")
Back to Journals » OncoTargets and Therapy » Volume 10
Primary pancreatic lymphoma: two case reports and a literature review
Received 4 September 2016
Accepted for publication 6 December 2016
Published 20 March 2017 Volume 2017:10 Pages 1687—1694
DOI https://doi.org/10.2147/OTT.S121521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ingrid Espinoza
Lili Yu,1,2,* Yajun Chen,1,2,* Ligang Xing2
1School of Medicine and Life Sciences, Shandong Academy of Medical Sciences, University of Jinan, 2Department of Radiation Oncology, Shandong Cancer Hospital Affiliated to Shandong University, Jinan, People’s Republic of China
*These authors contributed equally to this work
Abstract: Primary pancreatic lymphoma (PPL) is an extremely rare disease, with only a few cases reported in the literature. Clinical manifestations of PPL are often nonspecific and may mimic other pancreatic diseases. Because of the limited experience of PPL, clinicopathological features, differential diagnosis, optimal therapy, and outcomes are not well defined. We described two cases diagnosed as PPL and confirmed by histological examination and immunohistochemical analysis. Case 1 was a young man with obstructive jaundice and upper abdominal malaise mimicking a pancreatic adenocarcinoma. A computed tomography (CT) scan revealed a diffuse heterogeneous mass in the head of the pancreas along with dilated bile ducts, no dilated pancreatic duct, no liver or splenic involvement, or evident retroperitoneal adenopathies. The patient underwent a pancreatico-duodenectomy, and the postoperative histopathology confirmed diffuse large B-cell non-Hodgkin lymphoma. Postoperatively, he received six courses of the CHOP regimen (cyclophosphamide, doxorubicin, vincristine, and prednisolone). Case 2 was an older man with left flank pain. A CT confirmed a mass with irregular margins at the tail of the pancreas and the hilum of the spleen. The mass was heterogeneous, with no clear boundary between lesions, spleen, stomach, and pancreas, with nearby blood vessels wrapped around it, and multiple enlarged lymph nodes in the abdominal cavity. A CT-guided biopsy was performed. The immunohistological findings of the specimen revealed a diffuse large B-cell lymphoma. The size of the tumor was significantly reduced after four cycles of the CHOP chemotherapy regimen. These two cases were different in clinical manifestation, location, and treatment. We reviewed the literature and discussed the clinicopathological features, differential diagnosis, optimal therapy, and outcomes of this neoplasm.
Keywords: primary pancreatic lymphoma, pancreatic malignant tumor, diffuse large B-cell lymphoma, diagnosis, treatment policy, survival and prognosis
Introduction
Primary pancreatic lymphoma (PPL) is an extremely rare entity, accounts for <0.5% of malignant pancreatic tumors,1–3 and is difficult to preoperatively diagnose even with various modalities. Clinically, the presentation of patients with PPL arising from the pancreatic head can often overlap that of patients with pancreatic adenocarcinoma and is difficult to radiographically distinguish. However, due to a much better prognosis for PPL, proper diagnosis is very important. To confirm the diagnosis of PPL, histological analysis is required. Management of PPL involves chemotherapy and radiotherapy,4 yet uncertainty regarding the diagnosis of a mass in the pancreatic head usually results in surgical resection as well. For patients with symptoms caused by obstruction of the biliary tract, surgical intervention is effective.4 Furthermore, the combination of surgical treatment and adjuvant chemotherapy has a better survival benefit than chemotherapy alone.5,6 Despite these efforts, surgical intervention is still controversial. We present two cases of PPL that were different in clinical manifestation, location, and treatment, and reviewed this neoplasm. Written informed consent was obtained from both patients to publish the case reports and any accompanying images.
Case 1
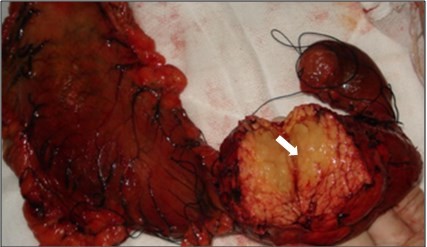
A 32-year-old man was admitted to our hospital after feeling ill for 10 days with complaints of jaundice and upper abdominal malaise. A physical examination revealed jaundice. Organomegaly or lymphadenopathy was not detected. Laboratory test results revealed indirect hyperbilirubinemia (total bilirubin: 154.1 mg/dL, conjugated bilirubin: 90.9 mg/dL). The liver function enzymes showed that alanine transaminase, aspartate transaminase, and lactate dehydrogenase (LDH) levels were increased to 465, 268, and 248 U/L, respectively. The β2-microglobulin level of the patient was 1.89 mg/L. Both his carbohydrate antigen 19-9 (CA 19-9) and carcinoembryonic antigen (CEA) were within the normal limits. Abdominal computed tomography (CT) showed an irregular enlargement of the pancreas and a 3.0×3.5 cm heterogeneous enhanced mass arising from the pancreatic head (Figure 1). The main pancreatic duct was of normal caliber, and there was no connection between the mass and the pancreatic duct. The bile duct was in distention because of the compression by the mass in the head of the pancreas. No focal enhanced mass was observed in the liver, and there were no enlarged lymph nodes in the abdominal cavity. The initial diagnosis was pancreatic head cancer or a neuroendocrine tumor of the pancreas. The patient underwent a pancreatico-duodenectomy. Macroscopically, the mass had arisen from the pancreatic head and was limited to the pancreas. The cut surface of the tumor was yellowish (Figure 2). A histological report confirmed diffuse large B-cell lymphoma (Figure 3A). Immunohistochemical staining showed the cells to be positive for CD20 and CD79a (Figure 3B and C) and negative for CD3, CD10, and BCL-2. The histology also confirmed a proliferative index of over 50%–60% (Figure 3D). Finally, based on pathological and immunohistochemical findings, the tumor was diagnosed as PPL. Postoperatively, the patient received six cycles of treatment with doxorubicin, cyclophosphamide, vincristine, and prednisolone (CHOP regimen). He is still alive and asymptomatic after 16 months of follow-up. The last positron emission tomography-CT did not detect any signs of disease recurrence (Figure 4).
