")
Back to Journals » OncoTargets and Therapy » Volume 9
Primary yolk sac tumor of the gluteus: a case report and literature review
Authors Li B, Jiang Q, Zhang S, Zhou Y, zhang Q, Ouyang L
Received 22 February 2016
Accepted for publication 20 May 2016
Published 2 August 2016 Volume 2016:9 Pages 4715—4719
DOI https://doi.org/10.2147/OTT.S106924
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Bo Li,1 Qianqian Jiang,1 Shitai Zhang,1 Yang Zhou,1 Qing-Fu Zhang,2 Ling OuYang1
1Department of Obstetrics and Gynecology, Shengjing Hospital of China Medical University, Shenyang, 2Department of Pathology, the First Affiliated Hospital and College of Basic Medical Sciences, China Medical University, Shenyang, People’s Republic of China
Abstract: Yolk sac tumor (YST) is a common malignant primitive germ cell tumor that often exhibits differentiation into endodermal structures. They most commonly occur in childhood and adolescence and are rare after the age of 40 years. Derived from the yolk sac during the embryonic period, YSTs can occur in the gonads and germ cells because the tumor cells migrate from the yolk sac toward the gonads. Here, we present a rare case of primary gluteus YST in a 3-year-old girl. She received BEP chemotherapy (bleomycin + etoposide + cisplatin) after surgical resection. There was no evidence of recurrence 7 months after primary treatment.
Keywords: immunohistochemistry, yolk sac tumor, germ cell tumor
Background
Yolk sac tumors (YSTs) are believed to originate from a selective differentiation of the yolk sac structure by an undifferentiated or multipotential embryonal carcinoma. YSTs are most common in the gonads (ovaries and testicles), and only 10%–15% of cases are extragonadal, occurring in the median line of the human body (such as the presacral and sacrococcygeal region, mediastinum, intracranial region [mostly pineal], vagina, corpus uteri, prostate glands, presacral region, retroperitoneum, liver, fundus of the stomach, orbit, face, and umbilical region of the abdominal wall).1 However, YSTs that present in the subdermal soft tissue of the gluteus are rarely reported in the literature. We describe an unusual case of an extragonadal primary YST in the gluteus and review the literature.
Case presentation
A 3-year-old Asian girl was brought to the hospital with a history of an aggravating claudication that first appeared 2 months previously. Physical examination revealed that the skin color of the left gluteus was normal. A solid, tender mass, ~7×3 cm in size, was palpated in the deep gluteal region. It was associated with radiating pain to the left lower limb when the mass was pressed. On palpation, there was no fluctuation and little mobility of the mass.
Magnetic resonance imaging showed a mass with strong T1 and T2 signals in the left inferior intermuscular plane of the gluteus maximus muscle. The mass was ~7.4×2.4 cm and had a markedly heterogeneous hyperintense signal intensity on T2-weighted imaging, with a low signal linear lace and high signal in the sac variable region (Figure 1A). The mass showed a low signal intensity and local cystic degeneration on T1-weighted imaging (Figure 1B). An enhanced scan showed that the solid component of the mass showed diffuse enhancement and the cystic component demonstrated no enhancement (Figure 1C and D).
