Back to Journals » OncoTargets and Therapy » Volume 7
Primary concomitant EGFR T790M mutation predicted worse prognosis in non-small cell lung cancer patients
Authors Li H, Hu H, Wang R, Pan Y, Wang L, Li Y, Zhang Y, Ye T, zhang Y, Li B, Shen L, Sun Y, Chen H
Received 5 January 2014
Accepted for publication 1 February 2014
Published 3 April 2014 Volume 2014:7 Pages 513—524
DOI https://doi.org/10.2147/OTT.S60122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Hang Li,1,2,* Haichuan Hu,1,2,* Rui Wang,1,2 Yunjian Pan,1,2 Lei Wang,1,2 Yuan Li,2,3 Yang Zhang,1,2 Ting Ye,1,2 Yiliang Zhang,1,2 Bin Li,1,2 Lei Shen,2,3 Yihua Sun,1,2 Haiquan Chen1,2
1Department of Thoracic Surgery, 2Department of Oncology, 3Department of Pathology, Shanghai Medical College, Fudan University Shanghai Cancer Center, Shanghai, People's Republic of China
*These authors contributed equally to this work
Purpose: We performed this analysis to improve the understanding of the clinicopathological characteristics and clinical outcome of non-small cell lung cancer (NSCLC) patients harboring the primary epidermal growth factor receptor (EGFR) T790M mutation along with activating EGFR mutation.
Methods: Resected tumors from 1903 NSCLC patients were analyzed for mutation in EGFR, as well as KRAS (Kirsten rat sarcoma viral oncogene homolog), BRAF (v-raf murine sarcoma viral oncogene homolog B), HER2 (human epidermal growth factor 2), PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha), and EML4 (echinoderm microtubule associated protein like 4)-ALK (anaplastic lymphoma receptor tyrosine kinase) fusion. Fluorescence in situ hybridization was performed to define EGFR and c-MET (met proto-oncogene gene amplification. Expression of PIK3CA and p-Akt (phosphorylated protein kinase B) were tested using immunohistochemistry. Clinical and pathological data, including sex, age at diagnosis, stage, tumor differentiation, smoking history, histological subtype, relapse-free and overall survival, were further analyzed.
Results: In all, 16 NSCLC patients were found to harbor primary EGFR T790M mutation, including 14 adenocarcinomas and two adenosquamous carcinomas, accounting for 2.04% of all the EGFR mutant cases and 0.84% of the total. No c-MET amplification was found to coexist with primary EGFR T790M. Fewer EGFR copy-number variations were found in samples harboring EGFR T790M mutations compared with those in patients with exon 19 deletions and L858R. Overall survival was significantly shorter for patients harboring EGFR T790M mutation than it was for patients with exon 19 deletions (logrank P=0.008). When taking patients harboring EGFR L858R or exon 19 deletions as one group, the overall survival was also significantly longer than that in patients with T790M mutation (logrank P=0.012). There was no significant difference in relapse-free survival among three subgroups of patients.
Conclusion: Our study described the clinicopathological and molecular characteristics of NSCLC patients harboring primary EGFR T790M mutations. Its value of being a predictor for worse prognosis was established. Primary EGFR T790M mutation is a rare event in NSCLC cases, but the therapeutic strategies for this subtype of patients should be precisely considered.
Keywords: driver mutation, survival, clinicopathological profile, EGFR tyrosine kinase inhibitor, acquired resistance
Introduction
The presence of mutations in the kinase domain of epidermal growth factor receptor (EGFR) gene in non-small cell lung cancer (NSCLC) patients has been regarded as a predictive marker of good response to tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib. Nearly 90% of these mutations occur as either multinucleotide in-frame deletions in exon 19 or as single missense mutations that result in substitution of arginine for leucine at position 858 (L858R) in exon 21. However, after a median of approximately 10 months from the onset of TKI treatment, some patients whose tumors were initially sensitive to EGFR-TKIs have eventually developed acquired resistance.1−3
Test of biopsied tumor after disease progression has shown that half of these TKI-resistant cases contained a second site mutation in the EGFR kinase domain.4−6 The most common alteration involves a C-T (cytosine to thymine) change at nucleotide 2369 in exon 20, which results in substitution of methionine for threonine at position 790 (T790M). Cell lines with EGFR T790M mutation have been shown to have a proliferative advantage over wild type (WT) EGFR due to its increased kinase activity and downstream signaling.7 Mulloy et al8 showed the EGFR T790M/L858R double mutant exhibits a substantial increase in phosphorylation compared with the EGFR L858R mutant alone. Nevertheless, Oxnard et al9 and Arcila et al10 also observed lower growth rate in cell lines harboring both the EGFR exon 19 deletion and T790M mutation compared with cells harboring only the exon 19 deletion. Considering that the current in vitro results have been inconsistent, the impact of primary EGFR T790M mutation on the prognosis of NSCLC patients needs to be studied.
