Back to Journals » International Journal of Nanomedicine » Volume 16
Preparation, Characterization, Pharmacokinetic, and Therapeutic Potential of Novel 6-Mercaptopurine-Loaded Oral Nanomedicines for Acute Lymphoblastic Leukemia
Authors Zou Y, Mei D ![]() , Yuan J, Han J, Xu J, Sun N, He H, Yang C, Zhao L
, Yuan J, Han J, Xu J, Sun N, He H, Yang C, Zhao L ![]()
Received 10 November 2020
Accepted for publication 7 January 2021
Published 12 February 2021 Volume 2021:16 Pages 1127—1141
DOI https://doi.org/10.2147/IJN.S290466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ebrahim Mostafavi
Yaru Zou,1,2,* Dong Mei,1,* Jinjie Yuan,1,2 Jiaqi Han,1 Jiamin Xu,1 Ning Sun,1 Huan He,1 Changqing Yang,2 Libo Zhao1
1Clinical Research Center, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, 100045, People’s Republic of China; 2School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing, 211198, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Libo Zhao
Clinical Research Center, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, People’s Republic of China
Tel +86-010-59617018
Email [email protected]
Background: Acute lymphoblastic leukemia (ALL) is the most common hematologic malignancy in children. It requires a long and rigorous course of chemotherapy treatments. 6-Mercaptopurine (6-MP) is one of the primary drugs used in chemotherapy. Unfortunately, its efficacy has been limited due to its insolubility, poor bioavailability and serious adverse effects. To overcome these drawbacks, we constructed 6-mercaptopurine (6-MP)-loaded nanomedicines (6-MPNs) with biodegradable poly(lactide-co-glycolide) (PLGA) to enhance the anticancer efficacy of 6-MP.
Methods: We prepared the 6-MPNs using a double-emulsion solvent evaporation method, characterizing them for the physicochemical properties. We then investigated the plasma, intestinal region and other organs in Sprague Dawley (SD) rats for pharmacokinetics. Additionally, we evaluated its anticancer efficacy in vitro on the human T leukemia cell line Jurkat and in vivo on the ALL model mice.
Results: The 6-MPNs were spherical in shape with uniform particle size and high encapsulation efficiency. The in vitro release profile showed that 6-MPNs exhibited a burst release that a sustained release phase then followed. The apoptosis assay demonstrated that 6-MPNs could improve the in vitro cytotoxicity in Jurkat cells. Pharmacokinetics profiles revealed that 6-MPNs had improved oral bioavailability. Tissue distribution experiments indicated that 6-MPNs increased the duodenum absorption of 6-MP, at the same time having a low accumulation of the toxic metabolites of 6-MP. The in vivo pharmacodynamics study revealed that 6-MPNs could prolong the survival time of the ALL model mice. The prepared 6-MPNs, therefore, have superior properties in terms of anticancer efficacy against ALL with reduced systemic toxicity.
Conclusion: Our nanomedicines provide a promising delivery strategy for 6-MP; they offer a simple preparation method and high significance for clinical translation.
Keywords: 6-mercaptopurine, nanomedicines, Jurkat cells, acute lymphoblastic leukemia, ALL, bioavailability
Introduction
Acute lymphoblastic leukemia (ALL) is a common hematological malignancy that accounts for 70%–80% of all cases of leukemia in children. It is also the most frequent cause of death among cancer patients who are younger than 20 years old.1,2 Although treatments for pediatric ALL have resulted in a cure rate of about 85%-90%, it recurs in about 15%–20% of children and it is ultimately fatal.3 At present, chemotherapy is one of the main treatments for children with ALL; it includes three phases of remission induction, post-remission consolidation, and long-term maintenance.4 Induction treatment is the most critical phase, as it carries the highest risk of serious toxicity.5 Moreover, the maintenance period is particularly important to control the relapse of ALL, but it can also result in side effects of a low severity in cases of long-term use of chemotherapeutic agents.6
6-Mercaptopurine (6-MP) is one of the most commonly used antitumor drugs and immunosuppressants with demonstrated efficacy against childhood ALL in clinical medicine. It is used either independently or in combination with other drugs in induction and maintenance phases.7 The oral medication maintenance period lasts approximately 2–3 years in pediatric patients; this timeframe is necessary for decreasing relapse risk.3 6-Thioguanine nucleotides (6-TGNs) are the main active metabolites derived from 6-MP. They can disrupt the DNA’s replication and transcription processes, inducing cytotoxicity for cancer cells.8 However, the oral bioavailability of 6-MP is low (16%–50%) due to its insolubility (0.22 mg/mL) and its short half-life (0.5–1.5 h).9 Moreover, there is also considerable inter-individual variability in the clinical response. The non-selective distribution of 6-MP and genetic polymorphisms can cause serious side effects, including life-threatening effects like liver toxicity and myelosuppression – this greatly affects the clinical efficacy of 6-MP.10
In recent years, researchers have made several attempts to overcome these limitations. Among such attempts, nanotechnology-based approaches have been particularly useful to mitigate issues.11 The currently reported nanocarriers for 6-MP have been mainly prepared by chemical coupling of 6-MP or 6-MP derivatives with polymers, including chitosan,9 carboxymethyl chitosan (CMCS),12 and dendrimer,13,14 which inevitably use crosslinking agents. In addition, experts have used metal vector,15,16 mesoporous silica17 and magnetic materials such as iron oxide18 to prepare 6-MP nanoparticles. Some researchers have also modified these nanoparticles with hyaluronic acid19,20 and folate21 to improve the nanomedicine’s ability to target tumors and cells. While nanotechnology has enhanced the solubility of 6-MP, the preparation processes of the above techniques are complex. They also involve safety concerns caused by toxic crosslinkers, surfactants or organic solvents. Additionally, these 6-MP nanomedicines generally have low encapsulation efficiency and they have been typically administrated by intravenous injection, which is an unacceptable administration route for long-term clinical application. It is, therefore, necessary to develop oral biodegradable nanoformulations to improve the bioavailability and curative effect, while also mitigating side effects.
There have been few studies on the oral nanomedicines for 6-MP. Poly(lactide-co-glycolide) (PLGA), one of the biodegradable, biocompatible and nontoxic polymers, has recently been approved as a material for efficient drug loading by the Food and Drug Administration (FDA).22 In turn, the preparation methods of PLGA nanomedicines are relatively simple and suitable for oral administration drugs. PLGA nanomedicines display enhanced bioavailability of poorly water-soluble antitumor drugs, good size control and high encapsulation efficiency.22 Moreover, the release could be controlled by selecting the molecular weight, the polymer ratio and the terminal groups of the PLGA.23 In this current study, we chose PLGA as the carrier material to prepare the 6-MP-containing nanomedicines (6-MPNs) using a water-in-oil-in-water (W/O/W) double-emulsion solvent evaporation method. An optimized formulation had been constructed and characterized for the physicochemical properties. Furthermore, we conducted an in-depth evaluation of the nanomedicine’s pharmacokinetics and anticancer efficacy in vitro and in vivo.
