")
Back to Journals » OncoTargets and Therapy » Volume 9
Primary renal carcinoid tumor: case report and review of the literature
Authors Li B, Cui T, Ban Z, Luo L, Sun L
Received 17 May 2015
Accepted for publication 2 November 2015
Published 23 February 2016 Volume 2016:9 Pages 741—743
DOI https://doi.org/10.2147/OTT.S88730
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Daniele Santini
Bin Li,1,* Tongyue Cui,2,* Ziqin Ban,3 Lei Luo,1 Lijiang Sun1
1Department of Urology, Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, 2Department of Urology, People’s Hospital of Changle County, Changle, 3Community Health Center, Qingdao, Shandong, People’s Republic of China
*These authors contributed equally to this work
Objective: The aim of this case report is to discuss the clinicopathological features of a patient with a primary renal carcinoid tumor.
Methods: We report on the clinical and pathological information of one case of a patient with a primary renal carcinoid tumor as well as review relative literature.
Results: The patient was diagnosed with a renal tumor when she received physical examination, and exhibited no positive symptoms. The diameter of tumor was 5 cm, the cross surface of the tumor was light yellow and firm, and the central part was soft with hemorrhage and necrosis. Immunohistochemical staining revealed strong and diffuse staining with synaptophysin, chromogranin A, and neuron-specific enolase.
Conclusion: A primary renal carcinoid tumor is extremely rare. Surgical resection is a preferred therapeutic method.
Keywords: renal, carcinoid tumors, parenchyma
Introduction
Carcinoid tumors, which are commonly located in the digestive tract and respiratory system, are rarely found in the genitourinary system. Primary renal carcinoid tumors are extremely rare. As of 2013, there were fewer than 100 cases of primary renal carcinoid tumors reported in English literature.1 We herein report a patient with a primary renal carcinoid tumor and review the related literature.
Case report
A 27-year-old woman was found to have a kidney tumor when she underwent a routine physical examination. The patient exhibited no history of hematuria, back pain, or abdominal mass. She also had no paraneoplastic syndromes, such as fever, hypertension, or a higher erythrocyte sedimentation rate. She had no family history of renal carcinoma and was a nonsmoker.
A high echolocation, high resolution, color Doppler ultrasound revealed a 5.3×6.0 cm tumor in the upper pole of the left kidney. The tumor was well demarcated from the adjacent renal parenchyma and received blood supply.
On computed tomography (CT), the attenuation value of the tumor was approximately +38 HU and +56 HU on routine and enhanced CT, respectively. The contrast-enhanced CT exhibited heterogeneous density and showed that the renal capsule was invaded by the tumor (Figure 1). Calcification was also observed in the tumor. There was no evidence of a tumor in the renal vein, the inferior vena cava, or in the respiratory or digestive systems. The patient was treated with laparoscopic left radical nephrectomy under general anesthesia in our surgical center.
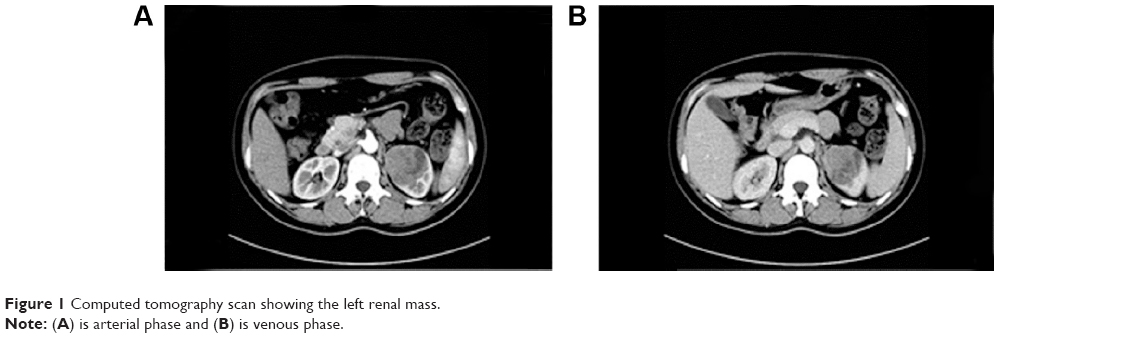 | Figure 1 Computed tomography scan showing the left renal mass. |
Macroscopically, a solid tumor of 5 cm in diameter was found to originate from the renal parenchyma in the upper pole of the left kidney. The cross surface of the tumor was light yellow and firm, and the central part was soft with hemorrhage and necrosis. The tumor had invaded the capsule of the kidney, but the renal vein and left adrenal gland were found to be free of tumor invasion.