Bell et al11 reported a family with multiple cases of NSCLC associated with germline EGFR T790M mutation, where four of the six available samples from two patients showed an activating EGFR mutation (L858R, in-frame deletions in exon 19 or G719A). But in another series, EGFR T790M mutation was never found among 237 NSCLC family probands.7 Thus, the finding of an NSCLC patient with primary concomitant T790M mutation in the kinase domain of EGFR was rare event in the available series. Not much is known about this subset of cases. Here we report the clinicopathological characteristics and clinical outcome of NSCLC patients whose tumor harbored T790M along with an activating EGFR kinase domain mutation, with the aim of identifying the molecular profile of this subset of NSCLC patients.
Materials and methods
Patients and specimen collection
We consecutively enrolled patients with newly diagnosed primary NSCLC between October 2007 and October 2012. All patients provided written informed consent and underwent radical surgery. Eligible patients for this study had to meet the following criteria: review of pathological diagnosis of NSCLC by morphological features and immunohistochemistry staining, confirmed by two pathologists (L Shen and Y Li); sufficient tumor tissue and corresponding normal tissue available for analysis; and patient had not received neoadjuvant treatment. This study was approved by the institutional review board of Shanghai Cancer Center, Fudan University, Shanghai, People’s Republic of China.
Mutation analysis
After the surgery, samples were divided into two parts: one part was fixed using formalin and embedded with paraffin to make pathological sections; the other part was snap-frozen in liquid nitrogen at the time of resection and stored in liquid nitrogen. Ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) were extracted from tumors and distant histological normal lung, after frozen specimens were dissected into TRIzol® (Life Technologies Corp, Carslbad, CA, USA). Total RNA samples were reverse transcribed into complementary DNA (cDNA).
EGFR (exons 18 to 21), KRAS (Kirsten rat sarcoma viral oncogene homolog) (exons 2 to 3), HER2 (human epidermal growth factor 2) (exons 18 to 21), BRAF (v-raf murine sarcoma viral oncogene homolog B) (exons 11 to 15), PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha) (exon 9 and exon 20) were amplified by polymerase chain reaction (PCR) (Eppendorf Mastercycler® nexus; Eppendorf, Hamburg, Germany) using complementary (c) DNA. For detection of EML4 (echinoderm microtubule associated protein like 4)−ALK (anaplastic lymphoma receptor tyrosine kinase) fusions (multiple 5′ primers, including EML4 E2F, EML4 E13F, EML4 E18F and fixed 3′ primer located to ALK E20R, were designed to amplify all known fusions variants, as previously described).12
cDNA PCR products were assessed using the Sanger direct sequencing method13 in the forward and reverse directions. All mutations were verified by analysis of an independent PCR isolate. When combined EGFR mutations were found in the cDNA PCR product, separated genotyping of each exon was performed using DNA PCR product.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) testing (ThermoBrite StatSpin®; Abbott Molecular, Abbott Park, IL, USA) was performed on the corresponding formalin-fixed paraffin-embedded specimens matching the frozen specimens used for the RNA/DNA extraction, for all patients. We used the Vysis LSI EGFR SpectrumOrange/CEP 7 SpectrumGreen Probe (Abbott Laboratories, Abbott Park, IL, USA), and c-met/CEN7q FISH Probe (Abnova Corp, Taipei, Taiwan) to assess the EGFR and c-MET (met proto-oncogene) gene copy number variations. At least 50 cells were assessed for each case by two pathologists and were classified, in line with the published criteria, as disomy, low polysomy (<4 copies in >40% of cells), high polysomy (≥4 copies in >40% of cells), or gene amplification (homogenously staining regions with ≥15 copies in ≥10% of cells or a gene/chromosome ratio ≥2:1 per cell).14,15
Immunohistochemistry
Corresponding paraffin-embedded tissue sections were analyzed for the protein expression of PIK3CA (1:400 dilution) (PI3 Kinase p110 [C73F8] Rabbit mAb #4249; Cell Signaling Technology, Inc., Danvers, MA, USA) and p-Akt (phosphorylated protein kinase B) (1:50 dilution) (Phospho-Akt [Ser473] [736E11] Rabbit mAb #3787; Cell Signaling Technology, Inc.). Immunostaining was evaluated by two blinded observers. Immunoreactivity to any antibody was scored positive if more than 10% cells showed membranous staining of any intensity.15
Clinical and pathological variables
Clinical and pathological data collected for the analyses included sex, age at diagnosis, smoking history, pathological TNM stage, tumor differentiation, and histological type. If a sample was confirmed to be lung adenocarcinoma (immunohistochemistry staining showed thyroid transcription factor [TTF]-1 and cytokeratin [CK]7 positive), the histological subtypes, according to the new International Association for the Study of Lung Cancer, American Thoracic Society, and European Respiratory Society (IASLC/ATS/ERS) multidisciplinary classification were collected.16 Pathological TNM stages were evaluated in accordance with the seventh edition of the lung cancer staging classification system.17 Patients were interrogated about disease recurrence and survival information, either in clinic or by telephone every 3 months after the date of diagnosis. Relapse-free survival (RFS) was defined as the time elapsed between diagnosis and the date of relapse or last follow-up visit. Overall survival (OS) was calculated from the date of diagnosis to date of death or last follow-up visit.
