Back to Journals » Clinical Epidemiology » Volume 6
Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors
Authors Chia PL, Mitchell P, Dobrovic A, John T
Received 20 June 2014
Accepted for publication 3 September 2014
Published 20 November 2014 Volume 2014:6 Pages 423—432
DOI https://doi.org/10.2147/CLEP.S69718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Vera Ehrenstein
Puey Ling Chia,1 Paul Mitchell,1 Alexander Dobrovic,2–4 Thomas John1,2,4
1Department of Medical Oncology, Olivia-Newton John Cancer and Wellness Centre, Victoria, Australia; 2Ludwig Institute for Cancer Research, Austin Health, Victoria, Australia; 3Department of Pathology, University of Melbourne, Victoria, Australia; 4School of Cancer Medicine, La Trobe University, Victoria, Australia
Abstract: Improved understanding of molecular drivers of carcinogenesis has led to significant progress in the management of lung cancer. Patients with non-small-cell lung cancer (NSCLC) with anaplastic lymphoma kinase (ALK) gene rearrangements constitute about 4%–5% of all NSCLC patients. ALK+ NSCLC cells respond well to small molecule ALK inhibitors such as crizotinib; however, resistance invariably develops after several months of treatment. There are now several newer ALK inhibitors, with the next generation of agents targeting resistance mutations. In this review, we will discuss the prevalence and clinical characteristics of ALK+ lung cancer, current treatment options, and future directions in the management of this subset of NSCLC patients.
Keywords: anaplastic lymphoma kinase (ALK), gene rearrangements, lung cancer, kinase inhibitors, lung adenocarcinoma
Introduction to the gene mutations in patients with non-small-cell lung cancer
Lung cancer is not only the most common cancer worldwide with 1.8 million people diagnosed per year, but is also the deadliest with 1.6 million annual deaths.1,2 Although the majority of cases are detected in current or ex-smokers, increasingly patients with minimal or no smoking history are being diagnosed.3 Recent efforts to improve earlier detection of non-small-cell lung cancer (NSCLC) through screening4,5 may improve the stage of diagnosis and cure rates; however, currently most patients are diagnosed with locally advanced or metastatic disease.
The management of cancer has undergone significant evolution over the last decade due to improvement in the understanding of molecular drivers of carcinogenesis. The discovery of oncogenes, such as the epidermal growth factor receptor (EGFR), Kirsten rat sarcoma viral oncogene (KRAS), and V-raf murine sarcoma viral oncogene homolog B1 (BRAF), and the development of medications that specifically target these mutations or the wild-type receptor have led to the ability to personalize therapy (Figure 1).6–9 In a recent study, an actionable driver mutation was detected in 64% of tumors from patients with lung adenocarcinoma and oncologists can now employ specific therapies based on molecular profiling.10
 | Figure 1 Incidence and variety of oncogenic drivers in lung adenocarcinoma. |
Although the importance of anaplastic lymphoma kinase (ALK) as an oncogene in lymphoma has been recognized for many years, it has only recently become targetable using small molecule inhibitors. The success of these therapies has resulted in dramatic improvements in survival without significant toxicities. Here, we summarize the current literature and future directions in targeting tumors harboring ALK gene rearrangements (ALK+). We will detail the types of rearrangements present, correlation with clinicopathological features, therapeutic strategies to abrogate these pathways in addition to resistance mechanisms, and the novel agents being trialed at present.
Identification and prevalence of ALK rearrangements
ALK+ NSCLC represents approximately 4%–5% of all NSCLC patients in both Caucasian and Asian populations.11–13 Although this population is a small fraction of the overall NSCLC population, given the worldwide prevalence of NSCLC, this still represents potentially 40,000 new cases worldwide each year.14,15
The ALK gene was initially discovered in 1994 via the cloning of the t (2; 5) (p23; q35) translocation found in a subset of anaplastic large-cell lymphomas.16 Although the ALK gene is known to be an important determinant of prognosis in lymphoma, its association with NSCLC was only reported in 2007 when a small inversion within chromosome 2p that juxtaposes the 5′ end of the echinoderm microtubule-associated protein-like 4 (EML4) gene with the 3′ end of the ALK gene, resulting in the novel fusion oncogene EML4-ALK in NSCLC cells, was reported (Figure 2).6,17 Multiple EML4-ALK variants have been identified with variations in truncations of EML4 on different exons, but the ALK gene in all of them includes the exon 20 kinase domain.6,18
 | Figure 2 Illustration of EML4-ALK fusion oncogene in non-small-cell lung cancer and the detection by FISH. |
Confirmation of EML4-ALK as an oncogenic driver in NSCLC was demonstrated by the insertion of the fusion protein into NIH 3T3 fibroblasts that were then implanted subcutaneously into nude mice. All (eight out of eight) of the implanted mice developed lung adenocarcinomas,19 whereas those injected without the translocation did not form tumors.