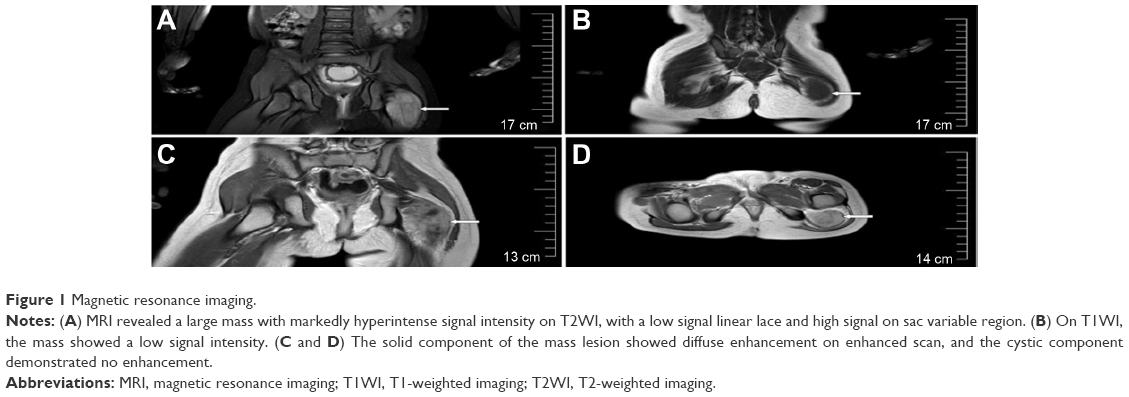 | Figure 1 Magnetic resonance imaging. |
The typical sites of YSTs, such as the vulva, corpus uteri, coccyx, and ovaries, were not involved. The femoral heads, articular surfaces, and joint spaces on both sides did not show any abnormalities. Three-dimensional color Doppler ultrasonography showed multiple enlarged retroperitoneal lymph nodes with apparently clearly defined margins; the maximum diameter was 1.2 cm. The liver, spleen, uterus, and adnexa showed normal signals. Computed tomography findings of the lungs and mediastinum were normal. Serum levels of alpha-fetoprotein (AFP) were significantly increased to 1,238 μg/L. The levels of other markers such as β-human chorionic gonadotropin, serum lactate dehydrogenase, carbohydrate antigen 125, and carcinoembryonic antigen were within normal limits.
During the operation, the deep fascia was incised longitudinally; then, we made an incision along the gluteus maximus to expose the deep mass, which was ~7×4×3 cm in size, yellow, soft, and smooth with a clear border. The tumor was located in the epineurium and adhered tightly to the sciatic nerve. The tumor was totally excised.
The tumor had negative margins on both gross and microscopic findings. The patient had no recurrence 7 months after the surgical treatment, with two cycles of BEP chemotherapy (bleomycin 15 U/m2 on day 1 + etoposide 167 mg/m2 on days 1–3 + cisplatin 33.3 mg/m2 on days 1–3) given every 3 weeks.
Materials and methods
The resected specimen was fixed in 10% buffered formalin and embedded in paraffin; 4 μm thick sections were cut from the paraffin block and stained with hematoxylin and eosin. Immunohistochemical staining was performed by using the streptavidin–peroxidase procedure (SP kit, MaiXin Inc, Fuzhou, People’s Republic of China) according to the instructions of the manufacturer. Positive and negative controls were included to evaluate the procedure.
Pathologic findings
Histologically, the tumor was composed of a loose network of flat to cuboid cells forming microcystic structures (Figure 2A). Occasional rosettes were also seen (Figure 2B). A few pink, hyaline droplets were seen adjacent to the tumor cells. Nuclear atypia is obviously in nuclear enlargement, and the tumor cells have a significant mitotic activity (~5 mitoses/10 high-power fields, Figure 2C).