Materials and Methods
Materials
In this study, 6-mercaptopurine (assay purity 98.0%) was purchased from Toronto Research Chemicals Inc (Toronto, Canada). PLGA with a 50/50 for lactic/glycolic ratio (Resomer® RG 503H, acid terminated, molecular weight (Mw) 24,000–38,000; Resomer® RG 502H, acid terminated, Mw 7000–17,000; Resomer® RG 502, ester terminated, Mw 7000–17,000), Polyvinyl alcohol (PVA, 87%–89% hydrolyzed, Mw 31,000–50,000), dichloromethane (DCM), ethyl acetate (EA) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich Chemical Co., Ltd. Pluronic F68 was purchased from Yuanye Bio-Technology Co., Ltd. Ammonia water (NH3.H2O) was obtained from Macklin Co., Ltd (Shanghai, China). Phosphate buffered saline (PBS) and penicillin-streptomycin were obtained from Solarbio Technology Co., Ltd. In addition, cell culture media RPMI1640 and trypsin were bought from Macgene (Beijing, China). Fetal bovine serum (FBS) was supplied by Gibco Invitrogen Co. (Carlsbad, USA). The Cell Counting Kit-8 (CCK8) was purchased from Dojindo (Shanghai, China). The Annexin V-FITC/PI Apoptosis Detection Kit was bought from YEASEN (Shanghai, China). All other reagents were of analytical grade.
Preparation of 6-MPNs
Nanomedicines containing 6-MP were formulated using a modified double-emulsion solvent evaporation method.24,25 The 6-MP was dissolved in aqueous ammonia containing PVA to form the inner water phase. The drug solution was added dropwise in the organic phase containing the PLGA so as to get the primary emulsion under magnetic stirring. Subsequently, using a probe-type sonifier, the primary emulsion was sonicated in an ice water bath and quickly injected into the outer aqueous phase (1% w/v pluronic F68 solution) to achieve the 6-MP-loaded PLGA double emulsion with ultrasonication for 2 minutes. The double emulsion was evaporated at 40°C for 1 h to remove the organic phase. In turn, this formed the nanomedicines, which were collected through centrifugation, then washed with deionized water and dispersed into a 10% w/v mannitol solution. The nanomedicines were lyophilized using freeze-drying for storage. Table 1 lists the formulation variables and identifier codes.
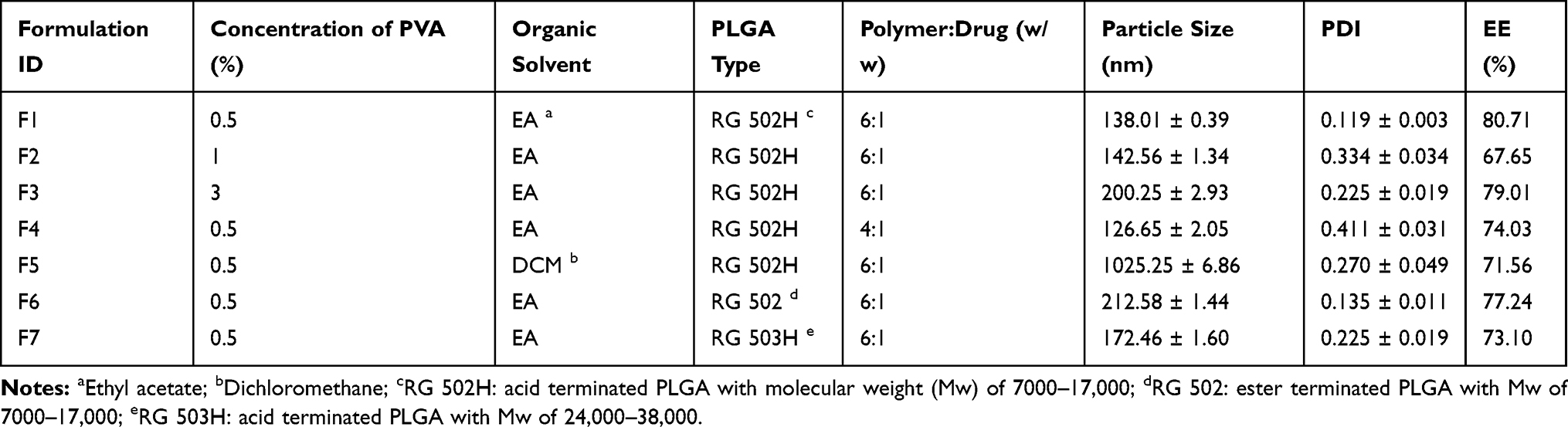 |
Table 1 Particle Size, Polydispersity Index (PDI) and Encapsulation Efficiency (EE) of 6-MPNs Prepared with Various Formulations |
Preparation of 6-MP Suspensions
For the control formulation, 6-MP suspensions were prepared with 0.5% (w/v) sodium carboxymethyl cellulose solution (CMC-Na). Briefly, a certain amount of 6-MP powder was made by grinding commercially available tablets and suspended in 0.5% CMC-Na to obtain suspensions (6-MPCs) at a concentration of 2 mg/mL.
Physicochemical Properties and the Morphology Characterization of 6-MPNs
The current study determined the particle size and polydispersity index (PDI) of 6-MPNs using dynamic light scattering (DLS). The Zeta potential was measured through Laser Doppler Electrophoresis using a Malvern Zeta-sizer. The surface morphology of 6-MPNs was characterized through transmission electron microscopy (TEM, JOEL, JEM 2100) that operated at an accelerating voltage of 80 kV; this occurred after negative staining via a 1% uranyl acetate solution. The 6-MPNs were dispersed in different media, including PBS (pH = 7.4), RPMI1640 and RPMI1640 with 10% FBS, before being incubated at 37°C. Their physical stability was assessed by measuring their particle size at different incubation times.
Determination of Loading Drug and Encapsulation Efficiency (EE)
Drug loading refers to the ratio of the drug weight in nanomedicines to the total weight of nanomedicines. In this study, a certain amount of lyophilized powder of 6-MPNs was weighed and dissolved in methanol/water (1:1, v/v) to determine the encapsulated 6-MP of 6-MPNs. Calculating the difference between the initial added amount of 6-MP and the non-entrapped free drug that remained in the supernatant after centrifugation allowed determination of the EE of the 6-MPNs. The free drug was quantified by HPLC (Shimadzu, Kyoto, Japan). A Diamonsil C18 column (150 × 4.6 mm, 5 µm) was used for chromatographic separation at a flow rate of 1 mL/min with UV detection (325 nm). The column oven temperature was set to 40°C, accompanied by an injection volume of 20 µL. Isocratic elution was used with a mobile phase of phosphate buffer (pH = 3.31) and acetonitrile (75%: 25%, v/v).26
In vitro Drug Release
Using the dialysis method, we studied the in vitro release of 6-MP from the prepared 6-MPNs. 6-MPNs (2 mg/mL) were added into dialysis bags (molecular weight cut off = 3500 Da), then dialyzed against 50 mL of PBS (pH 4.8 and 7.4) containing 0.02% (v/v) tween 20 at 37 ± 2°C, with gentle stirring at 100 rpm. At predetermined intervals, we withdrew aliquots of the PBS (1.0 mL) and refilled them with equal amounts of fresh PBS. The samples were centrifuged (12,000 rpm, 15 min) to collect the supernatants, which were determined through HPLC (described above).