There are now over 20 ALK fusion partners identified in NSCLC. EML4 represents the commonest fusion partner with 29%–33% of gene fusions identified to date.20 After EML4, the commonest are TFG and KIF5B, although other partners include NPM, TPM3, TPM4, ATIC, CLTC, MSN, MYH9, ALO17, IMT, SEC31A, and SQSTM1.16,21–34 This has led to some researchers developing panels of single and multiplex reverse transcription polymerase chain reaction (RT-PCR) assays suitable for rapid and accurate detection of the more common ALK+ variants, which are a useful adjunct to fluorescence in situ hybridization (FISH) assay of tumor specimens.35
Detection of ALK gene rearrangements
Several diagnostic platforms have been developed to detect ALK+ cells. Due to its ability to visualize rearrangements using dual color, FISH with break-apart probes has become accepted as a reference standard in the assessment of ALK+ NSCLC.36
Immunohistochemistry (IHC) for ALK can also detect ALK fusion proteins; however, it is reliant on increased cellular protein levels that may not always accompany the ALK fusion. Several different antibodies have been developed, including the murine monoclonal ALK1 clones, SP8, 5A4, ZAL4, P80, 5A4, or the rabbit D5F3.37 Although some groups have found IHC to be specific but relatively poor in sensitivity to detect ALK rearrangements,38 a recent report by Wynes et al found the ALK IHC assay was highly sensitive (90%), specific (95%), and accurate relative (93%) to the ALK FISH results.39 A recent analysis by Cabillic et al reviewed 3,244 consecutive NSCLC cases with parallel FISH and IHC ALK testing via the primary ALK monoclonal antibody clone 5A4 (Abcam, Cambridge, UK). A significant level of discrepancy was found with 70/150 (47%) found to be discordant. This study supports the need to combine testing to optimize the selection of eligible patients to be treated with ALK inhibitors, given that some patients with discordant testing were also found to respond to crizotinib.40 However, IHC remains a reliable screening tool for identification of ALK rearrangements and is certainly more cost-effective than FISH,41 with the caveat that occasional fusions will be missed. IHC detection of the ALK protein can be affected by a number of factors including variations in antigen retrieval, tissue fixatives, and fixation methods; varying sensitivities of reagents; and intra and interobserver variations.
Recently, RT-PCR of cDNA was reported as another useful tool that is sensitive and specific in the identification of ALK rearrangements,42,43 and it also allows the fusion partner of ALK or EML4-ALK variants to be identified if the partner is screened for.44 However, this methodology runs the risk of false negative results as RT-PCR requires high-to-moderate quality RNA that can be difficult to extract from the paraffin-embedded specimens used in daily clinical practice. It is therefore less appealing as a primary screening tool for ALK+ NSCLC but may be an adequate test for confirming results of IHC or FISH analysis.35 Furthermore, the need for RNA may limit this platform for routine medical testing.
Clinical features, natural history, and prognosis of patients with ALK+ NSCLC
The median age of patients with ALK rearrangements is 52 years, which is younger than most NSCLC patients either with an EGFR mutation or an unselected NSCLC population. There is a male preponderance11 and most patients have a never or light (<10 pack years) smoking history.45 Shaw et al reviewed 141 patients with two or more of the following characteristics: female sex, Asian ethnicity, never/light smoking history, and adenocarcinoma histology, and found the frequency of ALK+ NSCLC to be 22%. Although the cohort of screened patients was enriched for women, a greater percentage of men were ALK+ (23% vs 9%). Within the same group of patients who did not have an EGFR mutation, the frequency of ALK+ was 33% underscoring the importance of testing patients with this phenotype.11 Most ALK+ patients have advanced disease at time if diagnosis, which may reflect the aggressiveness of these tumors and their predilection for cerebral and hepatic metastases in addition to pleural and pericardial effusions.46 The anatomical location of ALK+ lung cancers appears to be more central and subsequently bronchoscopic cytology positivity is more common in the ALK+ group of patients.47
The majority of ALK+ NSCLCs are adenocarcinomas with only a few reports of squamous cell pathology.48 The solid growth pattern with signet ring cell component, a feature not often seen with EGFR mutant or wild-type NSCLC, and mucinous cribriform pattern with extracellular mucinous materials are the major histological findings.49,50 Although rare reports of coexistent oncogenic driver mutations exist, most patients are wild type for other common drivers such as EGFR, BRAF, and KRAS.51–53
There are varying opinions on the implications for survival with ALK+ NSCLC with most concluding ALK positivity as a poor prognostic factor in NSCLC.11,54–58 In a study by Shaw et al, the median overall survival (OS) for ALK+, crizotinib-naive patients compared with ALK−, and EGFR wild-type patients did not differ significantly (20 months vs 15 months; hazard ratio [HR] =0.77, 95% confidence interval [CI] =0.50–1.19; P=0.244). In comparison, ALK+ patients who were given second-line or third-line crizotinib had significantly better survival than patients with ALK−, EGFR wild-type tumors (HR =0.49, 95% CI =0.27–0.91, P=0.020). Fallet et al reviewed 116 French patients who were prospectively screened for ALK rearrangements (14.6% ALK+) and the median OS was not reached for ALK+ patients (94.1% of ALK+ patients received crizotinib), which was significantly longer than ALK− patients (HR for death =2.98, 95% CI =1.29–6.90, P=0.01).59 Therefore, in the absence of crizotinib or ALK inhibitor therapy, ALK rearrangements are not a favorable prognostic factor for survival. Kulig et al reviewed published data between July 2007 and November 2013 and also concluded ALK+ was a negative prognostic factor in NSCLC in studies controlling for known confounding factors.60
Zhang et al reviewed a group of patients without ALK inhibitor treatment and did not detect a survival difference according to ALK status after adjusting for disease stage, histology, and EGFR/KRAS mutation status.61 However, other groups have subsequently found ALK+ NSCLC to be associated with improved survival outcomes. Wu et al62 found that patients with ALK+ NSCLC (naive to ALK inhibitor treatment) identified from pleural effusion cytology had a significantly improved survival outcome compared with patients without EML4-ALK (median 14.7 vs 10.3 months, P=0.009, HR =0.53, 95% CI =0.32–0.87). Takeuchi et al63 found that the presence of a kinase fusion (ALK, ROS 1, and RET fusion) was an independent favorable prognostic factor after taking into consideration age, sex, stage, and smoking status, and found no significant difference in OS between kinase positive and EGFR mutant groups (P=0.32). The difference in survival outcomes and effects could be related to several factors such as small sample size of ALK+ patients in the studies, heterogeneous patient population between different countries, particularly the background EGFR mutant rate that has been noted to be higher in Asian populations and the variability of treatment regimes, including the use of ALK inhibitors in some studies.