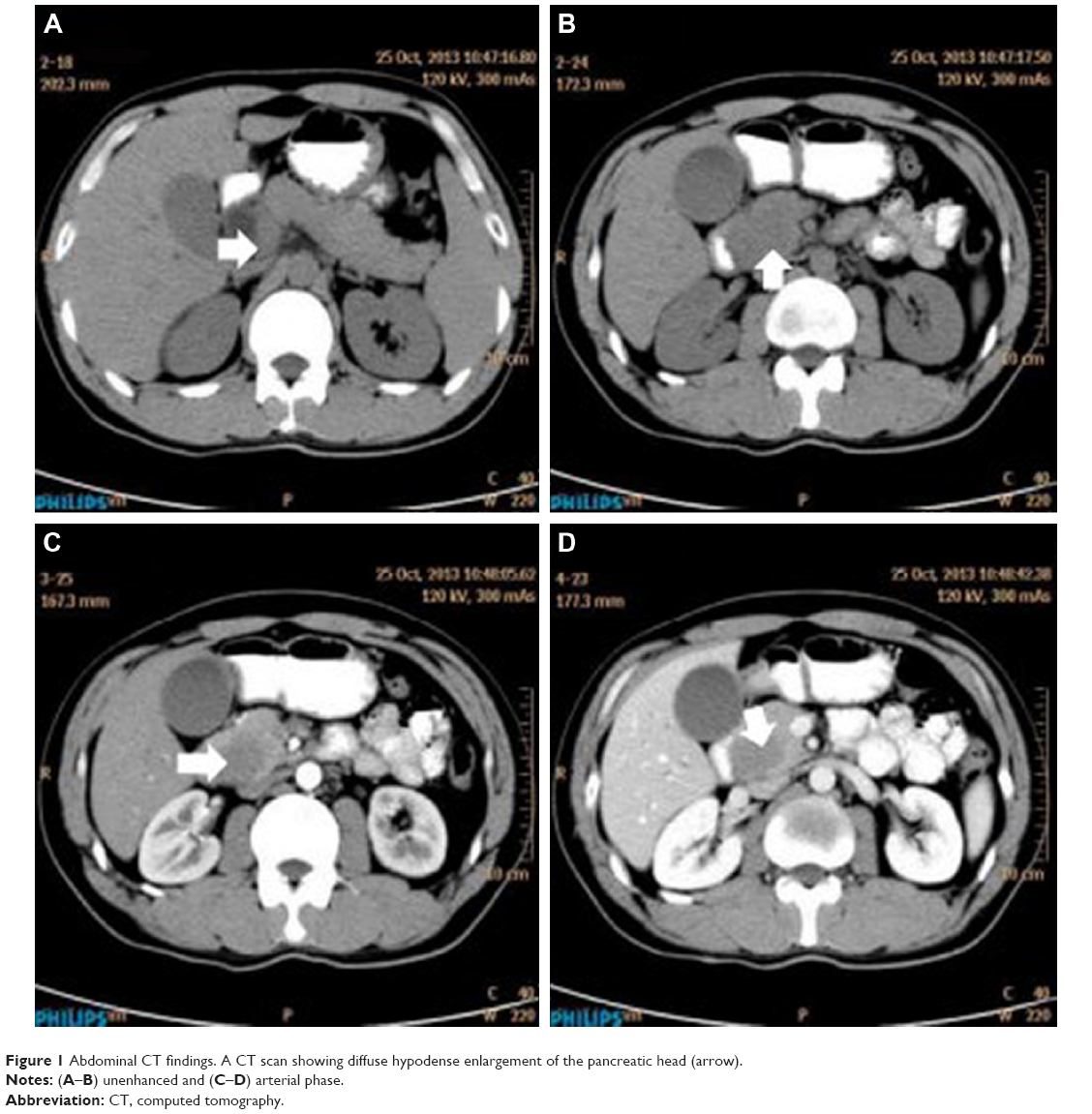 | Figure 1 Abdominal CT findings. A CT scan showing diffuse hypodense enlargement of the pancreatic head (arrow). |
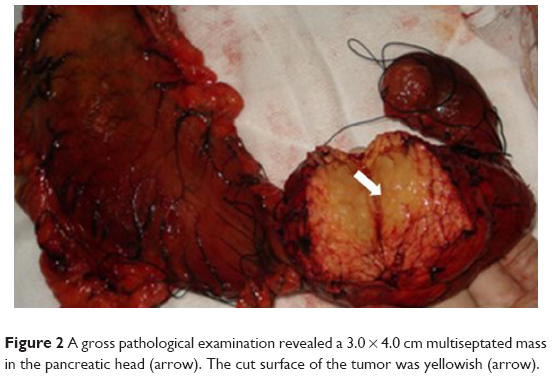 | Figure 2 A gross pathological examination revealed a 3.0 × 4.0 cm multiseptated mass in the pancreatic head (arrow). The cut surface of the tumor was yellowish (arrow). |
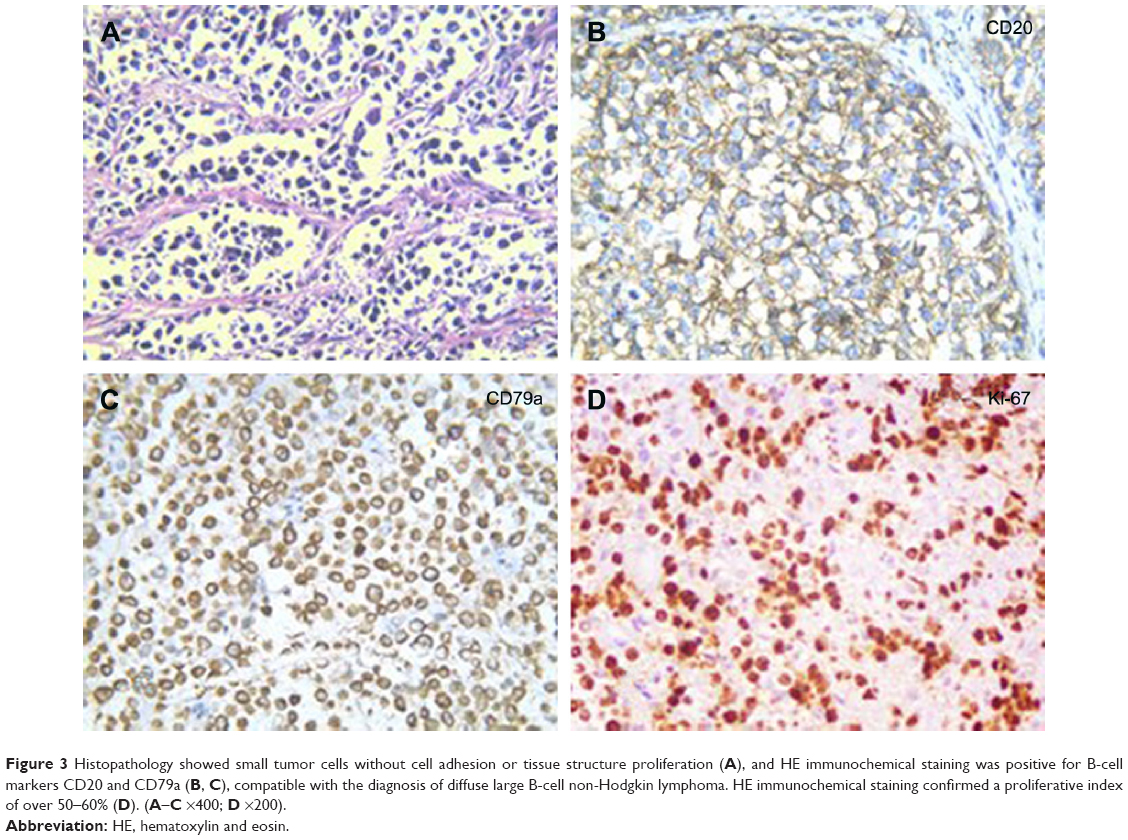 | Figure 3 Histopathology showed small tumor cells without cell adhesion or tissue structure proliferation (A), and HE immunochemical staining was positive for B-cell markers CD20 and CD79a (B, C), compatible with the diagnosis of diffuse large B-cell non-Hodgkin lymphoma. HE immunochemical staining confirmed a proliferative index of over 50–60% (D). (A–C ×400; D ×200). |
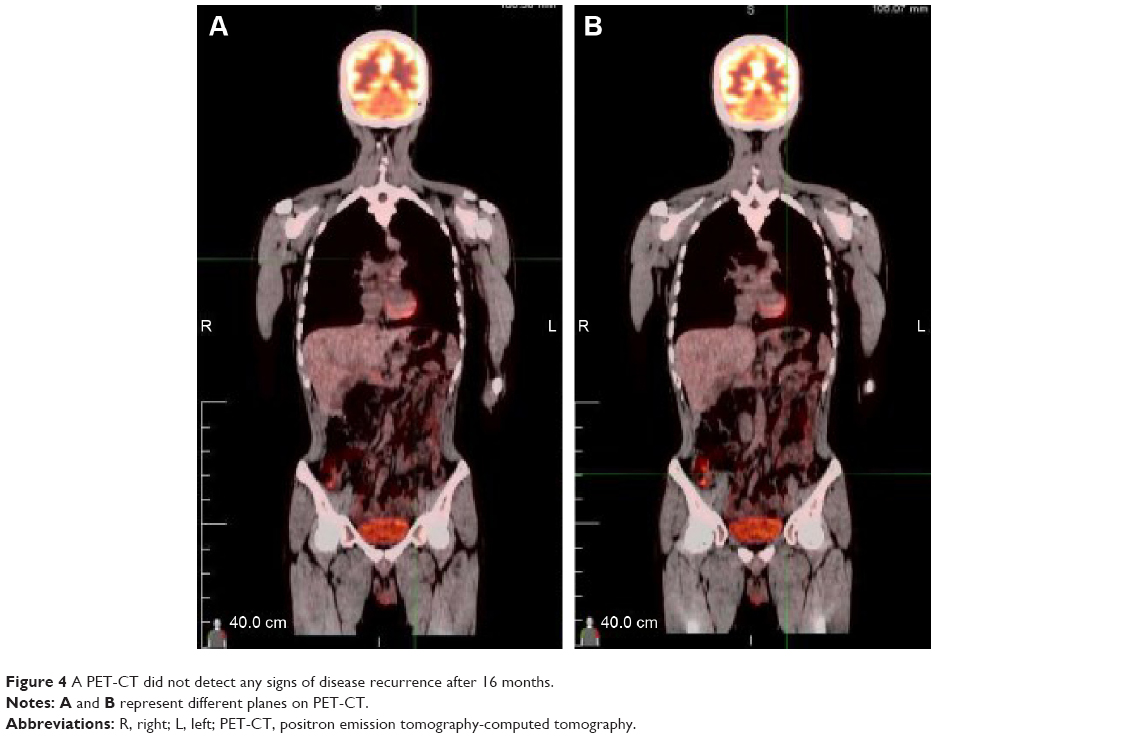 | Figure 4 A PET-CT did not detect any signs of disease recurrence after 16 months. |
Case 2
A 62-year-old man presented with a 1-month history of left flank pain. He presented to another hospital and had a CT which revealed a lesion in the tail of the pancreas. He was then transferred to our institution for further workup and evaluation. His past medical history was unremarkable. A