Statistical analysis
We used percentages for qualitative variables, and mean and standard deviation for quantitative variables. Pearson Χ2 test or Fisher’s exact tests were used to assess the association between two categorical variables. RFS and OS were compared using the Kaplan−Meier method and logrank test. Cox regression was performed for multivariate analysis of survival. All the statistical analyses were performed using SPSS for Windows (version 16.0) (SPSS Inc., Chicago, IL, USA). P values were two tailed for all the tests, and statistical significance was set as P<0.05.
Results
Prevalence of EGFR T790M mutation in NSCLC
In all, 1,903 patients were eligible for this analysis, including 1,213 adenocarcinomas, 539 squamous carcinomas, 57 adenosquamous carcinomas, 27 large cell carcinomas, and 67 other NSCLC subtypes. A total of 783 patients were found to harbor EGFR mutation, accounting for 41.2% of all the NSCLC cases. Patients harboring EGFR T790M accounted for 2.04% of all the EGFR mutant cases and 0.84% of the total. There were 14 adenocarcinoma patients who were found to harbor a primary concomitant EGFR T790M mutation, accounting for 1.1% of all lung adenocarcinoma cases. Two patients with adenosquamous carcinoma harbored a EGFR T790M mutation, accounting for 3.5% of all adenosquamous cases. Ten patients harbored an EGFR T790M mutation and L858R mutation concurrently. Five patients harbored EGFR T790M mutation and exon 19 in-frame deletions concurrently. One patient harbored EGFR T790M, G719S mutations, and PIK3CA E542K mutation concurrently. None of the 16 patients had any other oncogenic driver mutations, including KRAS, HER2, BRAF, and EML4-ALK fusions.
Clinicopathological characteristics of NSCLC patients harboring EGFR T790M
Primary EGFR T790M mutation occurred in 13 females and three males, ranging in age at diagnosis from 45 to 66 (mean age: 59.3) years. Of these, 93.7% (15 of 16) patients had never smoked. Three tumors were poorly differentiated; the other 13 tumors were moderately differentiated. The number of patients in pathological TNM stages I−IV was 8, 2, 5, and 1, respectively. Among the 16 patients, two had adenosquamous carcinomas, and the other 14 had adenocarcinomas. The majority subtype of adenocarcinoma was acinar (n=6), followed by papillary (n=4), micropapillary (n=2), solid, and adenocarcinoma in situ. Details of the 16 patients harboring the EGFR T790M mutation are listed in Table 1. There were 54 patients with a tumor harboring EGFR L858R or exon 19 deletions consecutively enrolled. These samples underwent FISH to detect EGFR gene copy number variations. We compared the clinicopathological characteristics between these patients and patients harboring EGFR T790M (Table 2).