Anticancer Effects in vitro
Cell Line and Culture
Human T cell leukemia cell lines (Jurkat) were provided by the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). We maintained the cells in RPMI 1640 media and supplemented them with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FBS at 37°C in an environment of 5% CO2. The media were changed daily and cells were passaged once every 2–3 days.
In vitro Cytotoxicity of 6-MPNs to Jurkat Cells
We analyzed the cytotoxicity studies using a Cell Counting Kit-8 (CCK8) assay. The Jurkat cells were seeded in a 96-well plate (Corning®, USA) at a cell density of approximately 2 × 104 cells/well in 100 µL. The cells were treated with a graded concentration of drugs, including 6-MP solution, 6-MPCs or 6-MPNs. Subsequently, the cells were incubated for 48 h at 37°C. The drug-containing medium was discarded carefully after centrifuging (1000 rpm, 5 min). Soon after, the cells were washed twice in a complete medium. Cell viability was measured by CCK8 in accordance with the manufacturer’s instructions. The optical density (OD) was read at 450 nm using a Spectra MAX 190 microplate reader.
Detection of Apoptosis by Flow Cytometry
We analyzed apoptosis using double staining via Annexin V-FITC and PI. The Jurkat cells were seeded in 6-well plates at a density of 4 × 105 cells/mL, and then exposed for 48 h to 6-MP solution, 6-MPCs and 6-MPNs at concentrations of 0.5 µM, 2 µM and 5 µM, respectively. The cells treated with PBS were used as the control. The cells were harvested at 1–5 × 105 cells/mL by centrifugation (300 g, 5 min) at 4°C before being washed three times with cold PBS. Subsequently, the cells were re-suspended in binding buffer and stained with 5 µL Annexin V-FITC and 10 µL PI following the manufacturer’s instructions. The apoptotic/necrotic populations were analyzed using a FACS Calibur cell analyzer (BD Biosciences).
Animal Studies
Experimental Animals
We purchased one-month-old Sprague Dawley (SD) rats from Beijing Vital River Laboratory Animal Technology (Beijing, China). The SD rats (100 ± 10 g) were raised under standard animal feeding conditions with free access to water and food. Female immune-deficient NOD-Prkdcscid Il2rgnull (NPG) mice were procured from Beijing Vitalstar Biotechnology (Beijing, China) and housed under pathogen-free standard laboratory conditions with ad libitum access to water and food. All animals were maintained in accordance with the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals protocols. All animal studies were approved by the Animal Ethics Committee of Capital Medical University (Approval Number: AEEI-2020-072).
In vivo Pharmacokinetics in SD Rats
The SD rats were randomly placed in two groups (8 rats per group) who were then orally administered at a dose of 15.75 mg/kg of 6-MPCs and 6-MPNs, respectively. At specified time points after oral administration drugs, we collected blood samples (0.1 mL) from the retro-orbital plexus of the rats and centrifuged the samples at 5000 rpm for 10 min at 4°C to immediately separate the plasma. Subsequently, they were stored at −80°C until analysis. The plasma samples were deproteinized with methanol and centrifuged at 16,000 g for 10 minutes to collect the supernatants that were assayed by means of HPLC-MS/MS. An API 5500 triple-quadrupole mass spectrometer was used to perform the MS analysis (Applied Biosystems-Sciex, Toronto, Canada). The chromatographic separation was achieved by using a reverse-phase Atlantis T3 column (2.1 mm × 100 mm, 5 µm) with gradient elution. The mobile phase was composed of methanol containing 0.5% v/v formic acid and water. For all analyses, a flow rate of 0.4 mL/min, a sample injection volume of 3 µL and a run time of 5 min were employed. The column oven and autosampler were set at 37°C and 10°C. Multiple reaction monitoring transitions were performed for quantitation at m/z 153.0 > 118.9 for 6-MP, m/z 167.0 > 125.9 for 6-methylmercaptopurine (6-MMP), m/z 168.0 > 150.9 for 6-thioguanine (6-TG).27
Intestinal Absorption and Tissue Distribution
Food was withdrawn 16–18 h before administering the drug. Afterwards, 6-MPCs and 6-MPNs were orally administered to SD rats (n = 6) at a dose of 6-MP 15.75 mg/kg. The rats were euthanized at 10 min, 0.5 h, 1 h, 3 h, and 10 h post administration. We excised intestinal segments including the duodenum, jejunum, ileum and colon, as well as different organs such as the heart, liver, spleen, lungs, kidneys, and bone marrow. We rinsed these with PBS three times each. Sample tissues were weighed and homogenized in the appropriate amount of methanol [w (g)/v (mL) = 1/9]. The drug-containing supernatants were collected by centrifuging the tissue homogenates for HPLC-MS/MS analysis.
Anti-Leukemic Efficacy in Jurkat Cell Xenograft Mouse Model
To generate the human cell line xenograft in ALL mouse model, we injected Jurkat cells (1 × 106 cells) into the tail veins of the female NPG mice.28 After 7 days, we randomly placed the mice in three groups. Over the course of 2 weeks, 6-MPNs and 6-MPCs were administered at 20 mg/kg once daily by oral gavage (n = 6), and PBS was administered as the control. Throughout the research, we monitored the body weights and the survival of the mice on a daily basis. The mice with advanced leukemia (>20% weight loss, tachypnea, ruffled fur, hypothermia, hunched back, hind-limb paralysis, minimal activity) would be euthanized. We evaluated the livers and spleens of the mice for 6-MP-induced toxicity and a leukemia burden.
Statistical Analysis
Results are presented as mean ± SD for in vitro and in vivo studies. All data were calculated by GraphPad Prism Software (version 7.0) and analyzed utilizing the unpaired-samples Student’s t-test comparison of means. The P value < 0.05 was considered as statistical significance. The proportions of apoptotic/necrotic cells were analyzed automatically by FlowJo® X software. The total percentage of apoptotic cells was calculated as the sum of the early and late apoptosis rates based on the statistical data generated by the system.
Results and Discussion
Preparation and Characterization of 6-MPNs
As depicted in Figure 1, 6-MPNs were constructed via a double-emulsion solvent evaporation method. In brief, 6-MP and PVA were dissolved in aqueous ammonia to form an inner water phase (W1). Thereafter, W1 was added dropwise in the organic solvent containing PLGA (O) to get the primary emulsion (W1/O) under magnetic stirring and ultrasonication. W1/O was then dispersed in the F68 solution (W2) to obtain double emulsion (W1/O/W2) under ultrasonication. The W1/O/W2 was evaporated to completely remove the organic solvent. The final formulation was centrifuged and washed to purify the nanomedicines.