The role of ALK+ as a predictive marker for response to chemotherapy has been explored in a number of studies. For patients treated with cisplatin and pemetrexed first-line, the median time to progression was 9 months for ALK+ patients but 6.2 months among 32 ALK− patients.64 This is notably similar to another study, which also reported median progression free survival (PFS) of 9 months compared to 4 months (HR =0.36, 95% CI =0.17–0.73, P=0.0051) in EGFR, ALK, and KRAS negative patient group.65 PROFILE 1007 involved 347 ALK+ patients who had failed one prior platinum-based chemotherapy and were randomly assigned to receive crizotinib (250 mg twice daily) or single-agent chemotherapy with either docetaxel or pemetrexed. The response rate to pemetrexed was higher than expected – 29% as compared with 12.8% in the general population of patients with lung adenocarcinoma who had previously been treated with chemotherapy. Therefore, patients with ALK+ NSCLC may have higher response rates with pemetrexed than patients with ALK− NSCLC66 and pemetrexed may be a better chemotherapeutic option for ALK+ patients. Supporting this, ALK+ tumors have been shown to express significantly lower thymidylate synthase levels compared to ALK− adenocarcinomas. Increased thymidylate synthase expression in malignant tumors is associated with reduced sensitivity to pemetrexed.67 This may explain the observed improved responses and survival observed in ALK+ patients who received pemetrexed chemotherapy.68
Emerging treatment options for NSCLC targeting ALK rearrangements
Crizotinib was designed as a multitargeted receptor tyrosine kinase inhibitor (TKI) and entered early Phase I clinical development initially as an inhibitor of the mesenchyme to epithelial transition (MET) pathway. Attention was focused on ALK+ tumors once researchers discovered dramatic clinical benefit associated with crizotinib in the first two patients who were ALK+.
The role of crizotinib in the management of ALK+ lung cancer was first evaluated in an international, multicenter Phase I study (PROFILE 1001),69,70 where overall response rates of 57% (47 of 82 patients) were observed, with 27 patients (33%) achieving stable disease. Crizotinib was shown to be tolerable with a good safety profile. The main adverse effects included visual disturbances, gastrointestinal side effects (nausea, 56%; diarrhea, 50%; vomiting, 39%; and liver function abnormalities, 12%), and pneumonitis.70 Reduced levels of testosterone and potential hypogonadism have been observed and may also be related to the potential central effects of crizotinib on the hypothalamus/pituitary axis.71
In 2011, crizotinib received accelerated approval from the US Food and Drugs Administration (FDA) in view of its efficacy as a therapeutic modality for ALK+ NSCLC based on the results from the 2009 single-arm, global Phase II study of crizotinib (PROFILE 1005, NCT00932451). The study involved 136 patients with ALK+ NSCLC (as determined by a centralized FISH test) who had progressed after initial chemotherapy for advance disease. The objective response rate (ORR) was 51% and disease control rate at 12 weeks was 74%, which was impressive in a group of pretreated patients.72
The subsequent Phase III trial PROFILE 1007 demonstrated that when compared with second-line chemotherapy, crizotinib prolonged PFS, increased response rates, and improved the quality of life in patients with advanced, previously treated ALK+ NSCLC.66 PFS, which was the primary end point, was significantly improved in the crizotinib arm (median PFS =7.7 months vs 3.0 months, HR =0.49, 95% CI =0.37–0.64, P<0.001), but there was no OS benefit, which was likely due to 64% of patients crossing over to the crizotinib arm on progression.
