figure 6). TNFSF4 encodes OX40L that stimulates T cells via ligation of OX40, the member of tumor necrosis factor (TNF) receptor superfamily highly expressed in activated T cells.30 OX40L/OX40 is involved in various important regulatory functions associated with lymphocyte activation.31 Flow cytometry confirmed up-regulation of OX40L in AZA-treated ALL-2 cells, and increased OX40L expression remained in ALL-2 cells co-cultured with CAR T cells (Figure 9A).
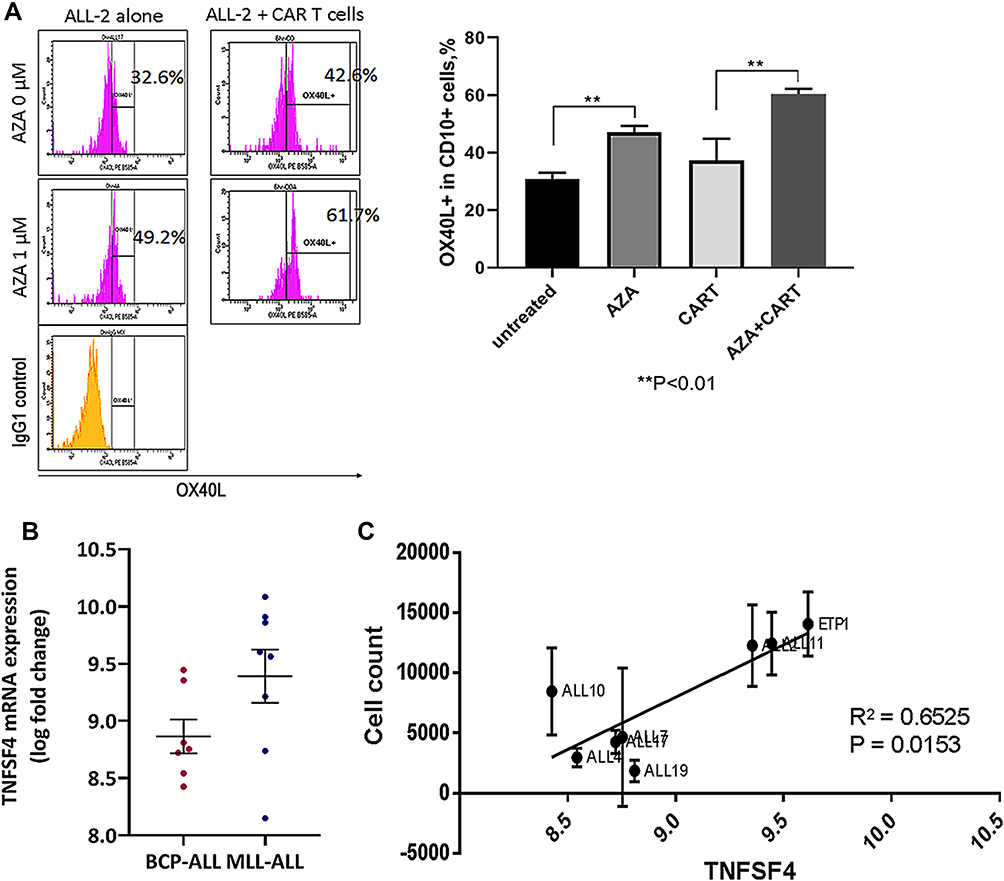 |
Figure 9 TNFSF4/OX40L up-regulation in AZA-primed leukemia cells promotes CAR T cell function. (A) Representative histograms showing OX40L expression in ALL-2 cells. ALL-2 cells were pre-treated with AZA (1 µM, 24 hrs) or remained untreated. Target cells were then co-cultured with CAR T cells for 6 hrs. Pre-treatment with AZA increased the proportion of OX40L+ cells. (B) TNFSF4 mRNA expression in 7 BCP-ALL and 8 MLL-ALL PDXs were determined using transcriptome analysis. (C) High TNFSF4 expression in PDXs is associated with high CAR T cell numbers identified in 24-hour co-cultures. |
CD19+ pediatric B cell precursor ALL (BCP-ALL) including mixed lineage leukemia ALL (MLL-ALL) demonstrate heterogeneous expression of TNFSF4, and regression analysis revealed that high TNFSF4 expression in PDXs is associated with high CAR T cell numbers identified in 24-hour co-cultures (Figure 9B and C). It is relevant that correlation analysis did not show any association between the expression of genes encoding other T cell co-stimulatory ligands such as ICOSL, CD70, CD86 and CD137/4-1BB, all differentially expressed in B-ALLs, and CAR T cell survival in co-cultures with individual B-ALL (data not shown).