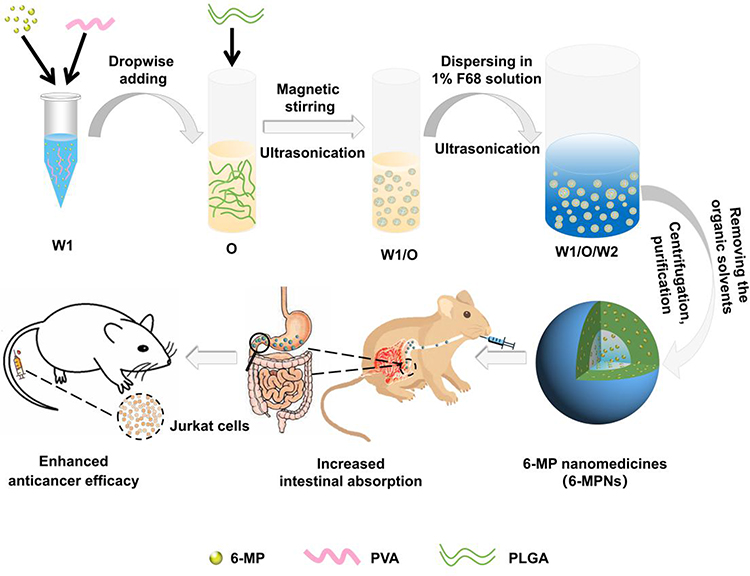 |
Figure 1 Preparation of 6-mercaptopurine-loaded nanomedicines (6-MPNs) and schematic of 6-MPNs augmenting the anticancer efficiency after oral administration. 6-MPNs were constructed via a double-emulsion solvent evaporation method. The inner water phase (W1) containing 6-MP and PVA was added dropwise in the oil phase containing PLGA (O) to get the primary emulsion (W1/O). Then the W1/O was dispersed in the outer water phase (W2) to obtain double emulsion (W1/O/W2), which was evaporated, centrifuged and washed to purify the nanomedicines. The 6-MPNs are capable of improving the solubility of 6-MP and increasing the intestinal absorption, thereby enhancing the anticancer effect and reducing chemotherapy-induced systemic toxicities. |
Table 1 presents the characteristics of 6-MPNs. The results revealed that, when the concentrations of PVA increased from 0.5% to 3%, the particle size of nanomedicines gradually increased. This is perhaps attributable to the incremental viscosity of the solution.25 All the encapsulation efficiencies (EEs) of 6-MP in different 6-MPNs formulations were above 60%. Based on the particle size and EEs, we considered 0.5% to be the optimal concentration of PVA in the internal water phase. Compared with ethyl acetate as the organic solvent, dichloromethane led to an apparent increase in nanoparticle size. The solubility of ethyl acetate in water is higher than that of dichloromethane in water. It is easy to spread in water. The polymer on the surface of the emulsion droplets therefore rapidly solidifies into balls without adhesion, leading to 6-MPNs having a smaller particle size, relatively speaking.29,30 Next, we evaluated the influence of the weight ratio of polymer to drug on the physicochemical properties of nanomedicines. The increment of PLGA-to-6-MP weight ratio slightly increased the particle size and it effectively enhanced the EEs. As polymer concentration in the organic phase increases, the viscosity of the organic phase heightens. As a result, the diffusional resistance of the drug increases. Therefore, the drug diffusion is restricted from the internal water phase to the continuous phase, entrapping more of the drug and leading to greater particle sizes.
In our study, we also prepared various 6-MPNs with PLGA polymers of different molecular weight and terminal groups, and accordingly observed that the molecular weight and terminal groups of PLGA dominantly affected the particle size.31 RG 502H with acid terminus and 7000–17,000 of Mw resulted in a smaller particle size of 6-MPNs than RG 502 and RG 503H. PLGA with lower Mw can lead to a smaller particle size compared to PLGA with high Mw.32 Moreover, PLGA with a higher hydrophilic terminal group could lead to smaller nanoparticle diameter. Thus, 6-MPNs prepared with PLGA containing hydrophilic carboxyl-terminal group (RG 502H and RG 503H) have slightly decreased particle size when compared with PLGA (RG 502) containing the alkyl end group.33
Based on the above results, we consider the formulation labeled F1 in Table 1 to be the optimal nanomedicine. In this formulation, 6-MP was dissolved in ammonia solutions containing 0.5% PVA, then added to ethyl acetate solutions of RG 502H PLGA with 6:1 of weight ratio for PLGA and 6-MP. F1 showed the highest encapsulation efficiency (80.71%) and drug loading (13.60%) with relatively small particle size (138.01 ± 0.39 nm) and narrow PDI (0.119 ± 0.003) (Figure 2A). We, therefore, selected this formulation of 6-MPNs for subsequent in vitro and in vivo studies.
 |
Figure 2 In vitro characterization and release of 6-MPNs. The particle size distribution determined by DLS (A), and morphology measured by TEM (B), scale bars: 100 nm. (C) The drug release of 6-MP from nanomedicines and suspensions in PBS (pH = 7.4) containing 0.02% Tween 20. The physical stability of 6-MPNs in different media including PBS (D), RPMI1640 (E) and RPMI 1640 with 10% FBS (F), monitored by particle size distribution over 48 h. |
The optimal formulation was slightly negative, with a zeta potential of −1.0 mV. The colloidal dispersion system of 6-MPNs appeared as light blue opalescence. TEM demonstrated that the nanomedicines were smooth-surfaced spheres with a core-shell structure (Figure 2B). We also evaluated the stability of 6-MPNs in different media. The 6-MPNs could maintain particle size in a proper range within 48 h in different media, including PBS (Figure 2D), RPMI 1640 (Figure 2E) and RPMI 1640 with 10% FBS (Figure 2F). This finding indicated that 6-MPNs were stable enough to use in the following cell experiments because the studies were all shorter than 48 h long. In general, the 6-MPNs we prepared had appropriate physicochemical properties and dispersion stability.
In vitro Release of 6-MP from 6-MPNs
As seen in Figures 2C and S1, 6-MPCs exhibited a gradual release up to 90% within 10 h at pH 7.4. 6-MP is more easily released from 6-MPCs at pH 7.4 than at pH 4.8 due to its higher solubility in an alkaline environment. In contrast, the drug release profile of 6-MPNs exhibited an initial burst release of the drug followed by a sustained release both at pH 4.8 and 7.4. The cumulative release percentage of 6-MPNs was 20% in the first 2 h and only achieved 50% within 96 h at pH 4.8. The release rate in the acidic medium was relatively slow. One could attribute this to the fact that the ester bond of PLGA is easily broken under basic conditions.34 However, compared with pH 4.8, we observed a higher and continuous release of 6-MP from the 6-MPNs incubated at pH 7.4. Specifically, the cumulative release percentage of 6-MPNs could achieve over 60% in the first 2 h. This stage was followed by a slow and sustained release of the remainder eventually up to 90% of cumulative released amount over the next 96 h. A possible reason for this biphasic release profile is that the 6-MP is encapsulated in the 6-MPNs and adsorbed on their surfaces.24 More specifically, the surface-adsorbed drug is expected to be released fast, while the drug entrapped within the stable core of nanomedicines will have a sustained release performance.35 We considered that the fast release would help achieve effective blood concentrations and promote rapid relief of diseases, while the sustained release would help maintain a stable blood concentration. In the release medium, we added the tween 20 to increase the solubility of 6-MP. This may enhance the release rate, but there was still a sustained release of 6-MPNs, possibly due to the slow degradation of PLGA skeleton in the absence of esterase in vitro.