The PROFILE 1014 trial randomized untreated stage IV ALK+ NSCLC patients to either crizotinib or a platinum/pemetrexed combination. Not surprisingly, these data showed that crizotinib was superior to standard doublet chemotherapy in prolonging PFS, the primary objective (median =10.9 months vs 7.0 months, HR =0.454, 95% CI =0.346−0.596, P<0.0001). The ORR was also significantly higher with crizotinib (74% vs 45%, P<0.0001).73 Table 1 details the summary of the Phase I to III studies on crizotinib for NSCLC with ALK rearrangements.
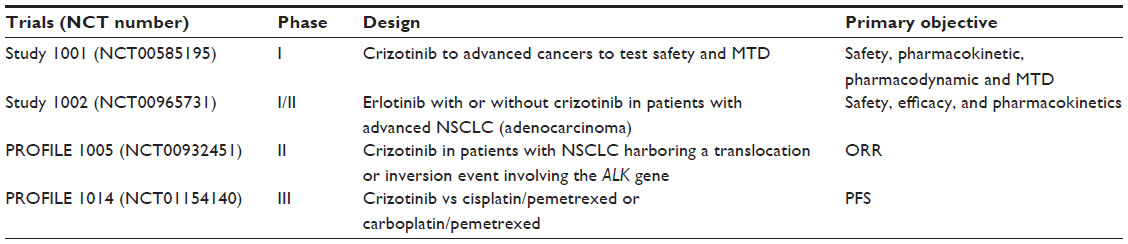 | Table 1 Summary of the Phase I–III studies on crizotinib for NSCLC with ALK rearrangements |
Although it is clear that crizoinib significantly improves response rates and survival, invariably the disease progresses at some point. Interesting recent data have also shown benefit for patients with advanced ALK+ lung cancer to continue ALK inhibition with crizotinib beyond progression in terms of maintaining ECOG performance status and may even prolong survival.74
Challenges, resistance patterns for ALK+ lung cancer to crizotinib
Targeted therapies directed at oncogene-addicted tumors often are efficacious for a limited period of time before the onset of acquired resistance. In the case of crizotinib, the median PFS in the PROFILE studies was 7.7 months. Similarly, in EGFR-mutated tumors, the efficacy of TKIs is limited by the development of resistance mechanisms, the commonest of which is the secondary T790M mutation within exon 20 of the EGFR gene.75,76 The presence of T790M gatekeeper resistance resulting in failure of treatment accounts for about 50% of secondary resistance77 with other mechanisms including activation of bypass mechanisms such as amplification of the MET proto-oncogene and transformation into small-cell lung cancer.78
To circumvent these mechanisms, researchers have now developed new second- and third-generation TKIs that are able to either intrinsically target the resistant mutation or bind irreversibly to the receptor tyrosine kinase.79,80
For fusion genes, however, it is apparent that many different mechanisms of resistance are possible given differences in the location of the fusion and the genes (Figure 3). Around one-third of secondary resistance mutations are located in the ALK TK domain with the most common mutation, the L1196M (22%–36%).81–83 The L1196M amino acid substitution is believed to hinder TKI binding through steric hindrance. Other amino acid substitutions observed include G1269A, G1202R, and S1206Y substitutions, as well as a 1151 threonine insertion. Another described resistance mechanism involves amplification of the ALK fusion gene and activation of other bypass mechanisms.84–86 KIT gene amplification and EGFR activation have also been reported as mechanisms of acquired crizotinib resistance.82,83,86
 | Figure 3 Potential mechanisms of resistance in ALK+ NSCLC. |
A number of next generation ALK inhibitors with the ability to bypass resistance mutations have recently been reported. Alectinib (CH5424802) is a potent, selective inhibitor of ALK that also potently inhibits the activity of ALK containing the L1196M gatekeeper mutation.87,88 This compound is also known to work in patients with central nervous system metastasis as it crosses the blood–brain barrier.89 Ceritinib (LDK378) is a second-generation ALK inhibitor that was granted an accelerated approval by FDA in April 2014 for treatment of patients with ALK+ metastatic NSCLC following treatment with crizotinib. Ceritinib demonstrated efficacy in Phase I trials with a remarkable ORR of 58% (95% CI =48–67) and the median PFS was 7.0 months (95% CI =5.6–9.5).90,91 Ceritinib was found to be efficacious in both ALK+ patients who have previously received crizotinib and crizotinib naive patients. The side effect profile was less favorable compared to crizotinib, but was still manageable with predominantly grade 1 or 2 gastrointestinal-related adverse effects. AP26113 is a dual inhibitor for both ALK and EGFR with activating mutations. It inhibits both L1196M in ALK+ NSCLC and the T790M in EGFR mutants.92 Phase I/II data for AP26113 have demonstrated efficacy in both crizotinib naive and resistant patients. It also has the ability to cross the blood–brain barrier and is active in ALK+ brain metastasis. Table 2 details the trials that are underway to study the efficacy of ceritinib.