The role of the OX40L/OX40 co-stimulatory pathway was examined in CAR T cells co-cultured with OX40+ Nalm-6 cells using OX40L-blocking antibodies. OX40 expression was up-regulated in CAR T cells co-cultured with target cells (Figure 10A), and the proportion of activated FSChigh T cells was higher in gated OX40+ compared to the OX40– subset, suggesting that OX40/OX40L signaling promotes CAR T cell activation (Figure 10B). Importantly, the proportion of OX40L+ Nalm-6 cells was reduced following co-culture with CAR T cells (Figure 10C), and OX40L blockade increased the proportion of OX40L+ target cells that survived CAR T cell killing (Figure 10C), suggesting that OX40L expression makes leukemia cells more susceptible to CAR T cell cytotoxicity. We propose that AZA-priming up-regulates OX40L in leukemia cells promoting OX40 co-stimulatory signaling in CAR T cells.
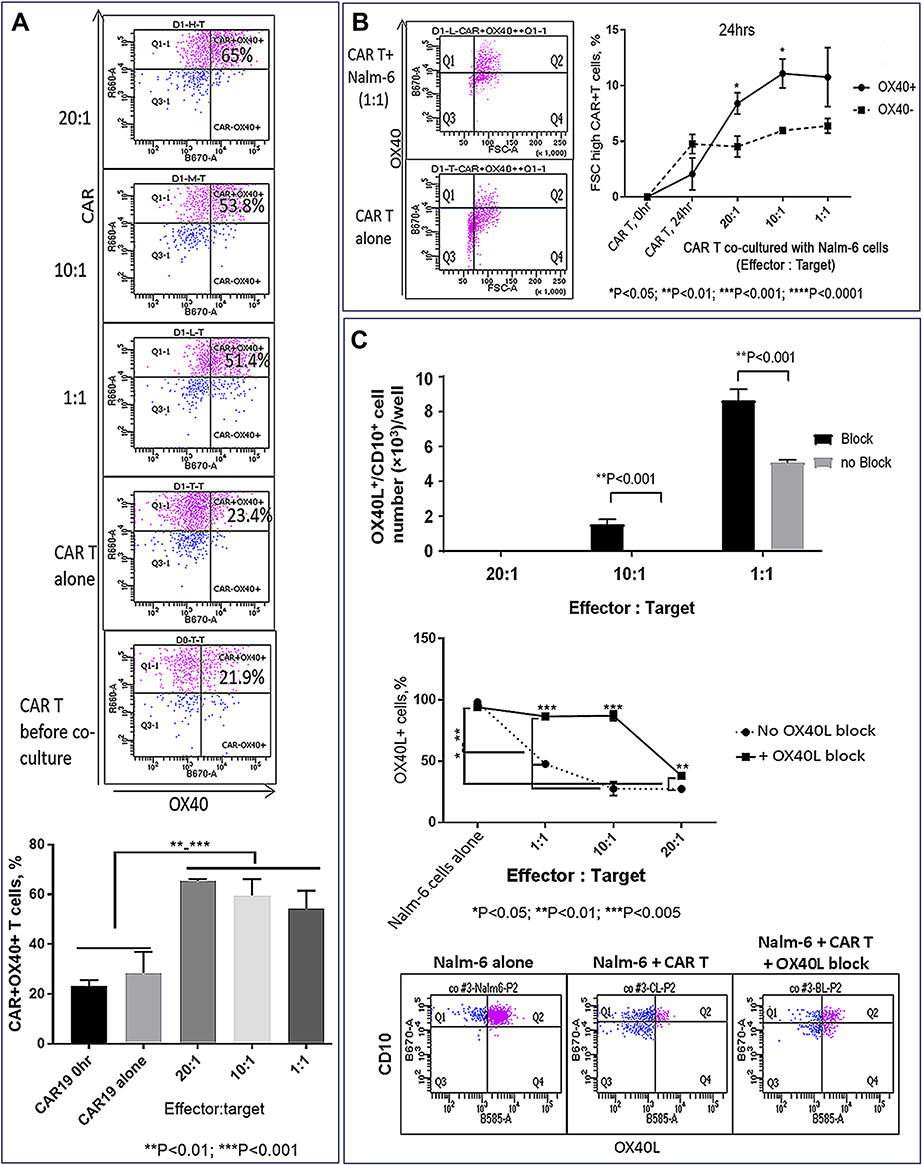 |
Figure 10 TNFSF4/OX40L up-regulation in AZA-primed leukemia cells promotes CAR T cell function (continued). (A) Co-culture with target cells increased proportion of OX40+ CAR T cells. OX40 and CAR expression were analyzed in CD3+ gate (top panel). Proportions of OX40+ CAR T cells in 24-hour co-cultures with ALL-2 cells (bottom panel). (B) High OX40 expression in CAR T cells is associated with the increased proportion of activated FSChigh cells (right panel). Representative dot plot analysis of OX40 versus FSC T cells in the CD3+ gate is shown (left panel). CAR T cells co-cultured with leukemia cells at E:T = 20:1, 10:1, 1:1 (24 hrs). (C) Reduced proportion of OX40L+ CD10+ target cells following co-culture with CAR T cells. OX40L blockade increased the proportion of OX40L+ target cells preserved from CAR T cell killing. Representative flow cytometry analysis for each condition is shown (bottom panel). OX40L-blocking antibodies and the antibodies used for detecting OX40L expression bound to different OX40L epitopes. |
Discussion
Our study reveals the capacity of AZA given upfront to improve the efficacy of CAR T cell therapy for CD19+ chemoresistant pediatric B-ALL patients. Single doses of AZA given 24 hrs prior to CAR T cell infusion delayed leukemia cell growth in PDX mouse models of B-ALL, promoted CAR T cell expansion and improved their effector function to increase INFγ+ cell numbers. Importantly, this effect was seen at large leukemia burden when both AZA and CAR T cells given as monotherapies were not effective, which suggests that priming patients with AZA prior to CAR T cell infusion may be beneficial for patients with advanced disease. The pro-immunogenic effects of AZA were reproduced in CAR T cells co-cultured with AZA-pre-treated leukemia cells. Pre-treatment of target cells with AZA increased CD19+ Nalm-6 leukemia cell/CAR T cell conjugation, promoted CAR T cell expansion, and increased INFγ+ effector cell numbers and leukemia cell killing. Thus, AZA appears to promote cytotoxicity (INFγ+ CAR T cells) and proliferation (higher CFSElow cells). However, it remains to be determined whether priming leukemia cells with AZA will modulate CAR T persistence in long-term experiments. It is relevant that excessive CAR T cell activation may lead to premature exhaustion.32–34