The Growth Inhibition Evaluation of 6-MPNs in Jurkat Cells
In vitro Cytotoxicity Assay
After successfully constructing the nanomedicines, we subsequently evaluated the in vitro therapeutic efficacy of 6-MPNs by using a human T cell leukemia Jurkat cell line. We determined the impact of 6-MP solution, 6-MPCs and 6-MPNs on the Jurkat cells’ proliferation and viability in vitro. As displayed in Figure 3, 6-MP solution, 6-MPCs and 6-MPNs all significantly reduced the cell viability in a dose-dependent manner. There were no significant differences in the effects of cell growth inhibition between the three formulations, except at the concentration of 1.0 µM. The percentages of cell viability at a concentration of 1.0 µM of 48 h exposure were 25.29 ± 1.28%, 20.90 ± 5.68%, and 52.18 ± 1.23%, for 6-MP solution, 6-MPCs and 6-MPNs, respectively. The 50% inhibitory concentrations (IC50) of the 6-MP solution, 6-MPNs and 6-MPCs for Jurkat cells after 48 h of exposure were 0.36 µM, 1.09 µM and 0.76 µM, respectively. Indeed, drug release may impact the cytotoxicity. As shown in Figure 2C, the cumulative release percentage of 6-MPNs could achieve over 90% at 48 h, while 6-MPCs exhibited a release of up to 90% at 10 h and about 100% at 48 h. At the end of the cytotoxic incubation period, 6-MP was released more from 6-MPCs than 6-MPNs. In the 6-MPNs group, 6-MP could enter cells as free drugs after being released from 6-MPNs, or enter cells directly as encapsulated nanoparticles. The endocytosis of 6-MPNs was relatively slower than the cellular uptake of free 6-MP in 6-MPCs and solution.36 We speculate that these two reasons may cause the cytotoxicity of 6-MPNs to be lower than 6-MPCs and solutions at 48 h. There may be other reasons to explore.
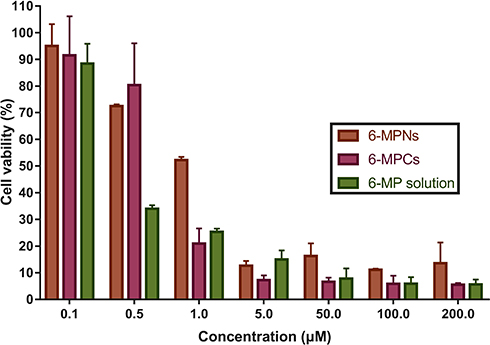 |
Figure 3 In vitro growth inhibition effect of 6-MPNs, 6-MPCs and 6-MP solution against Jurkat cells assessed by CCK8 assay after 48 h. |
Flow Cytometry Cell Apoptosis Analysis
We performed Annexin V-FITC and PI double staining to determine the apoptosis among Jurkat cells following treatment with the 6-MP solution, 6-MPCs and 6-MPNs at different concentrations for 48 h. With the rising concentrations of 6-MP, the apoptotic percentage of Jurkat cells increased. We also observed that 6-MPNs (Figure 4C) induced a significant late apoptosis effect at different concentrations in Jurkat cells compared with the 6-MP solution (Figure 4A) and 6-MPCs (Figure 4B). In the untreated control group, the apoptotic percentage was 5.89 ± 0.89%. In contrast, 6-MPNs treatment at different concentrations induced apoptosis rates of 14.56 ± 1.08%, 23.78 ± 1.22% and 30.58 ± 1.71% for 0.5 µM (Figure 4D), 2 µM (Figure 4E) and 5 µM (Figure 4F) respectively. The apoptotic percentages that the 6-MP solution treatment induced were 7.76 ± 0.82%, 16.30 ± 1.27% and 25.29 ± 0.80% at three concentrations, respectively. Similarly, the 6-MPCs treatment exhibited 5.14 ± 1.50%, 19.16 ± 0.59% and 23.04 ± 1.33%. These data revealed that 6-MPNs induced a significantly greater proportion of apoptotic cells than the 6-MP solution and 6-MPCs at all concentrations. One could speculate that compared to 6-MP solution and 6-MPCs, 6-MPNs entered cells relatively slowly and had a sustained release of 6-MP. The 6-MPNs group might therefore maintain a relatively higher concentration of 6-MP in the cells during the long exposure time, resulting in more DNA damage and further increasing cell apoptosis.37
 |
Figure 4 Flow cytometry analysis of Jurkat cells after staining with Annexin V-FITC/PI. Cells were treated with 6-MP solution (A), 6-MPCs (B) or 6-MPNs (C) at different concentrations of 0.5 μM, 2 μM and 5 μM, respectively. The quantitative apoptosis rates in 6-MPNs, 6-MP solution, and 6-MPCs groups were calculated as the sum of early and late apoptosis at different concentrations of 0.5 μM (D), 2 μM (E), 5 μM (F). The data are presented as the mean ± SD of three independent experiments (*P < 0.05; **P < 0.01). |
Pharmacokinetic Studies in One-Month-Old Rats
After SD rats were administered with 6-MPCs or 6-MPNs at a dose of 15.75 mg/kg, the mean plasma concentrations of 6-MP and its methylation metabolite, 6-MMP, were detected by HPLC-MS/MS. Figure 5 presents the mean plasma concentrations vs time profiles. The plasma concentrations of 6-MP for 6-MPNs reached a peak of 478.05 ± 233.00 ng/mL (Cmax) at 0.5 h. It was significantly greater than the peak for 6-MPCs (202.90 ± 94.29 ng/mL) (Figure 5A). The area under the curve (AUC) of 6-MP in the 6-MPNs group was 558.70 ± 110.80 mg/L·h, which was higher than that in the 6-MPCs group of 381.00 ± 71.20 mg/L·h. There was little difference in the half-life (t1/2) of 6-MP between the 6-MPNs and 6-MPCs groups (1.57 ± 1.09 h and 1.50 ± 0.94 h, respectively). 6-MPNs could thereby increase the Cmax and AUC of 6-MP in SD rats. This improvement in oral bioavailability could be explainable by the combination of the following effects. First, nanomedicines could improve the solubility of poorly water-soluble drugs.38 According to the biopharmaceutics classification system (BCS), 6-MP is a Class II drug with poor solubility and high permeation, wherein solubility limits absorption.39 In the current study, the nanomedicines we developed enhanced the solubility of 6-MP so that the 6-MP in 6-MPNs could be better absorbed compared to 6-MPCs. Second, orally administered nanomedicines can be absorbed by M cells of Peyer’s patches as long as the particle size is less than 200 nm. This leads to passive lymphatic targeting followed by increased systemic drug delivery.40
 |
Figure 5 Mean plasma concentrations (ng/mL) of 6-MP (A) and 6-MMP (B) vs Time (h) profiles after a single oral administration of 6-MPNs or 6-MPCs in SD rats (n = 8). |
Next, the metabolites of 6-MP formulate the main cause for the toxic effects of 6-MP. As a result, we further investigated the concentration of its metabolites, 6-MMP. As Figure 5B exhibits, the AUC of 6-MMP in the 6-MPNs group was 487.90 ± 74.25 mg/L·h, which was lower than that in the 6-MPCs group (568.80 ± 81.62 mg/L·h). The Cmax of 6-MMP did not differ between the two groups. There was no sustained release effect in vivo as the 6-MPNs did not evidently increase the half-life of the 6-MP. We can attribute this finding to the presence of esterase in vivo, which could degrade the PLGA skeleton via the hydrolysis of ester linkages.