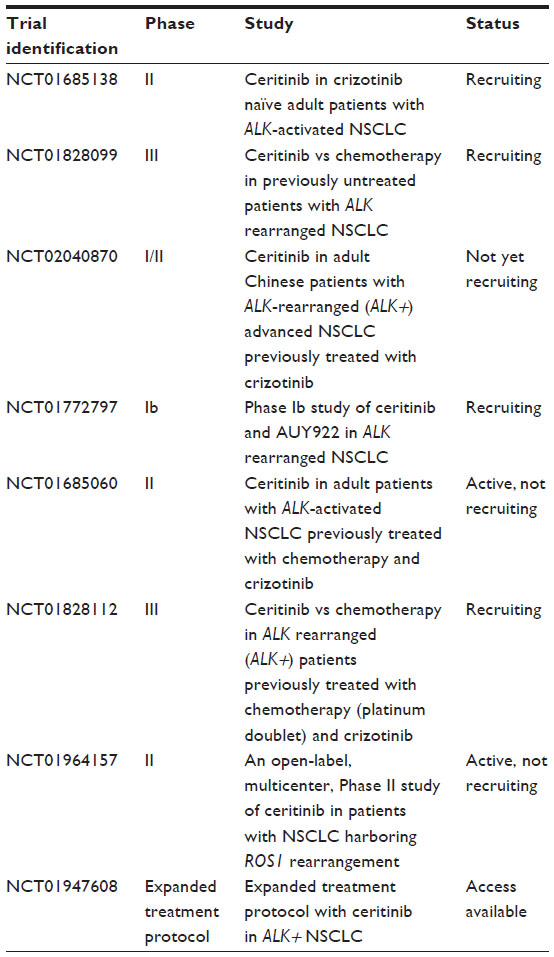 | Table 2 Eight available trials for ceritinib up to date from ClinicalTrials.gov |
Preclinical studies have shown that heat shock protein 90 (HSP90) inhibitors may have activity against patients with ALK+ lung cancer and this therapy may be useful alone or in combination with a TKI in the management of ALK resistance.93–96 HSP90 is required by certain oncogenic kinases such as AKT and phosphoinositide-dependent kinase-1(PDK1)97 for proper folding and can therefore be a useful target in cancers reliant on this mechanism. The drugs currently available and in development for ALK and HSP inhibitors are listed in Table 3.
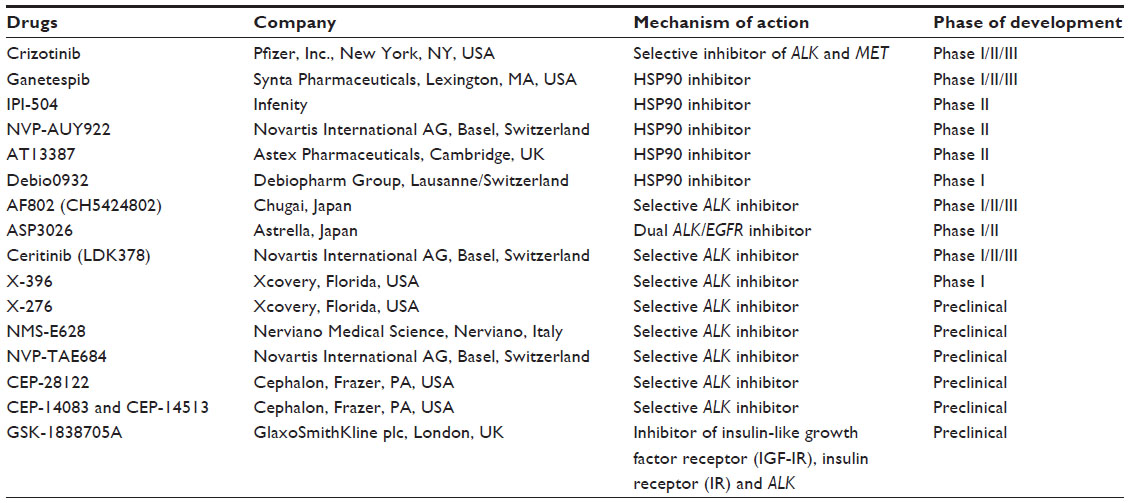 | Table 3 ALK and HSP inhibitors in development |
Clinical implications for enhanced patient care
Targeting ALK+ NSCLC has resulted in a paradigm shift in the management of advanced NSCLC. The use of crizotinib has demonstrated significant improvements in PFS and quality of life and appears to be beneficial in all lines of therapy, although resistance often develops. The development of novel inhibitors targeting these resistance mechanisms promises to expand the repertoire of therapies available, such that chemotherapy may be used as a last resort. Furthermore, combining ALK inhibitors with other pathway inhibitors promises to prolong the duration of therapy and leave cytotoxic chemotherapy as a last line of therapy.
Potential future immunotherapy management options in ALK+ NSCLC
Immunomodulatory drugs have recently generated significant interest in the management of advanced lung cancer with checkpoint pathway antagonistic antibodies that target cytotoxic T-lymphocyte antigen 4, the Programmed Death 1 (PD-1) receptor and its ligand (PD-L1) recently demonstrating activity in various cancers including NSCLC.98 These agents demonstrated response rates ranging from 23% to 67%99,100 with a potential for using PD-L1 expression to select the group of patients more likely to respond.