There are various means by which priming with AZA can affect CAR T cell function.32 Another demethylating agent, decitabine, was shown to increase the expression of CD19 on lymphoma cells and enhanced the cytotoxic effect of CAR19 T cells.35 In our experiments, however, AZA did not up-regulate CD19 cell surface expression in B-ALL cells. Additionally, neither PD-L1/PD-L2 expression in leukemia cells nor PD-1 expression in CAR T cells was affected by pre-treatment of leukemia/hosts with AZA, suggesting that AZA effects were not mediated through the modulation of the inhibitory PD-1 checkpoint. It is relevant that PD-1/PD-L1 axis was shown to be modulated by AZA in acute myeloid leukemia (AML).36–38
Host priming with AZA, however, modulated multiple immune genes and pathways in leukemia cells. AZA treatment enriched genes that encode HLAs key for antigen presentation. Leukemia-bearing mice primed with AZA showed up-regulated cytokine/chemokines, cancer testis antigens, integrins and immune stimulatory ligands. Pro-immunogenic modulation of gene expression in B-ALL cells aligns with previous studies that showed modulation of similar immune pathways in breast, colorectal and ovarian cancers treated with AZA.39 Importantly, AZA up-regulates TNFSF4 that encodes the T cell co-stimulatory ligand OX40L.30 OX40L binds its receptor OX40, the TNF receptor superfamily member that is highly specific for activated T cells. OX40L stimulates T cells to promote T cell proliferation, effector differentiation and long-term survival.31,40 OX40L/OX40 signaling was shown to promote Bcl-xL and Bcl-2 expression essential for long-term T cell survival and in vivo persistence of CAR.CD30T cells.41,42 AZA down-regulated IL-10 in ALL-2 cells, and OX40 co-stimulation was shown to abrogate IL-10 inhibitory effects on CAR T cells.19,43 Thus, we focused on exploring the effect of OX40L/OX40 signaling on CAR T cell function. PDXs from individual B-ALL patients express variable levels of TNFSF4, and high TNFSF4 expression in leukemia cells was associated with enhanced CAR T cell numbers in co-cultures. High OX40L expression seems to increase the susceptibility of leukemia Nalm-6 cells to CAR T cell killing, and OX40L blockade reduced CAR T cell-mediated killing of Nalm-6 cells. Additionally, higher proportions of activated T cells were seen in OX40+ T cell subsets, suggesting that up-regulation of OX40L in AZA-treated leukemia cells activates co-stimulatory OX40 signaling in CAR T cells. It is relevant that decitabine was shown to promote TCR-specific T cells through up-regulation of OX40L in colorectal cancer cells.44
Gene expression analysis revealed up-regulation of pro-apoptotic genes and down-regulation of anti-apoptotic genes in leukemia cells derived from AZA-primed mice. CAR T cell cytotoxicity depends on the expression of pro-apoptotic proteins in target cells.45 We have previously shown that treatment with AZA up-regulates caspase-3/cleaved caspase-3 in Nalm-6 cells and promotes CAR T cell-mediated cytolysis of target cells.23 Thus, direct activation of pro-apoptotic machinery by AZA may present an additional mechanism to promote CAR T cell therapy.