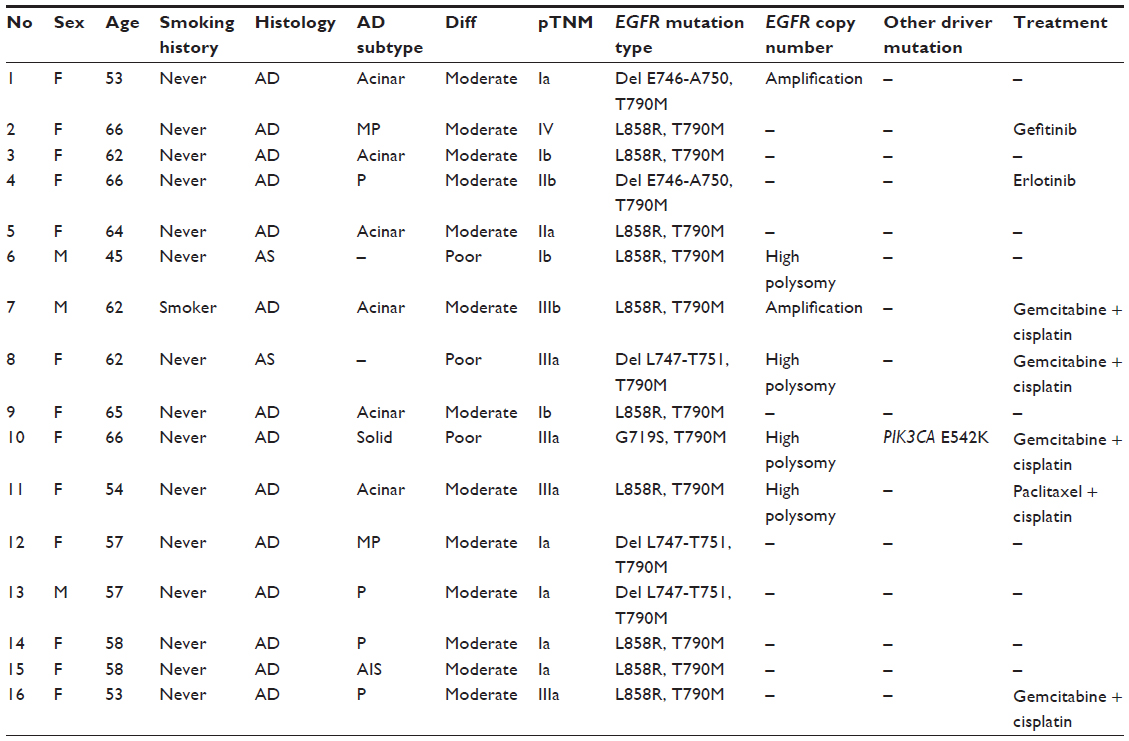 | Table 1 Clinicopathological and molecular characteristics of 16 patients harboring primary EGFR T790M mutation |
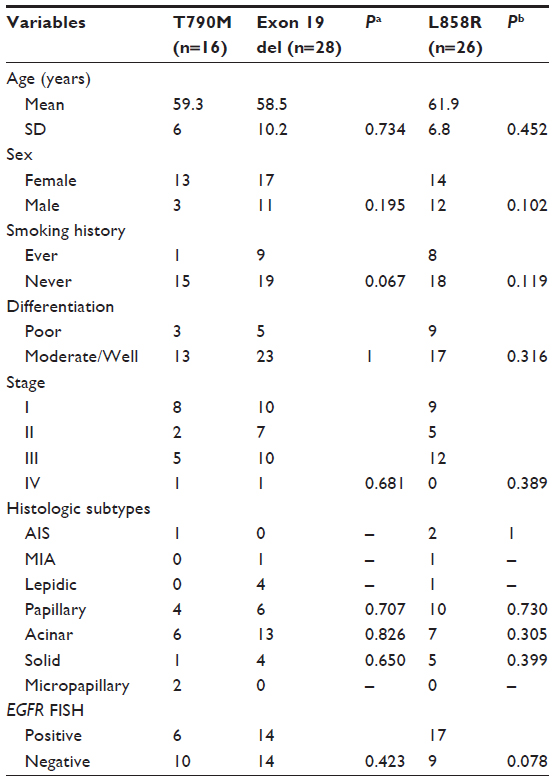 | Table 2 Comparison of clinicopathological and molecular characteristics between NSCLC patients harboring EGFR T790M mutations and those with classic activating mutations |
Clinical outcome
A total of 94 patients with only EGFR L858R mutation and 120 patients with only exon 19 in-frame deletions were consecutively enrolled from May 2009 to October 2012 (Table S1). The follow-up phase of these patients corresponded with that of the 16 patients harboring primary EGFR T790M. We analyzed the RFS and OS data of these three subgroups of patients. The median follow-up phase was 28.6 months for all the patients and 20.2 months for patients with EGFR T790M mutation. Two patients harboring EGFR T790M mutation had received EGFR-TKI treatment: one received gefitinib and had an occurrence of brain metastasis in the sixth month; the other received erlotinib to control the metastasis to the liver. The metastasis in the latter patient was assessed as progressive disease after 2 months of EGFR-TKI treatment. OS was significantly shorter for patients harboring EGFR T790M mutation than it was in patients with exon 19 deletions (logrank P=0.008). The OS was also shorter in patients with EGFR T790M mutation than that in patients harboring an L858R point mutation (logrank P=0.09). When taking patients with EGFR L858R or exon 19 deletions as one group, the OS was significantly longer than that in patients with T790M mutation (logrank P=0.012). There was no significant difference in RFS among three subgroups patients harboring different kinds of EGFR mutations (T790M versus exon 19 deletions [logrank P=0.387]; T790M versus L858R [logrank P=0.951]) (Figures 1 and 2).