The concept of immunotherapeutic agents with targeted therapy has not been fully investigated at this stage. D’Incecco et al tested 125 patients for the presence of mutations and their correlation with PD-L1 expression. They found 56 to harbor EGFR mutations, 29 KRAS mutations and 10 ALK translocations. PD-1 and PD-L1 expressions differed according to clinical and biological characteristics; PD-1 positive patients were generally male, smokers, with adenocarcinoma histology, and KRAS mutations; while PD-L1 positive patients were generally female, never/former smokers, with adenocarcinoma histology, EGFR mutated, or ALK+.101 Further investigation of PD-L1 expression and ALK translocation will likely be undertaken and reported in future research as the number of trials using immunomodulatory agents have grown significantly.
Conclusion
The detection of ALK fusion genes in NSCLC has led to significant progress in the clinical care of patients with advanced lung cancer. It is important to note that while phenotypic characteristics can aid in selecting patients with ALK+ tumors, the only way of truly determining ALK positivity is to perform an ALK-specific assay and relying on phenotype will potentially miss patients with an actionable target. While ALK+ NSCLC is associated with a poorer clinical outcome in the absence of treatment with a targeted agent, the emergence of several different inhibitors and the development of therapeutic strategies to abrogate resistance promise to improve clinical outcome in these patients. Finally, the promise of combining targeted therapies with immunotherapies may provide a mechanism for more durable remissions, given the inevitable development of resistance to current small molecule inhibitors.
Acknowledgment
AD has received funding from Cancer Australia and the Cancer Council of Victoria. TJ is an NHMRC early career fellow and receives funding through the Victorian Cancer Agency and Cancer Council.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Ervik M, et al. Cancer Incidence and Mortality Worldwide. GLOBOCAN 2012 v1.0. 2012. Available from: http://www.iarc.fr/en/media-centre/pr/2013/pdfs/pr223_E.pdf. | |
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | |
Subramanian J, Govindan R. Lung cancer in never smokers: a review. J Clin Oncol. 2007;25(5):561–570. | |
Infante M, Lutman FR, Cavuto S, et al. Lung cancer screening with spiral CT: baseline results of the randomized DANTE trial. Lung Cancer. 2008;59(3):355–363. | |
National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365(5):395–409. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | |
Arteaga CL. The epidermal growth factor receptor: from mutant oncogene in nonhuman cancers to therapeutic target in human neoplasia. J Clin Oncol. 2001;19(Suppl 18):32S–40S. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | |
Marchetti A, Felicioni L, Malatesta S, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29(26):3574–3579. | |
Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998–2006. | |
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. | |
Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14(13):4275–4283. | |
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8(1):11–23. | |
Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31(8):1105–1111. | |
Jemal A, Clegg LX, Ward E, et al. Annual report to the nation on the status of cancer, 1975–2001, with a special feature regarding survival. Cancer. 2004;101(1):3–27. | |
Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284. | |
Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008;99(12):2349–2355. | |
Takeuchi K, Choi YL, Soda M, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14(20):6618–6624. | |
Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA. 2008;105(50):19893–19897. | |
Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. 2010;46(10):1773–1780. | |
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. | |
Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene. 1994;9(6):1567–1574. | |
Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood. 1999;93(9):3088–3095. | |
Meech SJ, McGavran L, Odom LF, et al. Unusual childhood extramedullary hematologic malignancy with natural killer cell properties that contains tropomyosin 4 – anaplastic lymphoma kinase gene fusion. Blood. 2001;98(4):1209–1216. | |
Colleoni GW, Bridge JA, Garicochea B, Liu J, Filippa DA, Ladanyi M. ATIC-ALK: a novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am J Pathol. 2000;156(3):781–789. | |
Hernández L, Pinyol M, Hernández S, et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood. 1999;94(9):3265–3268. | |
Touriol C, Greenland C, Lamant L, et al. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood. 2000;95(10):3204–3207. | |
Tort F, Pinyol M, Pulford K, et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab Invest. 2001;81(3):419–426. | |
Lamant L, Gascoyne RD, Duplantier MM, et al. Non-muscle myosin heavy chain (MYH9): a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer. 2003;37(4):427–432. | |
Cools J, Wlodarska I, Somers R, et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer. 2002;34(4):354–362. | |
Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000;157(2):377–384. | |