Conclusion
In summary, our findings provide a strong rationale to investigate AZA pre-treatment prior to adoptive CAR T cell therapy. Better understanding of the mechanisms of immunogenic modulation of AZA-treated leukemia cells will be required to determine how to best apply AZA to enhance CAR T cell therapy. Overall, our results suggest that AZA can be used to make leukemia cells more immunogenic through epigenetic modulation of genes stimulatory to T cells, and in particular, OX40L.
Acknowledgments
This work was funded by Kids Cancer Project grant. BT was supported by a PhD stipend from Australian Postgraduate Awards. The authors thank Professor Richard Lock from Children’s Cancer Institute for providing PDXs. The authors also thank Lauren Olaffson and Sylvie Shen from Children’s Cancer Institute, for the assistance in some experiments.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–3331. doi:10.1182/blood-2017-02-769208
2. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866
3. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
4. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi:10.1158/2159-8290.CD-12-0548
5. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019. doi:10.1038/s41571-019-0184-6
6. Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919
7. Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J. 2018;17:1–13. doi:10.1016/j.csbj.2018.11.004
8. Petersen CT, Hassan M, Morris AB, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kdelta inhibitors and VIP antagonists. Blood Adv. 2018;2(3):210–223. doi:10.1182/bloodadvances.2017011254
9. Piscopo NJ, Mueller KP, Das A, et al. Bioengineering solutions for manufacturing challenges in CAR T cells. Biotechnol J. 2018;13(2):1700095. doi:10.1002/biot.201700095
10. Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 2016;76(7):1683–1689. doi:10.1158/0008-5472.CAN-15-2125
11. Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986. doi:10.1016/j.cell.2015.07.011
12. Gang AO, Frøsig TM, Brimnes MK, et al. 5-azacytidine treatment sensitizes tumor cells to T-cell mediated cytotoxicity and modulates NK cells in patients with myeloid malignancies. Blood Cancer J. 2014;4(3):e197. doi:10.1038/bcj.2014.14
13. Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics. 2017;9(1):59. doi:10.1186/s13148-017-0358-y
14. Covre A, Coral S, Di Giacomo AM, Taverna P, Azab M, Maio M. Epigenetics meets immune checkpoints. Semin Oncol. 2015;42(3):506–513. doi:10.1053/j.seminoncol.2015.02.003
15. Covre A, Coral S, Nicolay H, et al. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. OncoImmunology. 2015;4(8):e1019978. doi:10.1080/2162402x.2015.1019978
16. Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non–small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi:10.1158/2159-8290.cd-11-0214
17. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249. doi:10.1038/nature15520
18. Wang L, Amoozgar Z, Huang J, et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol Res. 2015;3(9):1030–1041. doi:10.1158/2326-6066.CIR-15-0073
19. Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. OncoImmunology. 2012;1(4):458–466. doi:10.4161/onci.19855
20. Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29(3):187–201. doi:10.1615/CritRevImmunol.v29.i3.10
21. Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi:10.1172/jci46110
22. Carol H, Szymanska B, Evans K, et al. The anti-CD19 antibody–drug conjugate SAR3419 prevents hematolymphoid relapse postinduction therapy in preclinical models of pediatric acute lymphoblastic leukemia. Clin Cancer Res. 2013;19(7):1795–1805. doi:10.1158/1078-0432.ccr-12-3613
23. Dolnikov A, Shen S, Klamer G, et al. Antileukemic potency of CD19-specific T cells against chemoresistant pediatric acute lymphoblastic leukemia. Exp Hematol. 2015;43(12):1001–1014.e5. doi:10.1016/j.exphem.2015.08.006
24. Bishop DC, Xu N, Tse B, et al. PiggyBac-engineered T cells expressing CD19-specific CARs that lack IgG1 Fc spacers have potent activity against B-ALL xenografts. Mol Ther. 2018;26(8):1883–1895. doi:10.1016/j.ymthe.2018.05.007
25. Klamer G, Shen S, Song E, et al. GSK3 inhibition prevents lethal GVHD in mice. Exp Hematol. 2013;41(1):39–55.e10. doi:10.1016/j.exphem.2012.09.005
26. Boussiotis VA, Longo DL. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767–1778. doi:10.1056/NEJMra1514296
27. Zaballos A, Gutierrez J, Varona R, Ardavin C, Marquez G. Cutting edge: identification of the orphan chemokine receptor GPR-9-6 as CCR9, the receptor for the chemokine TECK. J Immunol. 1999;162(10):5671–5675.