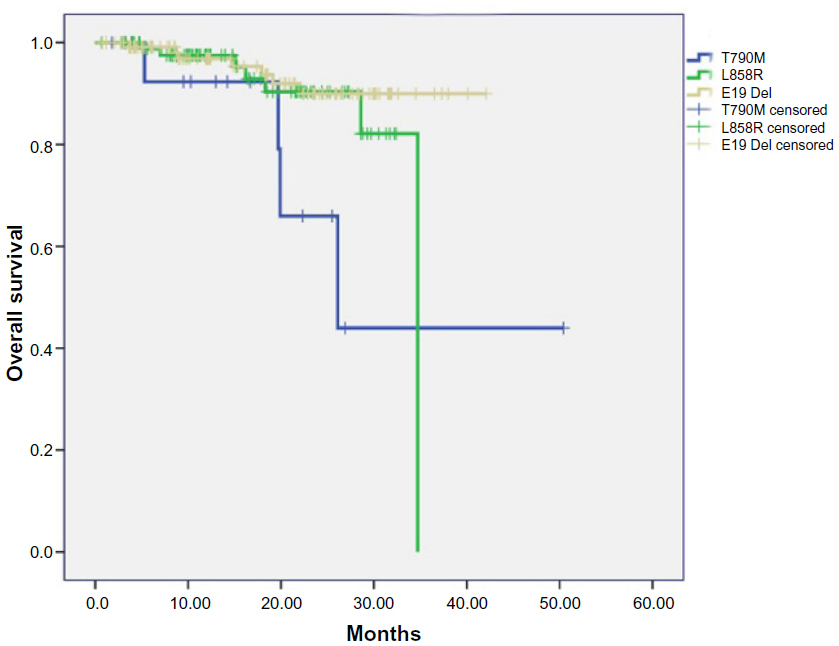 | Figure 1 Overall survival in patients harboring different kinds of EGFR mutation subtypes. |
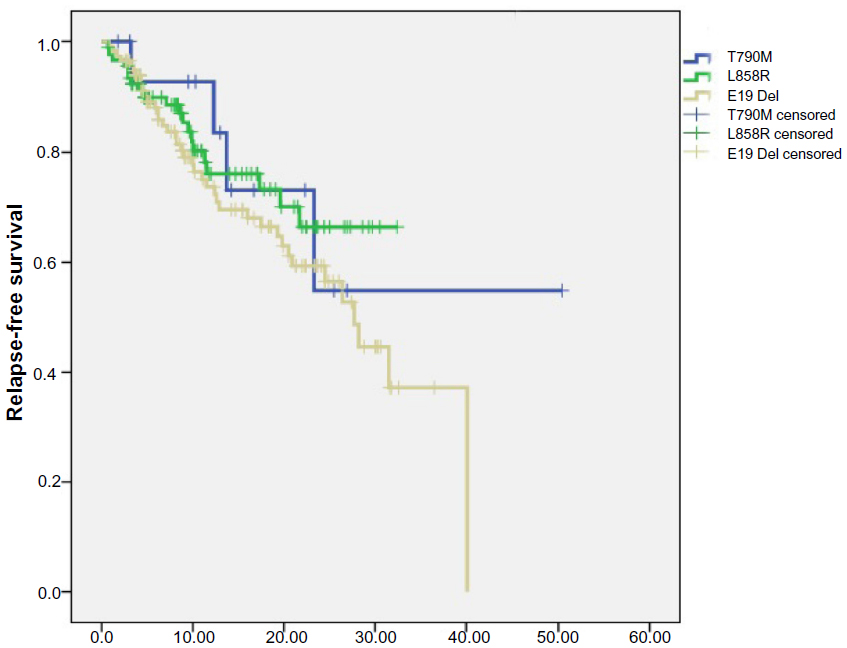 | Figure 2 Relapse-free survival in patients harboring different kinds of EGFR mutation subtypes. |
We also performed a multivariate analysis of survival significance of patients harboring a primary EGFR T790M mutation. Cox regression showed sex, age at diagnosis, smoking history, tumor differentiation, pathological type, EGFR gene copy number variations, and EGFR-TKI therapy had no significant influence on the OS or RFS of these patients.