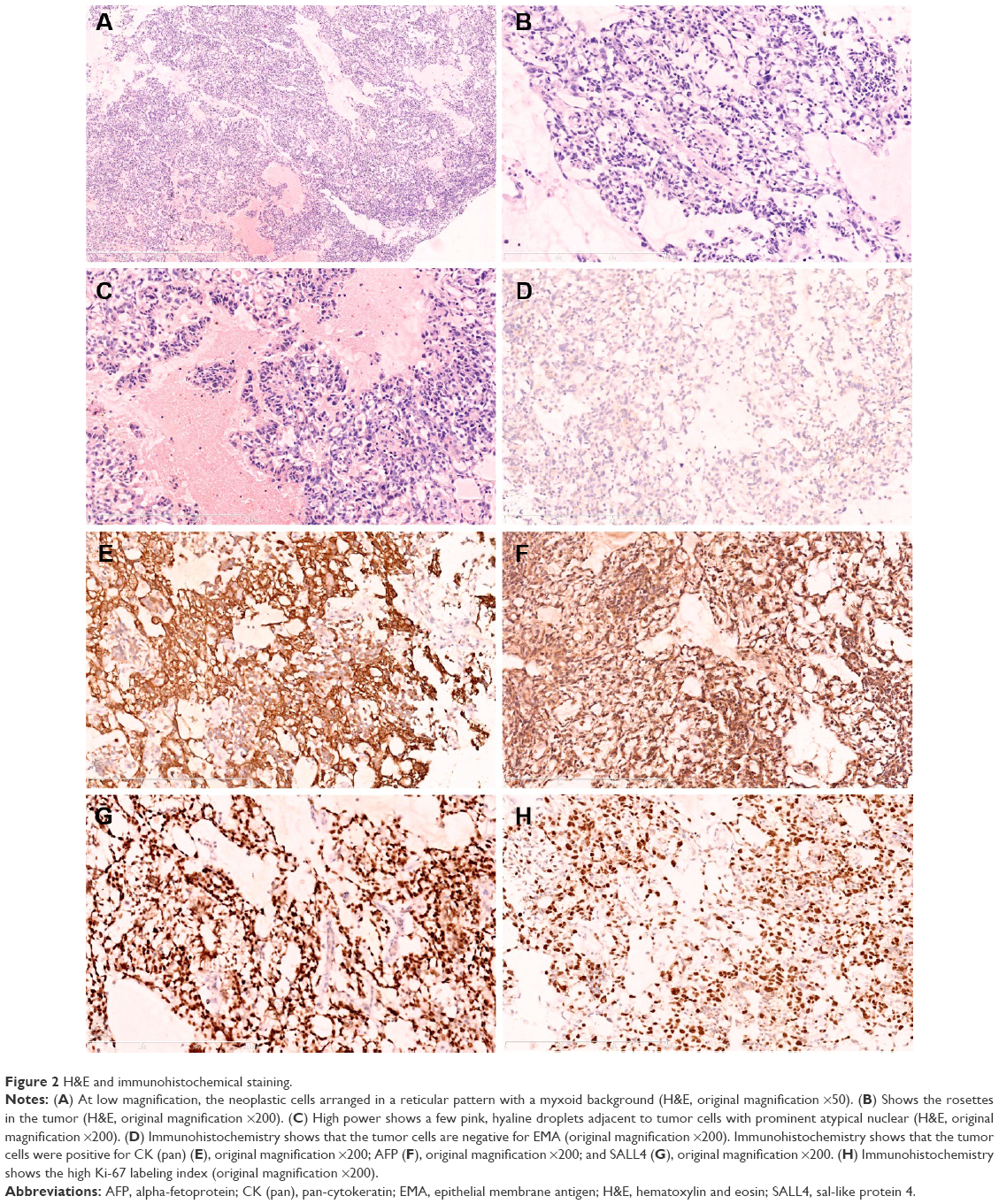 | Figure 2 H&E and immunohistochemical staining. |
Immunohistochemical findings
Immunohistochemistry was performed in this case demonstrating that the tumor cells were negative for epithelial membrane antigen (Figure 2D), S-100, myogenin, desmin, human placental alkaline phosphatase, actin (SMA), CD99, CD34, CD31, caudal-related homeobox transcription factor 2, glial fibrillary acidic protein, CD117, CD68, GATA-binding protein 3, octamer-binding transcription factor 3/4, and positive for pan-cytokeratin (Figure 2E), glypican-3, AFP (Figure 2F), and sal-like protein 4 (Figure 2G), which is consistent with the diagnosis of YST. Immunohistochemistry was positive for local expression of vimentin, synaptophysin, and CD30, The Ki-67 labeling index was ~60% (Figure 2H).
Discussion
At present, the etiology of YSTs remains controversial. Most experts consider that YSTs may derive from the false migration of embryonic primitive pluripotent germ cells or oncogenesis of the residual cells when they migrate from the genital ridge of the yolk sac endoderm.2 A few studies have shown that extragonadal YSTs express some embryonic stem cell-related proteins, suggesting that they are derived from primordial germ cells.3 Our case may be explained by the hypothesis that the tumor arose from misplaced primordial germ cells that entered the gluteus.