28. Jing X, Cui X, Liang H, et al. CD24 is a potential biomarker for prognosis in human breast carcinoma. Cell Physiol Biochem. 2018;48(1):111–119. doi:10.1159/000491667
29. Anwer F, Shaukat -A-A, Zahid U, et al. Donor origin CAR T cells: graft versus malignancy effect without GVHD, a systematic review. Immunotherapy. 2017;9(2):123–130. doi:10.2217/imt-2016-0127
30. Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol. 2010;28(1):57–78. doi:10.1146/annurev-immunol-030409-101243
31. Weiguang Y, Dalin L, Lidan X, et al. Association of OX40L polymorphisms with sporadic breast cancer in northeast Chinese Han population. PLoS One. 2012;7(8):e41277. doi:10.1371/journal.pone.0041277
32. Salter AI, Ivey RG, Kennedy JJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. 2018;11(544):eaat6753. doi:10.1126/scisignal.aat6753
33. Yang Y, Kohler ME, Chien CD, et al. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci Transl Med. 2017;9(417):eaag1209. doi:10.1126/scitranslmed.aag1209
34. Zhao Z, Condomines M, van der Stegen SC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi:10.1016/j.ccell.2015.09.004
35. Li S, Xue L, Wang M, et al. Decitabine enhances cytotoxic effect of T cells with an anti-CD19 chimeric antigen receptor in treatment of lymphoma. Onco Targets Ther. 2019;12:5627–5638. doi:10.2147/OTT.S198567
36. Daver N, Garcia-Manero G, Basu S, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, Phase II Study. Cancer Discov. 2018;9(3):370–383. doi:10.1158/2159-8290.CD-18-0774
37. El Khawanky N, Hughes A, Yu W, et al. Azacytidine sensitizes AML cells for effective elimination by CD123 CAR T-cells. Blood. 2019;134(Supplement_1):3904. doi:10.1182/blood-2019-124684
38. Mardiana S, Gill S. CAR T cells for acute myeloid leukemia: state of the art and future directions. Front Oncol. 2020;10:697. doi:10.3389/fonc.2020.00697
39. Saleh MH, Wang L, Goldberg MS. Improving cancer immunotherapy with DNA methyltransferase inhibitors. Cancer Immunol Immunother. 2016;65(7):787–796. doi:10.1007/s00262-015-1776-3
40. Huddleston CA, Weinberg AD, Parker DC. OX40 (CD134) engagement drives differentiation of CD4+ T cells to effector cells. Eur J Immunol. 2006;36(5):1093–1103. doi:10.1002/eji.200535637
41. Guercio M, Orlando D, Di Cecca S, et al. CD28. OX40 co-stimulatory combination is associated with long in vivo persistence and high activity of CAR.CD30 T-cells. Haematologica. 2020. doi:10.3324/haematol.2019.231183
42. Rogers P, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15(3):445–455. doi:10.1016/S1074-7613(01)00191-1
43. Benmebarek M-R, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. 2019;20(6):1283. doi:10.3390/ijms20061283
44. Kumari A, Cacan E, Greer SF, Garnett-Benson C. Turning T cells on: epigenetically enhanced expression of effector T-cell costimulatory molecules on irradiated human tumor cells. J Immunother Cancer. 2013;1(1):17. doi:10.1186/2051-1426-1-17
45. Chatterton Z, Morenos L, Mechinaud F, et al. Epigenetic deregulation in pediatric acute lymphoblastic leukemia. Epigenetics. 2014;9(3):459–467. doi:10.4161/epi.27585
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.