EGFR gene copy number variations, c-MET amplification, PIK3CA, p-Akt expression in NSCLC with EGFR T790M mutation
Among the 16 patients harboring EGFR T790M, no c-MET amplification was found. Six of the 16 samples (37.5%) were EGFR−FISH positive, including two (12.5%) with gene amplification and four (25%) with high polysomy. We compared the frequency of EGFR gene copy number variations in the 16 patients harboring EGFR T790M mutation with that in 54 patients, 28 with EGFR exon 19 deletions and 26 with EGFR L858R. A lower frequency of FISH-positive cases was found in samples harboring EGFR T790M mutation compared with those with exon 19 deletions or L858R (Table 2). Positive PIK3CA and p-Akt expression were observed in four (25%) and eight (50%) of the 16 patients, respectively (Figure 3). A total of 108 NSCLC patients without any mutation were consecutively enrolled from May 2009 to October 2012. We compared PIK3CA and p-Akt expression between these patients and the patients harboring EGFR T790M (Table 3).
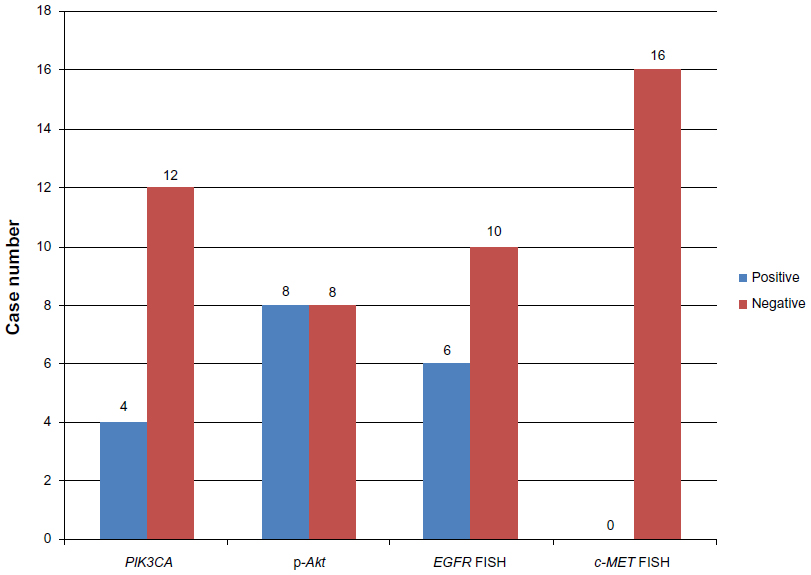 | Figure 3 Case number of PIK3CA, p-Akt-positive expression and EGFR, c-MET gene copy number variations. |
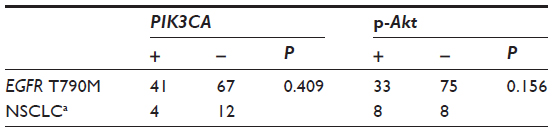 | Table 3 Comparison of PIK3CA, p-Akt expression between patients harboring EGFR T790M mutation and those without any mutation |
Discussion
Compared with classic activating EGFR mutations, such as L858R and exon 19 in-frame deletions, the clinicopathological and molecular characteristics of NSCLC patients harboring primary T790M mutations remain poorly defined. Somatic EGFR T790M is known to occur as a “secondary mutation” in more than 50% of patients in whom acquired resistance to EGFR-TKIs develops.5,6 Germline EGFR T790M mutation is rare and considered to be associated with genetic susceptibility to lung cancer, and may underlie familial predisposition to the disease.11 As far as we know, our study represents the largest investigation for the prevalence, clinicopathological features, molecular correlations, and clinical outcome in pretreated East Asian NSCLC patients harboring EGFR T790M mutation.