Extragonadal YSTs display remarkable differences in location of occurrence depending on the age of the child. For example, sacrococcygeal YSTs often occur in very young children. Vaginal YSTs occur in very young girls, commonly in those aged <2 years. On the other hand, mediastinal YSTs typically occur in young adult male patients.
Both extragonadal YSTs and YSTs of reproductive organs display a wide range of histological patterns. The most common patterns are microcystic, solid, glandular, and endodermal sinus (Schiller–Duval bodies); some less common patterns such as myxomatous, hepatoid, papillary, and sarcomatoid have also been observed.4 Several histological features are often seen in combination in the same tumor. The current case demonstrated predominantly microcystic, glandular–alveolar, and myxomatous patterns. Some areas contained small aggregates and microcysts within the myxomatous stroma. The tumor cells were flat and pleomorphic, with a predominantly clear cytoplasm with subnuclear vacuoles and large hyperchromatic nuclei. Increased mitotic figures were noted. In some areas, intracytoplasmic and extracellular hyaline globules were identified within the lesion.
Perivascular formation recapitulating endodermal sinuses (Schiller–Duval bodies), which are virtually diagnostic of YSTs, are present in only 30% of cases and were not identified in this case. Immunohistochemical staining for AFP, glypican-3, and sal-like protein 4 can usually help to confirm the diagnosis of YST. AFP and glypican-3 are specific markers for yolk sac differentiation during the embryonic stage into liver and digestive tract, respectively; they are also considered to be sensitive and specific markers of YST.5
Sal-like protein 4 is a zinc finger transcription factor that has been identified as a useful marker for the diagnosis of extratesticular germ cell tumors (GCTs).6,7 A recent study found that the zinc finger and BTB domain containing 16 (ZBTB16) is a novel, highly sensitive, and specific marker for distinguishing YSTs from other nonyolk sac GCTs.8
YSTs are not immunoreactive for epithelial membrane antigen and CK7 in contrast to endometrioid adenocarcinoma and other surface epithelial tumors of the ovary. DNA ploidy studies have shown that YSTs are almost invariably aneuploid.9
Patients with stage I YST only require follow-up measurement of serum AFP and β-human chorionic gonadotropin levels after initial surgery. The standard BEP chemotherapy regimen after resection is the preferred approach for intermediate-risk patients with YST.10 The use of radiotherapy for YST remains poorly defined. A study showed that the addition of radiotherapy to chemotherapy had no effect on improving survival.11
Almost all YST patients have an elevated serum AFP level and a negative serum human chorionic gonadotropin level. The serum AFP level is used to track and assess the therapeutic effect and detect clinical recurrence.12 In this case, the serum AFP levels returned to within the normal range after chemotherapy. At present, YST patients lack a specific targeted drug. Some studies suggest that the programmed death-1 (PD-1)/programmed death ligand-1 (PD-L1) pathway may be a novel therapeutic target in GCTs, but it requires further clinical study.13
Although YSTs are clinically aggressive, the 5-year overall survival rates are generally excellent because it is sensitive to chemotherapy. However, hepatoid differentiation of YST indicates a poor prognosis.14 Recently, a retrospective study of 519 patients with GCTs showed that the tumor site, age ≥11 years, and stage IV disease were significant predictors of poor survival. Elevated serum levels of AFP ≥10,000 ng/mL was associated with a worse outcome, while pure YST was associated with better outcomes.15 However, extragonadal YSTs are considered to have a poor prognosis.16
Conclusion
We describe a rare case of YST in the gluteus. Extragonadal YST tends to be ignored because of the lack of abnormalities in the gonads, but it should be considered when patients have characteristic histopathological changes and elevation of serum AFP levels.