physical examination revealed an upper abdominal mass but did not detect peripheral lymphadenopathy. His liver function enzymes were normal. However, LDH was 236 U/L. The β2-microglobulin level of the patient was 2.37 mg/L. Serum tumor markers CA 19-9, CEA, and chromogranin A were normal. An abdominal CT scan confirmed that a mass with irregular margins was in the hilum of the spleen. The mass was heterogeneous, with no clear boundary between lesions, spleen, stomach, and pancreas, nearby blood vessels were wrapped around it, and there were multiple enlarged lymph nodes in the abdominal cavity (Figure 5A and B). A CT-guided biopsy was taken from the mass. The pathological evaluation of the CT-guided biopsy revealed that the tumor was CD20-positive (Figure 6B) and CD3- and pancytokeratin-negative, with a high Ki-67 proliferation index (80%) (Figure 6C), and findings are consistent with diffuse large B-cell lymphoma (Figure 6A). Due to the ill-defined mass, it was difficult to judge whether the tumor originated from the pancreas or spleen. However, a CHOP chemotherapy regimen is the preferred regimen for diffuse large B-cell lymphoma regardless of the location of the primary site. He consented to chemotherapy with the CHOP regimen. Lesions were significantly reduced after two cycles of treatment. A CT confirmed that the lymph nodes of the splenic hilum are significantly narrowed. The spleen was normal, and a low-density lesion at the tail of the pancreas was still present. Under these circumstances, we confirmed our diagnosis of PPL arising from the pancreatic tail. The size of the tumor was significantly reduced after four cycles of the CHOP chemotherapy regimen (Figure 7), and further follow-up is being carried out.
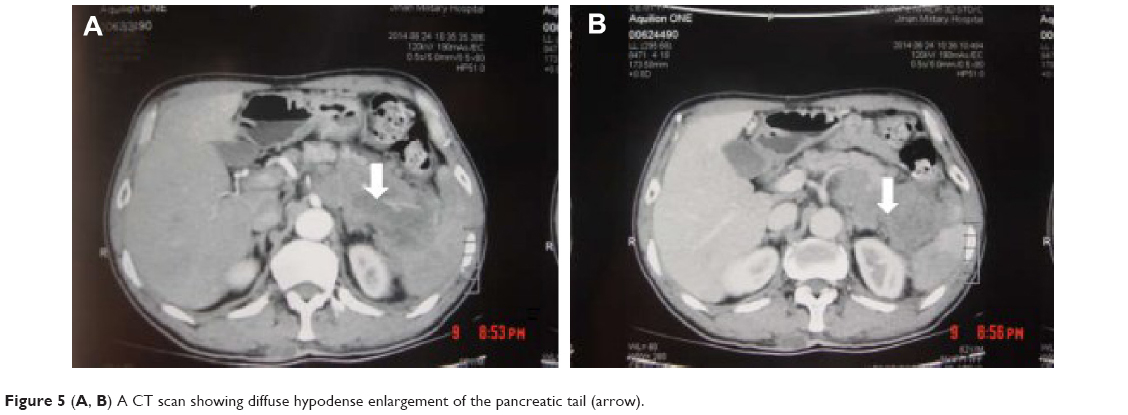 | Figure 5 (A, B) A CT scan showing diffuse hypodense enlargement of the pancreatic tail (arrow). |
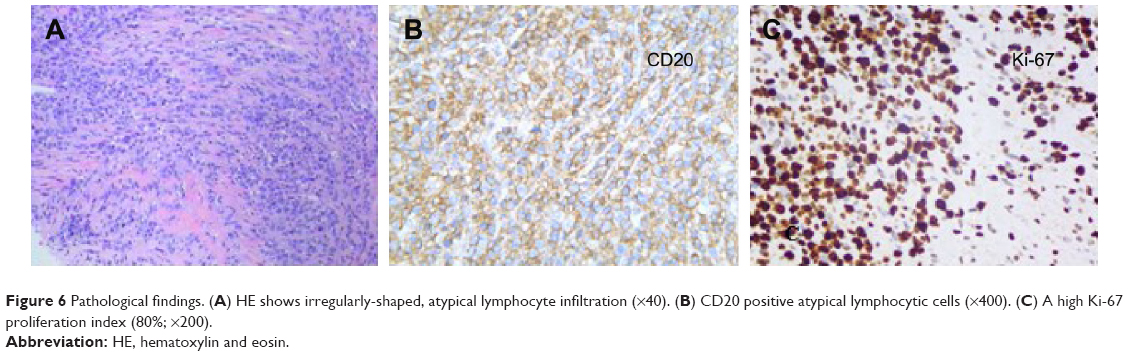 | Figure 6 Pathological findings. (A) HE shows irregularly-shaped, atypical lymphocyte infiltration (×40). (B) CD20 positive atypical lymphocytic cells (×400). (C) A high Ki-67 proliferation index (80%; ×200). |
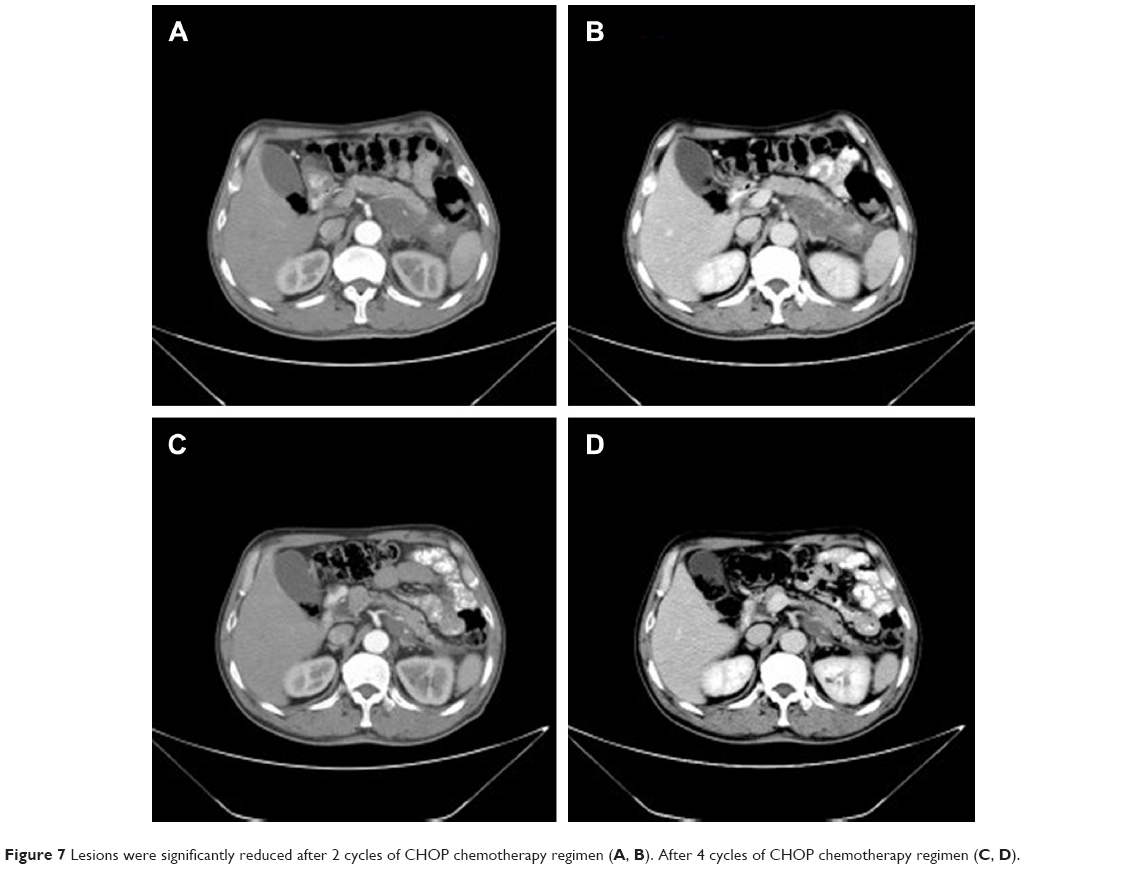 | Figure 7 Lesions were significantly reduced after 2 cycles of CHOP chemotherapy regimen (A, B). After 4 cycles of CHOP chemotherapy regimen (C, D). |
Discussion
PPL is extremely rare, comprising 1% of extra-nodal lymphomas and 0.5% of malignant pancreatic tumors.1–3,7 From our review of the English literature (Table 1),4,5,8–31 the most common lymphoma variant in the pancreas is diffuse large B-cell lymphoma, accounting in one study for as much as 80% of all the PPL cases.29 Abdominal pain, jaundice, acute pancreatitis, small bowel obstruction, and diarrhea are the most commonly presenting symptoms,7,30 but these are nonspecific and overlap those of pancreatic adenocarcinoma. Interestingly, the common presentations of non-Hodgkin lymphoma, such as fever, chills, and night sweats, are rare (2%) in PPL.7 The widely accepted criteria for diagnosis of PPL were established by Dawson et al.32 They include a dominant mass in the pancreas, the absence of superficial or mediastinal lymphadenopathy on chest imaging, a normal leukocyte count in the peripheral blood, and the absence of hepatic or splenic involvement. Most PPLs occur in the pancreatic head, although they may also be found in other parts of the pancreas.1,2,7 In our patients, case 1 presented with jaundice and upper abdominal malaise and the mass was located in the head of the pancreas, whereas case 2 presented with left flank pain and the mass was located at the tail of the pancreas.