Bridge JA, Kanamori M, Ma Z, et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am J Pathol. 2001;159(2):411–415. | |
Takeuchi K, Soda M, Togashi Y, et al. Identification of a novel fusion, SQSTM1-ALK, in ALK-positive large B-cell lymphoma. Haematologica. 2011;96(3):464–467. | |
Van RoosbroeckK, Cools J, Dierickx D, et al. ALK-positive large B-cell lymphomas with cryptic SEC31A-ALK and NPM1-ALK fusions. Haematologica. 2010;95(3):509–513. | |
Li T, Maus MK, Desai SJ, et al. Large-scale screening and molecular characterization of EML4-ALK fusion variants in archival non-small-cell lung cancer tumor specimens using quantitative reverse transcription polymerase chain reaction assays. J Thorac Oncol. 2014;9(1):18–25. | |
Tsao M-S, Hirsch FR, Yatabe Y. IASLC Atlas of ALK Testing in Lung Cancer. Aurora, CO, USA: IASLC; 2013. | |
Karachaliou N, Rosell R. Optimal detection of ALK rearranged lung adenocarcinomas. J Thorac Oncol. 2013;8(3):255–256. | |
Hofman P, Ilie M, Hofman V, et al. Immunohistochemistry to identify EGFR mutations or ALK rearrangements in patients with lung adenocarcinoma. Ann Oncol. 2012;23(7):1738–1743. | |
Wynes MW, Sholl LM, Dietel M, et al. An International Interpretation Study using the ALK IHC antibody D5F3 and a sensitive detection kit demonstrates high concordance between ALK IHC and ALK FISH and between evaluators. J Thorac Oncol. 2014; 9(5):631–638. | |
Cabillic F, Gros A, Dugay F, et al. Parallel FISH and immunohistochemical studies of ALK status in 3244 non-small-cell lung cancers reveal major discordances. J Thorac Oncol. 2014;9(3):295–306. | |
Conklin CM, Craddock KJ, Have C, Laskin J, Couture C, Ionescu DN. Immunohistochemistry is a reliable screening tool for identification of ALK rearrangement in non-small-cell lung carcinoma and is antibody dependent. J Thorac Oncol. 2013;8(1):45–51. | |
Wallander ML, Geiersbach KB, Tripp SR, Layfield LJ. Comparison of reverse transcription-polymerase chain reaction, immunohistochemistry, and fluorescence in situ hybridization methodologies for detection of echinoderm microtubule-associated protein like 4-anaplastic lymphoma kinase fusion-positive non-small cell lung carcinoma: implications for optimal clinical testing. Arch Pathol Lab Med. 2012;136(7):796–803. | |
Just PA, Cazes A, Audebourg A, et al. Histologic subtypes, immunohistochemistry, FISH or molecular screening for the accurate diagnosis of ALK-rearrangement in lung cancer: a comprehensive study of Caucasian non-smokers. Lung Cancer. 2012;76(3):309–315. | |
Sanders HR, Li HR, Bruey JM, et al. Exon scanning by reverse transcriptase-polymerase chain reaction for detection of known and novel EML4-ALK fusion variants in non-small cell lung cancer. Cancer Genet. 2011;204(1):45–52. | |
Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17(8):2081–2086. | |
Doebele RC, Lu X, Sumey C, et al. Oncogene status predicts patterns of metastatic spread in treatment-naive nonsmall cell lung cancer. Cancer. 2012;118(18):4502–4511. | |
Kang HJ, Lim HJ, Park JS, et al. Comparison of clinical characteristics between patients with ALK-positive and EGFR-positive lung adenocarcinoma. Respir Med. 2014;108(2):388–394. | |
Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol. 2008;3(1):13–17. | |
Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15(16):5216–5223. | |
Yoshida A, Tsuta K, Watanabe S, et al. Frequent ALK rearrangement and TTF-1/p63 co-expression in lung adenocarcinoma with signet-ring cell component. Lung Cancer. 2011;72(3):309–315. | |
Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19(15):4273–4281. | |
Solomon B, Varella-Garcia M, Camidge DR. ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J Thorac Oncol. 2009;4(12):1450–1454. | |
Wang J, Dong Y, Cai Y, et al. Clinicopathologic characteristics of ALK rearrangements in primary lung adenocarcinoma with identified EGFR and KRAS status. J Cancer Res Clin Oncol. 2014;140(3):453–460. | |
Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12(11):1004–1012. | |
Yang P, Kulig K, Boland JM, et al. Worse disease-free survival in never-smokers with ALK+ lung adenocarcinoma. J Thorac Oncol. 2012;7(1):90–97. | |
Kim HR, Shim HS, Chung JH, et al. Distinct clinical features and outcomes in never-smokers with nonsmall cell lung cancer who harbor EGFR or KRAS mutations or ALK rearrangement. Cancer. 2012;118(3):729–739. | |
Lee JK, Park HS, Kim DW, et al. Comparative analyses of overall survival in patients with anaplastic lymphoma kinase-positive and matched wild-type advanced nonsmall cell lung cancer. Cancer. 2012;118(14):3579–3586. | |
Koh Y, Kim DW, Kim TM, et al. Clinicopathologic characteristics and outcomes of patients with anaplastic lymphoma kinase-positive advanced pulmonary adenocarcinoma: suggestion for an effective screening strategy for these tumors. J Thorac Oncol. 2011;6(5):905–912. | |
Fallet V, Cadranel J, Doubre H, et al. Prospective screening for ALK: clinical features and outcome according to ALK status. Eur J Cancer. 2014;50(7):1239–1246. | |
Kulig K, Wang Y, Iyer S, Yang P. Predictive and prognostic value of ALK gene rearrangement in non- small cell lung cancer. Epidemiology. 2014;4:146. | |
Zhang XC, Blanckmeister CA, Yang J, et al. Retrospective study of clinicopathologic factors associated with ALK rearrangement and survival outcome in Chinese patients with NSCLC. Cancer Res. 2012; 72(8 Suppl 1):4504. | |