Microscopically, the tumor was composed mainly of trabeculae and anastomosing cords of tumor cells within a highly vascularized stroma. The tumor cells possessed eosinophilic cytoplasm with inconspicuous nucleoli, and mitosis was rare (Figure 2). Immunohistochemical staining showed strong and diffuse staining for synaptophysin, chromogranin A, and neuron-specific enolase (Figure 3).
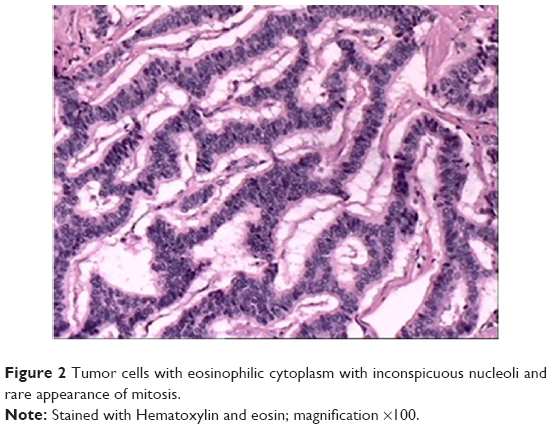 | Figure 2 Tumor cells with eosinophilic cytoplasm with inconspicuous nucleoli and rare appearance of mitosis. |
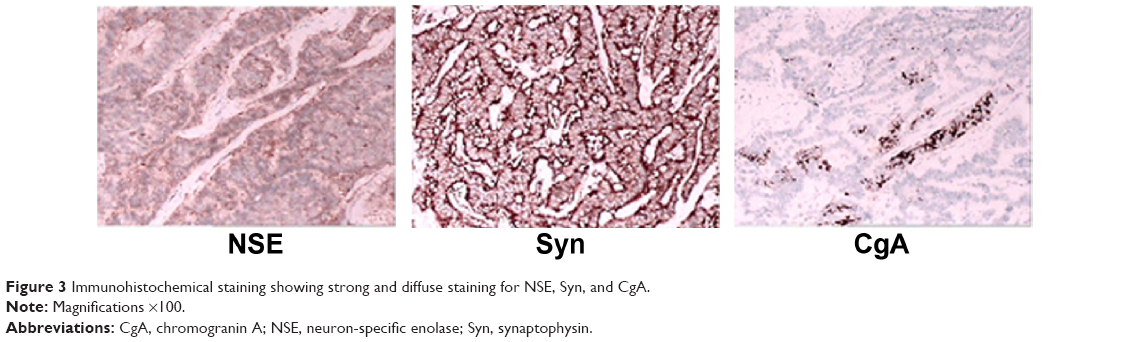 | Figure 3 Immunohistochemical staining showing strong and diffuse staining for NSE, Syn, and CgA. |
The institutional review board approval was not sought as per institutional guidelines around a clinical case report. The patient did provide written informed consent about this case report.
Discussion
Carcinoid tumors are neoplasms arising from specific neuroendocrine cells: amine precursor uptake and decarboxylation cells. It usually occurs in the lungs and gastrointestinal tract and is rarely seen in the genitourinary system. In 1966, Resnick et al reported the first case of a primary renal carcinoid tumor.2 Renal carcinoid tumors exhibit typical characteristics of carcinoid tumors seen elsewhere. The World Health Organization proposed a classification system for renal carcinoid tumors in 2004, which is similar to the classification of carcinoid tumors that arise in other anatomical locations.3 Neuroendocrine cells have not been found in the renal parenchyma, pelvis, or ureter, and the pathogenesis and origin of renal carcinoid tumors are still undefined. The congenital abnormality such as teratoma, horseshoe kidney, and polycystic kidney may play a part in its generation.
Korkmaz et al have summarized 82 renal carcinoid tumors until 2013, and analyzed the epidemiological, clinical, histopathological, therapeutic, and prognostic data.4 We also find no sex or side predilection in renal carcinoid tumors, and the age of patients ranges from 12 to 68 years (median 49 years).5 In our case, the patient was only 27 years old. As the kidneys are located in the retroperitoneal cavity, and many patients were asymptomatic at the time of diagnosis, the diameters of the tumors reported in the published literature range from 1.5 to 30 cm (median 8.4 cm).6 The diameter of our patient’s tumor was approximately 5 cm, but it had projected into the pericalyceal system.