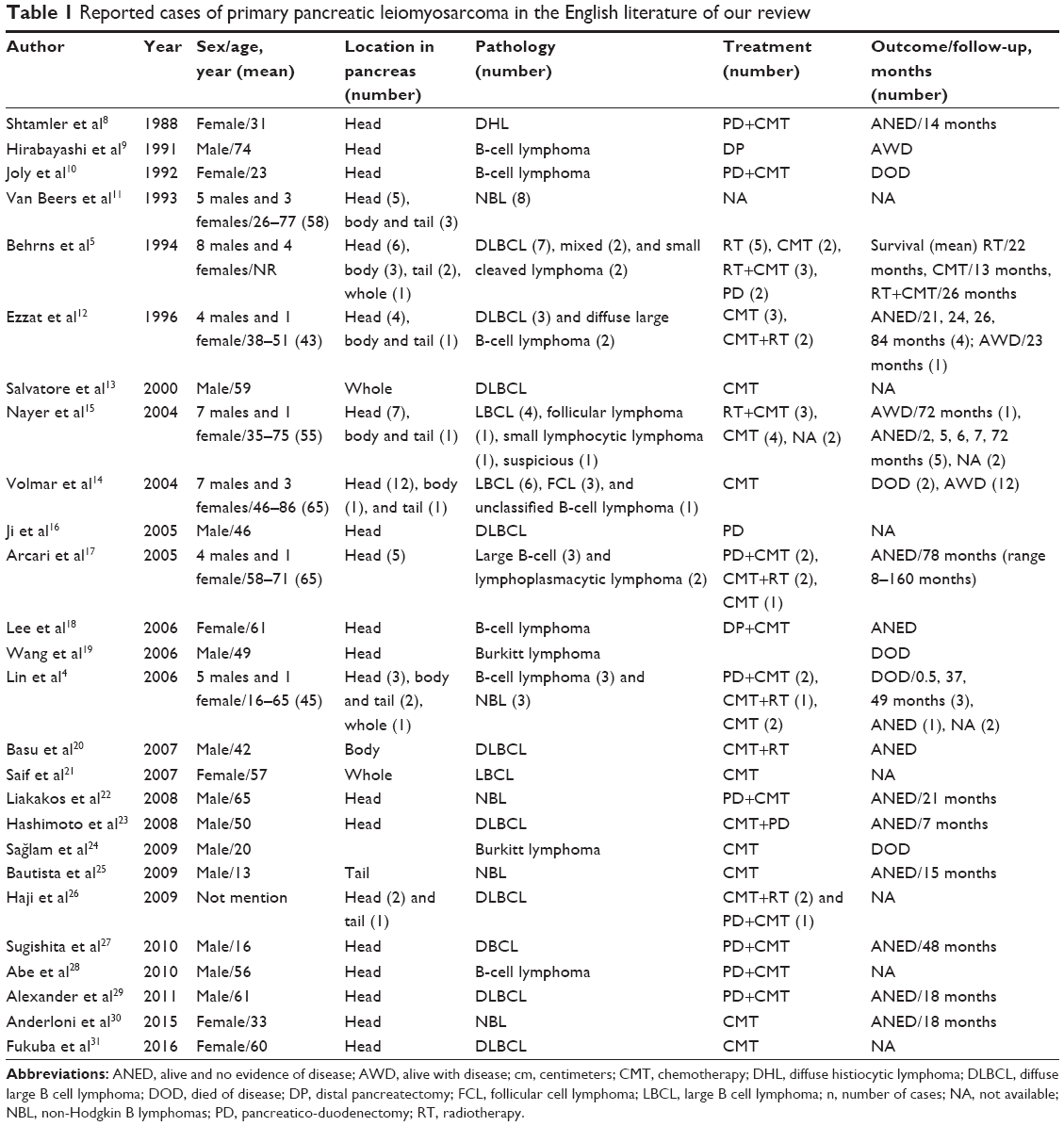 | Table 1 Reported cases of primary pancreatic leiomyosarcoma in the English literature of our review |
Nonspecific biochemical markers can help a physician to make an accurate diagnosis of PPL. CA 19-9, a serum tumor marker, is generally increased in patients with pancreatic adenocarcinoma,33 but it is rarely raised in patients with PPL. Lin et al reported that 80% of the patients with pancreatic adenocarcinoma have a high CA 19-9 level, whereas in patients with PPL, the serum CA 19-9 level is normal or only slightly elevated unless a biliary obstruction is presented.4 LDH and β2-microglobulin levels are thought to be elevated in lymphoproliferative disorders, but they are not necessarily elevated in PPL.34 However, a higher β2-microglobulin and LDH level are indicators of a poor prognosis for PPL patients.35
Various imaging modalities have been used in the evaluation of pancreatic mass lesions. PPL can be divided into two morphological groups on CT: the pancreas has a similar appearance to pancreatitis, with an infiltrative mass and peripancreatic fat stranding or the pancreas seems to have a well-circumscribed tumor.22 Some radiological findings that point to PPL (instead of adenocarcinoma) include a bulky, localized pancreatic head tumor; no significant pancreatic duct dilatation; no enlarged lymph nodes below the level of the renal veins; and no retroperitoneal, hepatic, or splenic involvement.36 Furthermore, the presence of calcification or necrosis can rule out PPL.7 With regard to the results of endoscopic ultrasonography (EUS), a retrospective analysis by Ramesh et al2 determined that none of the patients showed features of pancreatic ductal dilation. Surrounding lymphadenopathy is common with any lymphoma. However, this feature would not exclude adenocarcinoma. Concerning endoscopic retrograde cholangiopancreatography (ERCP) findings, moderate-to-severe ductal dilation that is often seen in pancreatic adenocarcinoma has not been reported in patients with PPL.36 Therefore, patients with moderate-to-severe ductal dilation may help to exclude the diagnosis of PPL. Magnetic resonance imaging is quite nonspecific.36
Certain clinical and imaging characteristics may help to suggest a diagnosis of PPL, but they are both nonspecific. Thus, pathology is necessary. Initially, the biopsy can be performed by fine-needle aspiration (FNA). However, EUS-guided FNA was shown to improve diagnostic accuracy, and the sensitivity of diagnosing PPL was a significant increase compared with that of FNA alone.15,37,38 CT-guided biopsy, which is minimally invasive, is another option for obtaining specimens.13 Alternatively, a laparoscopy or laparotomy can be performed to biopsy the pancreatic lesion or lymph nodes. In our cases, diagnoses were based on CT-guided biopsy and surgical biopsy.