Tissue Distribution of 6-MPNs in vivo
Figure 6 displays the 6-MP distribution in the duodenum (Figure 6A), jejunum (Figure 6B), ileum (Figure 6C) and colon (Figure 6D) of SD rats at various time points. In both the 6-MPNs group and 6-MPCs group, the concentrations of 6-MP in four intestine segments peaked at 10 min, then gradually decreased. These results indicate that 6-MP is rapidly distributed to the intestinal tissues after oral administration of 6-MPNs or 6-MPCs. The descending order of distribution of 6-MP in the four intestine segments was the ileum, jejunum, duodenum, and colon. The 6-MP absorption occurred mainly through the ileum and the jejunum. The duodenum distribution of 6-MP in the 6-MPNs group was significantly higher than that in the 6-MPCs group at 10 min, thereby confirming a higher penetration of nanomedicines in the duodenum compared with plain suspensions after oral delivery.40 In view of the poor oral absorption of 6-MP, the high intestinal permeability after nanoization is probable because the nanomedicines of small particle size are absorbed by intestinal enterocytes through endocytosis and transport, enhancing the drug’s paracellular passage and endocytotic uptake.41
 |
Figure 6 The absorption of 6-MP for 6-MPNs in duodenum (A), jejunum (B), ileum (C), and colon (D), compared with 6-MPCs (n = 6) (**P < 0.01). |
Figures 7 and S2 show the disposition of 6-MP in the bone marrow, heart, liver, spleen, lungs and kidneys. 6-MP distribution in SD rats was the highest in the spleen (Figure S2D), followed by the bone marrow (Figure 7A), kidney (Figure S2J), liver (Figure 7D), lung (Figure S2G) and heart (Figure S2A). The distribution of 6-MP mainly depended on the blood flow or perfusion rate of organs.42 There was no significant difference in the major organ distribution of 6-MP between the 6-MPNs and 6-MPCs groups. Furthermore, we evaluated the distribution of 6-MMP and 6-TG metabolites in the bone marrow (Figure 7B and C), heart (Figure S2B–C), liver (Figure 7E–F), spleen (Figure S2E–F), lungs (Figure S2H–I) and kidneys (Figure S2K–L). As Figure 7E illustrates, 6-MMP mainly accumulated in the liver. In the 6-MPNs group, the concentrations of 6-MMP in the liver were lower than those in the 6-MPCs group at 1 h, 3 h and 10 h (P < 0.01). 6-MMP is the methylated metabolite of the 6-MP. 6-MP could induce hepatotoxicity, likely because of the biotransformation of methylated mercaptopurine.43 Accordingly, we can infer that the risk of hepatotoxicity in the 6-MPNs group is lower than that in the 6-MPCs group. The 6-MP could cause myelotoxicity, possibly explained by the increased biotransformation of 6-TGNs.44 In the current study, we determined the concentrations of 6-TG in different organs to indirectly reflect the levels of the 6-TGNs in vivo, as 6-TG is the final product after the dephosphorylation of 6-TGNs. Figure 7C displays that there were large variabilities in 6-TG concentrations among different organs. 6-TG had the highest distribution in the bone marrow. The concentrations of 6-TG in the bone marrow at 1 h after 6-MPCs treatment were significantly higher than those after 6-MPNs treatment. We can speculate that the risk of myelotoxicity in the 6-MPNs group might be lower than in the 6-MPCs group. One possible cause for this concentration difference in bone marrow is that the direct metabolism of drugs by liver microsomal enzymes is avoidable following the encapsulation of 6-MP into the core of 6-MPNs, as nanomedicines could directly reach the systemic circulation.45,46
 |
Figure 7 The concentrations of 6-MP (A and D), 6-MMP (B and E), 6-TG (C and F) in bone marrow and liver, respectively, after treatment with 15.75 mg/kg of 6-MPNs and 6-MPCs in SD rats (n = 6) (*P < 0.05; ** P < 0.01). |
Overall, these results indicated that 6-MPNs could increase the intestinal absorption of 6-MP mainly through the duodenum, as well as reduce the accumulation of toxic metabolites in the liver and bone marrow.
Anticancer Efficacy of 6-MPNs in vivo
Finally, we performed the pharmacodynamic experiment of 6-MPNs against ALL as a way to verify the superior therapeutic efficacy in vivo. Firstly, we injected NPG mice with Jurkat cells to build an ALL model. After a week, mice were administered with 6-MPNs in doses of 20 mg of 6-MP equiv./kg every day for 14 days. PBS and 6-MPCs were used as the controls. Notably, not all the groups (treated and controls) exhibited a substantial loss in body weight within 16 days (Figure 8A), indicating that a lack of general toxicity resulting from the 6-MPCs or 6-MPNs. Comparatively, the PBS-treated group showed fast weight loss, while the 6-MPCs or 6-MPNs groups displayed slow weight loss. 6-MPNs significantly increased the survival rate of the mice compared to the 6-MPCs and PBS groups. The corresponding median survival time was 51 days versus 23.5 and 22.5 days, respectively (P < 0.05; Figure 8B). Interestingly, one mouse in the 6-MPNs group survived and returned to a normal state until the end of the 80-day observation period. These findings demonstrated that 6-MPNs could offer significantly better therapeutic efficacy on the human cell line xenograft in the ALL mouse model than 6-MPCs currently do. Methylated metabolite of 6-MP could cause liver toxicity, and liver and spleen are sites for leukemia cells’ blast accumulation and invasion. Therefore, we performed the H&E staining in livers and spleens to evaluate the toxicity and a leukemia burden in our different treatment groups. The histological analysis revealed no distinct damage to the livers and spleens in any of the three groups (Figure 9), while the PBS groups caused slight inflammatory cellular infiltration in the liver compared with the 6-MPCs and 6-MPNs groups. In conclusion, the enhanced anticancer effect of 6-MPNs is likely a result of its high bioavailability and low systemic toxicity.