Wu SG, Kuo YW, Chang YL, et al. EML4-ALK translocation predicts better outcome in lung adenocarcinoma patients with wild-type EGFR. J Thorac Oncol. 2012;7(1):98–104. | |
Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–381. | |
Altavilla G, Santarpia M, Arrigo C, et al. EML4-ALK fusion gene in lung adenocarcinoma: a retrospective analysis of the outcome of cisplatin plus pemetrexed treated patients. ASCO Proc. 2010;28:565S. (abstract 7610). | |
Camidge DR, Kono SA, Lu X, et al. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J Thorac Oncol. 2011;6(4):774–780. | |
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. | |
Sun JM, Han J, Ahn JS, Park K, Ahn MJ. Significance of thymidylate synthase and thyroid transcription factor 1 expression in patients with nonsquamous non-small cell lung cancer treated with pemetrexed-based chemotherapy. J Thorac Oncol. 2011;6(8):1392–1399. | |
Gandara DR, Huang E, Desai S, et al. Thymidylate synthase (TS) gene expression in patients with ALK positive (+) non-small cell lung cancer (NSCLC): implications for therapy. J Clin Oncol. 2012;30:(abstr 7582). | |
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | |
Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. | |
Weickhardt AJ, Rothman MS, Salian-Mehta S, et al. Rapid-onset hypogonadism secondary to crizotinib use in men with metastatic nonsmall cell lung cancer. Cancer. 2012;118(21):5302–5309. | |
Crinò L, Kim D, Riely GJ, et al. Initial phase 2 results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol. 2011;29(Suppl):7514. | |
Mok T, Kim D-W, Wu Y-L, et al. First-line crizotinib versus pemetrexed–cisplatin or pemetrexed–carboplatin in patients (pts) with advanced ALK-positive non-squamous non-small cell lung cancer (NSCLC): results of a phase III study (PROFILE 1014). J Clin Oncol. 2014;32:5s(Suppl; abstr 8002). | |
Ou SH, Janne PA, Bartlett CH, et al. Clinical benefit of continuing ALK inhibition with crizotinib beyond initial disease progression in patients with advanced ALK-positive NSCLC. Ann Oncol. 2014;25(2):415–422. | |
Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769. | |
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. | |
Suda K, Onozato R, Yatabe Y, Mitsudomi T. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol. 2009;4(1):1–4. | |
Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–673. | |
Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. | |
Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci USA. 2012;109(31):E2127–E2133. | |
Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31(31):3987–3996. | |
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | |
Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4(120):120ra117. | |
Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | |
Lovly CM, Pao W. Escaping ALK inhibition: mechanisms of and strategies to overcome resistance. Sci Transl Med. 2012;4(120):120s122. | |
Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71(18):6051–6060. | |
Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013;14(7):590–598. | |
Kaneda H, Yoshida T, Okamoto I. Molecularly targeted approaches herald a new era of non-small-cell lung cancer treatment. Cancer Manag Res. 2013;5:91–101. | |
Ou SH, Gadgeel S, Chiappori AA, et al. Consistent therapeutic efficacy of CH5424802/RO5424802 in brain metastases among crizotinib-refractory ALK-positive non-small cell lung cancer patients in an ongoing phase I/II study. Paper presented at: 15th World Conference on Lung Cancer 2013; October 29, 2013; Sydney, Australia. | |
Chen J, Jiang C, Wang S. LDK378: a promising anaplastic lymphoma kinase (ALK) inhibitor. J Med Chem. 2013;56(14):5673–5674. | |
Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197. | |
Ceccon M, Mologni L, Bisson W, Scapozza L, Gambacorti-Passerini C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res. 2013;11(2):122–132. | |
Sessa C, Shapiro GI, Bhalla KN, et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(13):3671–3680. | |
Chen Z, Sasaki T, Tan X, et al. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res. 2010;70(23):9827–9836. | |
Normant E, Paez G, West KA, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30(22):2581–2586. | |
Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28(33):4953–4960. | |
Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277(42):39858–39866. | |
Johnson DB, Rioth MJ, Horn L. Immune checkpoint inhibitors in NSCLC. Curr Treat Options Oncol. August 6, 2014. | |
Rizvi NA, Chow LQM, Dirix LY, et al. Clinical trials of MPDL3280A (anti-PDL1) in patients (pts) with non-small cell lung cancer (NSCLC). J Clin Oncol. 2014;32:5s[Suppl; Abstract TPS8123]. | |
Gettinger SN, Shepherd FA, Antonia SJ, et al. First-line nivolumab (anti-PD-1; BMS-936558, ONO-4538) monotherapy in advanced NSCLC: safety, efficacy, and correlation of outcomes with PD-L1 status. J Clin Oncol. 2014;32:5s[Suppl; Abstract 8024]. | |
D’Incecco A, Andreozzi M, Ludovini V, et al. PD-L1 and PD-1 expression in molecularly selected non-small-cell lung cancer. J Thorac Oncol. 2014;9(Suppl 9):S7–S52. | |
Gridelli C, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardiello F, ALK inhibitors in the treatment of advanced NSCLC, 300–306. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.