The strategy for PPL consists of surgery, chemotherapy, radiotherapy, or a combination of them. Most chemotherapy consists of CHOP and sometimes rituxan plus CHOP (R-CHOP). The majority of patients with PPL receiving only chemotherapy can achieve long-term disease remission.39 However, Behrns et al5 reported that it is difficult to obtain long-term survival by chemotherapy alone. Battula et al6 also reported that the 5-year survival rate of PPL treated with current chemotherapy was inferior to the combination of surgery and adjuvant chemotherapy. Surgical intervention, however, is still controversial. It has been proven that pancreatic resection alone does not improve the survival rate of PPL.22 For patients with jaundice, choledochojejunostomy is considered an effective treatment option.4,36,40 But, in recent years, various nonoperative strategies can relieve this complication with a high rate of success. The role of radiation therapy has also not yet been defined except as an adjuvant to chemotherapy. A study by Behrns et al showed some evidence that an initial surgical resection, when coupled with chemotherapy and radiotherapy, was associated with increased long-term survival of PPL.5 In case 1, the patient was not diagnosed with PPL preoperatively, but since there was obstruction of the biliary tract, a pylorus-preserving pancreatico-duodenectomy was performed. His postoperative course was uneventful, and six cycles of CHOP were performed with no signs of recurrence after 16 months. In case 2, the size of the tumor was significantly reduced after four cycles of CHOP chemotherapy. Therefore, it is important to choose the appropriate treatment depending on the disease progression and the patient’s condition.
Conclusion
We presented two rare cases of PPL. Certain clinical and imaging characteristics as well as biochemical markers are not specific for PPL. Since patients with PPL have a much better prognosis than those with adenocarcinoma and the treatment widely differs from those of other pancreatic neoplasms, a definite diagnosis should be sought based on the histological examination. Combined therapy may be the optimal treatment approach for PPL, but it needs further evaluation.
Acknowledgments
The authors thank Kai Cui for his valuable discussion. This work was supported by Natural Science Foundation of Shandong Province (Grant No ZR2015HZ004).
Disclosure
The authors report no conflicts of interest in this work.
References
Mishra MV, Keith SW, Shen X, Bar AV, Champ CE, Biswas T. Primary pancreatic lymphoma: a population-based analysis using the SEER program. Am J Clin Oncol. 2013;36(1):38–43. | ||
Ramesh J, Hebert-Magee S, Kim H, Trevino J, Varadarajulu S. Frequency of occurrence and characteristics of primary pancreatic lymphoma during endoscopic ultrasound guided fine needle aspiration: a retrospective study. Dig Liver Dis. 2014;46(5):470–473. | ||
Baylor SM, Berg JW. Cross-classification and survival characteristics of 5,000 cases of cancer of the pancreas. J Surg Oncol. 1973;5(4):335–358. | ||
Lin H, Li SD, Hu XG, Li ZS. Primary pancreatic lymphoma: report of six cases. World J Gastroenterol. 2006;12(31):5064–5067. | ||
Behrns KE, Sarr MG, Strickler JG. Pancreatic lymphoma: is it a surgical disease. Pancreas. 1994;9(5):662–667. | ||
Battula N, Srinivasan P, Prachalias A, Rela M, Heaton N. Primary pancreatic lymphoma: diagnostic and therapeutic dilemma. Pancreas. 2006;33(2):192–194. | ||
Saif MW. Primary pancreatic lymphomas. JOP. 2006;7(3):262–273. | ||
Shtamler B, Bickel A, Manor E, Ben Shahar M, Kuten A, Suprun H. Primary lymphoma of the head of the pancreas. J Surg Oncol. 1988;38(1):48–51. | ||
Hirabayashi K, Kawakami H, Aizawa T, et al. A case of the primary pancreatic lymphoma. Rinsho Ketsueki. 1991;32(4):414–418. | ||
Joly I, David A, Payan MJ, Sahel J, Sarles H. A case of primary non-Hodgkin lymphoma of the pancreas. Pancreas. 1992;7(1):118–120. | ||
Van Beers B, Lalonde L, Soyer P, et al. Dynamic CT in pancreatic lymphoma. J Comput Assist Tomogr. 1993;17(1):94–97. | ||
Ezzat A, Jamshed A, Khafaga Y, et al. Primary pancreatic non-Hodgkin’s lymphomas. J Clin Gastroenterol. 1996;23(2):109–112. | ||
Salvatore JR, Cooper B, Shah I, Kummet T. Primary pancreatic lymphoma: a case report, literature review, and proposal for nomenclature. Med Oncol. 2000;17(3):237–247. | ||
Volmar KE, Routbort MJ, Jones CK, Xie HB. Primary pancreatic lymphoma evaluated by fine-needle aspiration: findings in 14 cases. Am J Clin Pathol. 2004;121(6):898–903. | ||