We determined that primary EGFR T790M mutation was a rare event in NSCLC patients, and it was mutually exclusive with other well-identified molecular alterations, except for one PIK3CA E542K mutation. Prudkin et al18 identified two (0.83%) cases harboring the EGFR T790M mutation from 240 patients with lung adenocarcinomas who had never received treatment with EGFR TKIs. Inukai et al19 found one (0.54%) EGFR T790M mutation in 185 NSCLC patients without EGFR-TKI treatment, using mutant-enriched PCR analysis, but the findings could not be confirmed by direct sequencing. Considering the extremely low prevalence of primary EGFR T790M mutation in NSCLC patients, screening of this mutation in a large cohort is necessary to define the clinicopathological features and molecular correlations.
Gefitinib-sensitizing EGFR mutations, such as L858R and exon 19 in-frame deletions, selectively activate antiapoptosis signaling pathways.20 Thus, they can enhance the proliferation and the resistance of apoptosis of tumor cell and have been considered to be oncogenic. In our series, primary EGFR T790M mutation was found concomitant with either L858R or exon 19 deletions in 15 (93.8%) samples. And no single EGFR T790M mutation was found. Also, baseline EGFR T790M was proved to be associated with germline molecular alterations.21 This may indicate that primary T790M mutation alone cannot induce oncogenesis. It can occur before EGFR-TKI treatment because of tumor cell heterogeneity.
A valuable result we found is that patients harboring primary EGFR T790M mutation had significantly shorter OS than that did patients harboring EGFR exon 19 deletions. And the patients harboring EGFR L858R seem to have a longer OS than did the patients with EGFR T790M. It was proven that EGFR T790M may in fact provide a proliferative advantage by increasing kinase activity and downstream signaling.7,22 Our findings indicate that primary EGFR T790M mutation might be a predictor for poor prognosis. However, considering the different postoperative treatment strategies these patients received, this finding should be verified. That the RFS was not different among these mutation subtypes may be partly owing to the relatively short follow-up time.
c-MET amplification was considered to be a mechanism for acquired resistance to EGFR-TKIs. This molecular change had been found in samples with gefitinib or erlotinib resistance in vivo or in vitro.23 c-MET amplification has also occasionally been found to coexist with EGFR T790M mutation.24 Therefore, we detected this kind of molecular change, using FISH, in the 16 samples harboring EGFR T790M, with an attempt to define the association between these two kinds of TKI-resistant-related molecular changes. But we found no c-MET amplification in them. This result may indicate that concurrence of the two kinds of molecular change is a rare event in patients who have not undergone EGFR-TKI treatment.
Some former studies using FISH have proposed EGFR gene copy number variations as an important predictor of response to EGFR-TKIs.14,25,26 In our study, we found a lower frequency of EGFR gene copy number variations in patients harboring EGFR T790M than in patients harboring exon 19 deletions and L858R. This result may confirm that primary EGFR T790M mutation, as a molecular marker, can induce initial resistance to EGFR-TKIs.
To our knowledge, well-defined EGFR-TKI resistance mechanisms share the same potential concept: they enable the cancer cell to maintain its intracellular growth signaling pathways, especially the phosphatidylinositol 3-kinase (PI3K)–protein kinase B (AKT) pathway.20,27−30 In our series, we found 50% of samples had an overexpressed level of p-Akt, but a corresponding overexpression were not found in the upstream protein PIK3CA. Our results may indicate primary EGFR T790M mutation can induce p-Akt expression through other mechanism, which are yet to be revealed.
In summary, our study of a large cohort described the clinicopathological and molecular characteristics of NSCLC patients harboring primary EGFR T790M mutations. Its value of being a predictor for worse prognosis was established. Primary EGFR T790M mutation is indeed a rare molecular change in NSCLC cases, but the therapeutic strategies for this subtype of patients should be carefully considered.
Acknowledgments
The authors thank Qiong Lu, Jie Xu and Youjia Deng for technical support.
The authors received funding from: the Key Construction Program of the National “985” Project (Grant No 985III-YFX0102); the National Natural Science Foundation of China (Grants No 81172218 and 81101761); the Science and Technology Commission of Shanghai Municipality (Program of Shanghai Subject Chief Scientist, Grant No 12XD1402000); The Foundation of Shanghai Health Administration (Grant No 20114206); and the Shanghai Hospital Development Center (Grant No SHDC12012308).
Disclosure
The authors report no conflict of interest in this work.