 |
Figure 8 In vivo anticancer efficacy of 6-MPNs and 6-MPCs in ALL model mice. PBS was used as a control group. The drugs were given daily over 1–14 days (dosage: 20 mg of 6-MP equiv./kg). (A) Relative body weight changes (n = 6). (B) Survival rates of NPG mice. P = 0.0101, 6-MPNs vs PBS; P = 0.0316, 6-MPNs vs 6-MPCs. |
 |
Figure 9 Livers and spleens of mice from different groups were embedded with paraffin and stained with hematoxylin and eosin (H&E). Scale bar, 50 μm. |
Conclusions
In this study, we prepared the PLGA nanomedicines loaded with 6-MP using the double-emulsion solvent evaporation method. 6-MPNs characterization revealed that the nanomedicines had good physicochemical properties and dispersion stability. 6-MPNs exhibited an initial burst release followed by a sustained release of 6-MP. The in vitro apoptosis analysis in Jurkat cells demonstrated that 6-MPNs possessed enhanced a pro-apoptotic effect compared with 6-MPCs and 6-MP solution. The pharmacokinetics profiles showed that 6-MPNs could increase the Cmax and AUC of 6-MP in SD rats. Accordingly, in SD rats, the oral bioavailability of the 6-MPNs was better than that of 6-MPCs. The results of the tissue distribution demonstrated that 6-MPNs could improve the intestinal absorption of 6-MP, and reduced the accumulation of its toxic metabolites in the liver and bone marrow. The in vivo pharmacodynamic studies further confirmed its good treatment effect against ALL mice model with low systemic toxicity. In summary, 6-MPNs could be a valuable nanomedicine for 6-MP. Further research is required so as to explore the commercial potential of this novel drug delivery system.
Acknowledgments
The authors are grateful to the Children’s Health Research Institute at Beijing Children’s hospital. This work was supported by Beijing Natural Science Foundation, China (Grant No. 7204261, L202042, L202048 and 7171004), Beijing New-star Plan of Science and Technology (Grant No. Z201100006820009) and National Science and Technology Major Project (Grant No. 2018ZX09721003).
Disclosure
Yaru Zou and Dong Mei are co-first authors. The authors declare that they have no competing interests.
References
1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541–1552. doi:10.1056/NEJMra1400972
2. Wu C, Li W. Genomics and pharmacogenomics of pediatric acute lymphoblastic leukemia. Crit Rev Oncol Hematol. 2018;126:100–111. doi:10.1016/j.critrevonc.2018.04.002
3. Rudin S, Marable M, Huang RS. The promise of pharmacogenomics in reducing toxicity during acute lymphoblastic leukemia maintenance treatment. Genomics Proteomics Bioinformatics. 2017;15(2):82–93. doi:10.1016/j.gpb.2016.11.003
4. Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7(6):e577. doi:10.1038/bcj.2017.53
5. Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011;29(5):532–543. doi:10.1200/JCO.2010.30.1382
6. Inaba H, Pui C-H. Immunotherapy in pediatric acute lymphoblastic leukemia. Cancer Metastasis Rev. 2019;38(4):595–610. doi:10.1007/s10555-019-09834-0
7. Schmiegelow K, Nielsen SN, Frandsen TL, Nersting J. Mercaptopurine/Methotrexate maintenance therapy of childhood acute lymphoblastic leukemia: clinical facts and fiction. J Pediatr Hematol Oncol. 2014;36(7):503–517. doi:10.1097/MPH.0000000000000206
8. Gaynon PS. Mercaptopurine in childhood acute lymphoblastic leukaemia. Lancet Oncol. 2017;18(4):425–426. doi:10.1016/S1470-2045(17)30152-3
9. Kumar GP, Sanganal JS, Phani AR, et al. Anti-cancerous efficacy and pharmacokinetics of 6-mercaptopurine loaded chitosan nanoparticles. Pharmacol Res. 2015;100:47–57. doi:10.1016/j.phrs.2015.07.025
10. Moriyama T, Nishii R, Perez-Andreu V, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet. 2016;48(4):367–373. doi:10.1038/ng.3508
11. Ali I, Salim K, A Rather M, A Wani W, Haque A. Advances in nano drugs for cancer chemotherapy. Curr Cancer Drug Targets. 2011;11(2):135–146. doi:10.2174/156800911794328493
12. Wei X, Liao J, Davoudi Z, et al. Folate receptor-targeted and GSH-responsive carboxymethyl chitosan nanoparticles containing covalently entrapped 6-mercaptopurine for enhanced intracellular drug delivery in leukemia. Mar Drugs. 2018;16(11):439. doi:10.3390/md16110439
13. Wang X, Cai X, Hu J, et al. Glutathione-triggered “off-on” release of anticancer drugs from dendrimer-encapsulated gold nanoparticles. J Am Chem Soc. 2013;135(26):9805–9810. doi:10.1021/ja402903h
14. Neerman MF. The efficiency of a PAMAM dendrimer toward the encapsulation of the antileukemic drug 6-mercaptopurine. Anticancer Drugs. 2007;18(7):839–842. doi:10.1097/CAD.0b013e32809ef9d0
15. Viudez AJ, Madueno R, Pineda T, Blazquez M. Stabilization of gold nanoparticles by 6-mercaptopurine monolayers. Effects of the solvent properties. J Phys Chem B. 2006;110(36):17840–17847. doi:10.1021/jp062165l
16. Sierpe R, Noyong M, Simon U, et al. Construction of 6-thioguanine and 6-mercaptopurine carriers based on betacyclodextrins and gold nanoparticles. Carbohydr Polym. 2017;177:22–31. doi:10.1016/j.carbpol.2017.08.102
17. Wang W, Fang C, Wang X, et al. Modifying mesoporous silica nanoparticles to avoid the metabolic deactivation of 6-mercaptopurine and methotrexate in combinatorial chemotherapy. Nanoscale. 2013;5(14):6249–6253. doi:10.1039/c3nr00227f
18. Dorniani D, Hussein MZ, Kura AU, Fakurazi S, Shaari AH, Ahmad Z. Preparation and characterization of 6-mercaptopurine-coated magnetite nanoparticles as a drug delivery system. Drug Des Devel Ther. 2013;7:1015–1026. doi:10.2147/DDDT.S43035
19. Zhao Q, Geng H, Wang Y, et al. Hyaluronic acid oligosaccharide modified redox-responsive mesoporous silica nanoparticles for targeted drug delivery. ACS Appl Mater Interfaces. 2014;6(22):20290–20299. doi:10.1021/am505824d