Nayer H, Weir EG, Sheth S, Ali SZ. Primary pancreatic lymphomas: a cytopathologic analysis of a rare malignancy. Cancer. 2004;102(5):315–321. | ||
Ji Y, Kuang TT, Tan YS, Chen Y, Zeng HY, Jin DY. Pancreatic primary lymphoma: a case report and review of the literature. Hepatobiliary Pancreat Dis Int. 2005;4(4):622–626. | ||
Arcari A, Anselmi E, Bernuzzi P, et al. Primary pancreatic lymphoma. Report of five cases. Haematologica. 2005;90(2):ECR09. | ||
Lee MK, Jeon SW, Lee YD, et al. A case of primary pancreatic non-Hodgkin’s lymphoma. Korean J Intern Med. 2006;21(2):123–126. | ||
Wang YJ, Jeng CM, Wang YC, Chang PP, Wang TH. Primary pancreatic Burkitt’s lymphoma mimicking carcinoma with obstructive jaundice and very high CA19-9. Eur J Gastroenterol Hepatol. 2006;18(5):537–540. | ||
Basu A, Patil N, Mohindra P, et al. Isolated non-Hodgkin’s lymphoma of the pancreas: case report and review of literature. J Cancer Res Ther. 2007;3(4):236–239. | ||
Saif MW, Khubchandani S, Walczak M. Secondary pancreatic involvement by a diffuse large B-cell lymphoma presenting as acute pancreatitis. World J Gastroenterol. 2007;13(36):4909–4911. | ||
Liakakos T, Misiakos EP, Tsapralis D, Nikolaou I, Karatzas G, Macheras A. A role for surgery in primary pancreatic B-cell lymphoma: a case report. J Med Case Rep. 2008;2:167. | ||
Hashimoto M, Umekita N, Noda K. Non-Hodgkin lymphoma as a cause of obstructive jaundice with simultaneous extrahepatic portal vein obstruction: a case report. World J Gastroenterol. 2008;14(25):4093–4095. | ||
Sağlam M, Yilmaz MI, Mas MR, et al. A case of pancreatic Burkitt lymphoma: radiological findings. Diagn Interv Radiol. 2009;15(1):39–42. | ||
Bautista F, Moreno L, Andrés MM, Fernández-Navarro JM, Fernández-Sanmartín M, Verdeguer A. Abdominal pain as the first manifestation of primary pancreatic lymphoma. J Pediatr Hematol Oncol. 2009;31(3):222–223. | ||
Haji AG, Sharma S, Majeed KA, Vijaykumar DK, Pavithran K, Dinesh M. Primary pancreatic lymphoma: report of three cases with review of literature. Indian J Med Paediatr Oncol. 2009;30(1):20–23. | ||
Sugishita H, Watanabe Y, Yamamoto Y, et al. Primary pancreatic lymphoma: the role of surgical treatment. Case Rep Gastroenterol. 2010;4(1):104–110. | ||
Abe Y, Tamura K, Sakata I, et al. Unique intense uptake demonstrated by (18)F-FDG positron emission tomography/computed tomography in primary pancreatic lymphoma: a case report. Oncol Lett. 2010;1(4):605–607. | ||
Alexander RE, Nakeeb A, Sandrasegaran K, et al. Primary pancreatic follicle center-derived lymphoma masquerading as carcinoma. Gastroenterol Hepatol (N Y). 2011;7(12):834–838. | ||
Anderloni A, Genco C, Ballarè M, Carmagnola S, Battista S, Repici A. A case of primary pancreatic non-Hodgkin B-cell lymphoma mimicking autoimmune pancreatitis. J Gastrointestin Liver Dis. 2015;24(2):245–248. | ||
Fukuba N, Moriyama I, Ishihara S, et al. Primary pancreatic malignant lymphoma diagnosed from endoscopic ultrasound-guided fine-needle aspiration findings. Intern Med. 2016;55(1):31–35. | ||
Dawson IM, Cornes JS, Morson BC. Primary malignant lymphoid tumours of the intestinal tract. Report of 37 cases with a study of factors influencing prognosis. Br J Surg. 1961;49:80–89. | ||
Berger AC, Garcia M, Hoffman JP, et al. Postresection CA 19-9 predicts overall survival in patients with pancreatic cancer treated with adjuvant chemoradiation: a prospective validation by RTOG 9704. J Clin Oncol. 2008;26(36):5918–5922. | ||
Du X, Zhao Y, Zhang T, et al. Primary pancreatic lymphoma: a clinical quandary of diagnosis and treatment. Pancreas. 2011;40(1):30–36. | ||
Chen W, Luo RC, Fan WW, Ma SD. Clinical value of combined detection of LDH, TPS, CEA and beta2-MG in patients with non-Hodgkin’s lymphoma. Nan Fang Yi Ke Da Xue Xue Bao. 2006;26(2):227–228, 230. | ||
Merkle EM, Bender GN, Brambs HJ. Imaging findings in pancreatic lymphoma: differential aspects. AJR Am J Roentgenol. 2000;174(3):671–675. | ||
Khashab M, Mokadem M, DeWitt J, et al. Endoscopic ultrasound-guided fine-needle aspiration with or without flow cytometry for the diagnosis of primary pancreatic lymphoma – a case series. Endoscopy. 2010;42(3):228–231. | ||
McCauley AM, Gottlieb KT. Primary pancreatic lymphoma coexisting with chronic lymphocytic leukemia: EUS findings. Gastrointest Endosc. 2008;68(1):188–189. | ||
Tuchek JM, De Jong SA, Pickleman J. Diagnosis, surgical intervention, and prognosis of primary pancreatic lymphoma. Am Surg. 1993;59(8):513–518. | ||
Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6(10):715–727. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.