References
Riely GJ, Pao W, Pham D, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(3 Pt 1):839–844. | |
Jackman DM, Yeap BY, Sequist LV, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(13):3908–3914. | |
Morita S, Okamoto I, Kobayashi K, et al. Combined survival analysis of prospective clinical trials of gefitinib for non-small cell lung cancer with EGFR mutations. Clin Cancer Res. 2009;15(13):4493–4498. | |
Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769. | |
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. | |
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. | |
Vikis H, Sato M, James M, et al. EGFR-T790M is a rare lung cancer susceptibility allele with enhanced kinase activity. Cancer Res. 2007;67(10):4665–4670. | |
Mulloy R, Ferrand A, Kim Y, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67(5):2325–2330. | |
Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17(17):5530–5537. | |
Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17(5):1169–1180. | |
Bell DW, Gore I, Okimoto RA, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005;37(12):1315–1316. | |
Sun Y, Ren Y, Fang Z, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28(30):4616–4620. | |
Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–5467. | |
Hirsch FR, Varella-Garcia M, McCoy J, et al; Southwest Oncology Group. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study. J Clin Oncol. 2005;23(28):6838–6845. | |
Li C, Sun Y, Fang Z, et al. Comprehensive analysis of epidermal growth factor receptor gene status in lung adenocarcinoma. J Thorac Oncol. 2011;6(6):1016–1021. | |
Travis WD, Brambilla E, Noguchi M, et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6(2):244–285. | |
Goldstraw P, Crowley J, Chansky K, et al; International Association for the Study of Lung Cancer International Staging Committee; Participating Institutions. The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol. 2007;2(8):706–714. | |
Prudkin L, Tang X, Wistuba II. Germ-line and somatic presentations of the EGFR T790M mutation in lung cancer. J Thorac Oncol. 2009;4(1):139–141. | |
Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66(16):7854–7858. | |
Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–1167. | |
Oxnard GR, Miller VA, Robson ME, et al. Screening for germline EGFR T790M mutations through lung cancer genotyping. J Thorac Oncol. 2012;7(6):1049–1052. | |
Godin-Heymann N, Bryant I, Rivera MN, et al. Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation. Cancer Res. 2007;67(15):7319–7326. | |
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | |
Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–20937. | |
Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97(9):643–655. | |
Takano T, Ohe Y, Sakamoto H, et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23(28):6829–6837. | |
Engelman JA, Jänne PA, Mermel C, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102(10):3788–3793. | |
Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Jänne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004;64(20):7241–7244. | |
Mukohara T, Engelman JA, Hanna NH, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97(16):1185–1194. | |
Amann J, Kalyankrishna S, Massion PP, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65(1):226–235. |
Supplementary materials
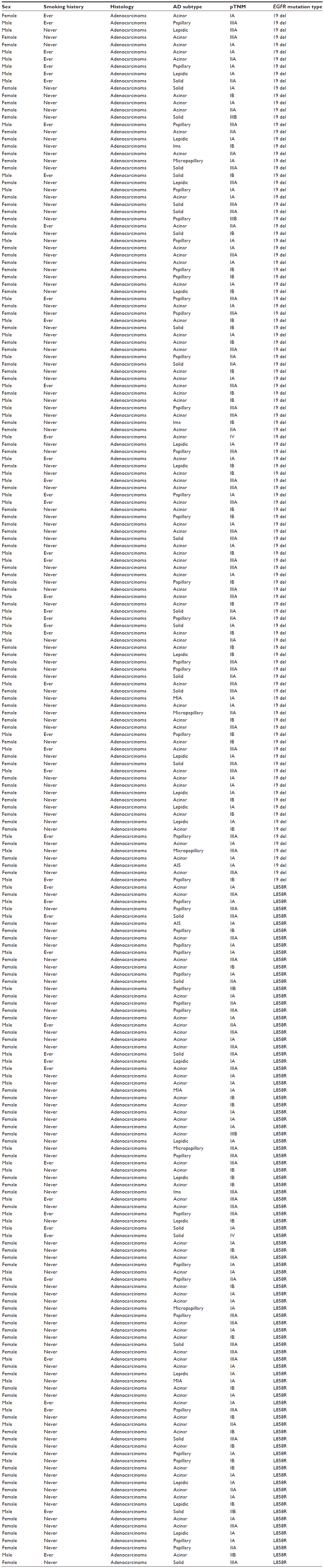 | Table S1 Details of NSCLC patients harboring EGFR L858R mutation or exon 19 deletions |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.