The clinical manifestations of renal carcinoid tumors are atypical, and most patients were found to have a renal tumor on a routine physical examination. Routine auxiliary examinations such as ultrasound, CT, and magnetic resonance imaging cannot distinguish renal carcinoid tumor from renal cell carcinoma. The diagnostic gold standard is the pathological examination of the tumor after surgical resection. Immunohistochemical staining, particularly strong and diffuse staining with synaptophysin, chromogranin A, and/or neuron-specific enolase supports the diagnosis. In our case, the ultrasound result shows well-defined and high echogenic mass in the left kidney, and arterial signal can also be detected in the mass. The contrast-enhanced CT reveals heterogeneous density in mass, no enlarged lymph nodes were found.
The primary treatment of localized primary renal carcinoid tumors is radical nephrectomy. Nephron sparing surgery is an alternative depending on the diameter and location of the tumor. Resection of regional lymph nodes enables the pathological TNM staging of the tumor, but has no beneficial impact on the patient’s overall survival or progression-free survival.1 The summarized data which testify the benefit of radical nephrectomy or cytoreductive surgery in metastatic neuroendocrine tumors, are not available. Gedaly et al7 reported a case with isolated hepatic metastatic tumor. The patient received radical nephrectomy and formal left hepatectomy. No evidence of tumor recurrence was found in the patient 12 months after the operation. Cisplatin, fluorouracil and cyclophosphamide have been used in advanced carcinoid. As this type of renal carcinoid tumor is rare the effect of chemotherapy on overall survival of metastatic renal carcinoids has not been shown to date.
Conclusion
Due to the rarity of the tumor, there is a lack of papers describing long-term follow-up after treatment, and any prognostic factors are still uncertain.8 However, some histological and clinical parameters may play an important part in evaluating the prognosis of primary renal carcinoid tumors. For patients above 40 years this was found to be significantly associated with a more severe presentation and poorer outcome. Tumors with a diameter of no more than 4 cm and limited to the renal parenchyma may indicate a lower possibility of metastasis and a better prognosis. Serum 5-hydroxytryptamine, uric 5-hydroxyindoleacetic acid, and octreotide scintigraphy may show some positive value in diagnosis and follow-up. A mitotic rate higher than 2/10 high-power fields may also indicate a worse prognosis. Periodic reviews of our patient 12 months after surgery have revealed no sign of recurrence. Although, as metastases may appear as long as 5 years after the surgery,9 long-term follow-up is necessary.
Disclosure
The authors report no conflicts of interest in this work.
References
Korkmaz T, Seber S, Yavuzer D, Gumus M, Turhal NS. Primary renal carcinoid: treatment and prognosis. Crit Rev Oncol Hematol. 2013;87(3):256–264. | ||
Resnick ME, Unterberger H, McLoughlin PT. Renal carcinoid producing the carcinoid syndrome. Med Times. 1966;94(8):895–896. | ||
Eble JN, Sauter G, Epstein JL. World Health Organization classification of tumors. In: Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Volume 7. 3rd ed. Lyon: IARC Press; 2004:81–82. | ||
Korkmaz T, Seber S, Yavuzer D, Gumus M, Turhal NS. Primary renal carcinoid: treatment and prognosis. Crit Rev Oncol Hematol. 2013;87: 256–264. | ||
Canacci AM, MacLennan GT. Carcinoid tumor of the kidney. J Urol. 2008;180(5):2193. | ||
Romero FR, Rais-Bahrami S, Permpongkosol S, et al. Primary carcinoid tumors of the kidney. J Urol. 2006;176(6 Pt 1):2359–2366. | ||
Gedaly R, Jeon H, Johnston TD, McHugh PP, Rowland RG, Ranjan D.Surgical treatment of a rare primary renal carcinoid tumor with livermetastasis. World Journal of Surgical Oncology. 2008;22;6:41. | ||
Yoo J, Park S, Jung Lee H, Jin Kang S, Kee Kim B. Primary carcinoid tumor arising in a mature teratoma of the kidney: a case report and review of the literature. Arch Pathol Lab Med. 2002;126(8):979–981. | ||
Hasteh F, Pu R, Michael CW. A metastatic renal carcinoid tumor presenting as breast mass: a diagnostic dilemma. Diagn Cytopathol. 2007;35(5):306–310. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.