Acknowledgments
This study was supported by Program for Shenyang Science Foundation and Natural Science Foundation (No:F15-199-1-34) of Liaoning Province of China (No:L2015598, No:L2015585). Written informed consent was obtained from the guardian for the use of patient data and imaging for this case report.
Disclosure
The authors report no conflicts of interest in this work.
References
Young RH. The yolk sac tumor: reflections on a remarkable neoplasm and two of the many intrigued by it-Gunnar Teilum and Aleksander Talerman-and the bond it formed between them. Int J Surg Pathol. 2014;22(8):677–687. | ||
Mckenney J, Heerema-Mckenney AR. Extragonadal germ cell tumors: a review with emphasis on pathologic features, clinical prognostic variables, and differential diagnostic considerations. Adv Anat Pathol. 2007;14(2):69–92. | ||
Hoei-Hansen CE, Sehested A, Juhler M, et al. New evidence for the origin of intracranial germ cell tumours from primordial germ cells: expression of pluripotency and cell differentiation markers. J Pathol. 2006;209(1):25–33. | ||
Cornejo KM, Frazier L, Lee RS, Kozakewich HP, Young RH. Yolk sac tumor of the testis in infants and children: A clinicopathologic analysis of 33 cases. Am J Surg Pathol. 2015;39(8):1121–1131. | ||
Zynger DL, Mccallum JC, Chunyan L, Chou PM, Yang XJ. Glypican 3 has a higher sensitivity than alpha-fetoprotein for testicular and ovarian yolk sac tumour: immunohistochemical investigation with analysis of histological growth patterns. Histopathology. 2010;56(6):750–757. | ||
Dengfeng C, Shuangping G, Allan RW, Molberg KH, Peng Y. SALL4 is a novel sensitive and specific marker of ovarian primitive germ cell tumors and is particularly useful in distinguishing yolk sac tumor from clear cell carcinoma. Am J Surg Pathol. 2009;33(6):894–904. | ||
Camparo P, Comperat EM. SALL4 is a useful marker in the diagnostic work-up of germ cell tumors in extra-testicular locations. Virchows Arch. 2013;462(3):337–341. | ||
Xiao GQ, Li F, Unger PD, et al. ZBTB16: a novel sensitive and specific biomarker for yolk sac tumor. Mod Pathol. 2016;29(6):591–598. | ||
Paolo C, Sergio C. An up-date on newly discovered immunohistochemical biomarkers for the diagnosis of human testicular germ cell tumors. Histol Histopathol. 2014;29(8):999–1006. | ||
Satoh T, Aoki Y, Kasamatsu T, et al. Administration of standard-dose BEP regimen (bleomycin + etoposide + cisplatin) is essential for treatment of ovarian yolk sac tumour. Eur J Cancer. 2015;51(3):340–351. | ||
Gilligan T. Decision making in a data-poor environment: management of brain metastases from testicular and extragonadal germ cell tumors. J Clin Oncol. 2016;34(4):303–306. | ||
Guo YL, Zhang YL, Zhu JQ. Prognostic value of serum α-fetoprotein in ovarian yolk sac tumors: A systematic review and meta-analysis. Mol Clin Oncol. 2015;3(1):125–132. | ||
Cierna Z, Mego M, Miskovska V, et al. Prognostic value of programmed-death-1 receptor (PD-1) and its ligand 1 (PD-L1) in testicular germ cell tumors. Ann Oncol. 2016;27(2):300–305. | ||
Rittiluechai K, Wilcox R, Lisle J, Everett 1, Wallace HJ 3rd, Verschraegen CF. Prognosis of hepatoid yolk sac tumor in women: what’s up, Doc? Eur J Obstet Gynec Reprod Biol. 2014;175(1): | ||
Lindsay F, Hale JP, Carlos RG, Nicholson JC. Revised risk classification for pediatric extracranial germ cell tumors based on 25 years of clinical trial data from the United Kingdom and United States. J Clin Oncol. 2015;33(2):195–201. | ||
Sano K. Pathogenesis of intracranial germ cell tumors reconsidered. J Neurosurg. 1999;90(2):258–264. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.