20. Qiu J, Cheng R, Zhang J, et al. Glutathione-sensitive hyaluronic acid-mercaptopurine prodrug linked via carbonyl vinyl sulfide: a robust and CD44-targeted nanomedicine for leukemia. Biomacromolecules. 2017;18(10):3207–3214. doi:10.1021/acs.biomac.7b00846
21. Park J, Jeon WI, Lee SY, et al. Confocal Raman microspectroscopic study of folate receptor-targeted delivery of 6-mercaptopurine-embedded gold nanoparticles in a single cell. J Biomed Mater Res A. 2012;100(5):1221–1228. doi:10.1002/jbm.a.33294
22. Swider E, Koshkina O, Tel J, Cruz LJ, de Vries I, Srinivas M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018;73:38–51. doi:10.1016/j.actbio.2018.04.006
23. Rezvantalab S, Drude NI, Moraveji MK, et al. PLGA-based nanoparticles in cancer treatment. Front Pharmacol. 2018;9:1260. doi:10.3389/fphar.2018.01260
24. Lee YS, Johnson PJ, Robbins PT, Bridson RH. Production of nanoparticles-in-microparticles by a double emulsion method: a comprehensive study. Eur J Pharm Biopharm. 2013;83(2):168–173. doi:10.1016/j.ejpb.2012.10.016
25. Haggag YA, Osman MA, El-Gizawy SA, et al. Polymeric nano-encapsulation of 5-fluorouracil enhances anti-cancer activity and ameliorates side effects in solid Ehrlich Carcinoma-bearing mice. Biomed Pharmacother. 2018;105:215–224. doi:10.1016/j.biopha.2018.05.124
26. Hawwa AF, Millership JS, Collier PS, McElnay JC. Development and validation of an HPLC method for the rapid and simultaneous determination of 6-mercaptopurine and four of its metabolites in plasma and red blood cells. J Pharm Biomed Anal. 2009;49(2):401–409. doi:10.1016/j.jpba.2008.10.045
27. Mei S, Li X, Gong X, et al. LC-MS/MS analysis of erythrocyte thiopurine nucleotides and their association with genetic variants in patients with neuromyelitis optica spectrum disorders taking azathioprine. Ther Drug Monit. 2017;39(1):5–12. doi:10.1097/FTD.0000000000000362
28. Wu X, Wang L, Qiu Y, Zhang B, Hu Z, Jin R. Cooperation of IRAK1/4 inhibitor and ABT-737 in nanoparticles for synergistic therapy of T cell acute lymphoblastic leukemia. Int J Nanomedicine. 2017;12:8025–8034. doi:10.2147/IJN.S146875
29. Haque S, Boyd BJ, McIntosh MP, Pouton CW, Kaminskas LM, Whittaker M. suggested procedures for the reproducible synthesis of poly(d,l-lactide-co-glycolide) nanoparticles using the emulsification solvent diffusion platform. Curr Nanosci. 2018;14(5):448–453. doi:10.2174/1573413714666180313130235
30. Ekanem EE, Nabavi SA, Vladisavljević GT, Gu S. Structured biodegradable polymeric microparticles for drug delivery produced using flow focusing glass microfluidic devices. ACS Appl Mater Interfaces. 2015;7(41):23132–23143. doi:10.1021/acsami.5b06943
31. Ding D, Zhu Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater Sci Eng C Mater Biol Appl. 2018;92:1041–1060. doi:10.1016/j.msec.2017.12.036
32. Rafiei P, Haddadi A. A robust systematic design: optimization and preparation of polymeric nanoparticles of PLGA for docetaxel intravenous delivery. Mater Sci Eng C Mater Biol Appl. 2019;104:109950. doi:10.1016/j.msec.2019.109950
33. Kranz H, Bodmeier R. A novel in situ forming drug delivery system for controlled parenteral drug delivery. Int J Pharm. 2007;332(1–2):107–114. doi:10.1016/j.ijpharm.2006.09.033
34. Singh V, Singh S, Das S, Kumar A, Self WT, Seal S. A facile synthesis of PLGA encapsulated cerium oxide nanoparticles: release kinetics and biological activity. Nanoscale. 2012;4(8):2597–2605. doi:10.1039/c2nr12131j
35. Wright L, Rao S, Thomas N, Boulos RA, Prestidge CA. Ramizol(®) encapsulation into extended release PLGA micro- and nanoparticle systems for subcutaneous and intramuscular administration: in vitro and in vivo evaluation. Drug Dev Ind Pharm. 2018;44(9):1451–1457. doi:10.1080/03639045.2018.1459676
36. Daglioglu C, Okutucu B. Synthesis and characterization of AICAR and DOX conjugated multifunctional nanoparticles as a platform for synergistic inhibition of cancer cell growth. Bioconjug Chem. 2016;27(4):1098–1111. doi:10.1021/acs.bioconjchem.6b00080
37. Anand P, Nair HB, Sung B, et al. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol. 2010;79(3):330–338. doi:10.1016/j.bcp.2009.09.003
38. Shekhawat PB, Pokharkar VB. Understanding peroral absorption: regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm Sin B. 2017;7(3):260–280. doi:10.1016/j.apsb.2016.09.005
39. Wang JR, Yu X, Zhou C, et al. Improving the dissolution and bioavailability of 6-mercaptopurine via co-crystallization with isonicotinamide. Bioorg Med Chem Lett. 2015;25(5):1036–1039. doi:10.1016/j.bmcl.2015.01.022
40. Joshi G, Kumar A, Sawant K. Bioavailability enhancement, Caco-2 cells uptake and intestinal transport of orally administered lopinavir-loaded PLGA nanoparticles. Drug Deliv. 2016;23(9):3492–3504. doi:10.1080/10717544.2016.1199605
41. Joshi G, Kumar A, Sawant K. Enhanced bioavailability and intestinal uptake of Gemcitabine HCl loaded PLGA nanoparticles after oral delivery. Eur J Pharm Sci. 2014;60:80–89. doi:10.1016/j.ejps.2014.04.014
42. Kurowski V, Iven H. Plasma concentrations and organ distribution of thiopurines after oral application of azathioprine in mice. Cancer Chemother Pharmacol. 1991;28(1):7–14. doi:10.1007/BF00684949
43. Umrethia M, Ghosh PK, Majithya R, Murthy RS. 6-mercaptopurine (6-MP) entrapped stealth liposomes for improvement of leukemic treatment without hepatotoxicity and nephrotoxicity. Cancer Invest. 2007;25(2):117–123. doi:10.1080/07357900701224862
44. Gunnarsdottir S, Elfarra AA. Distinct tissue distribution of metabolites of the novel glutathione-activated thiopurine prodrugs cis-6-(2-acetylvinylthio)purine and trans-6-(2-acetylvinylthio)guanine and 6-thioguanine in the mouse. Drug Metab Dispos. 2003;31(6):718–726.
45. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan W. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. doi:10.1016/j.jconrel.2016.01.020
46. Govindappa PK, Joladarashi D, Hallur R, Sanganal JS, Phani AR. Toxicity evaluation of 6-mercaptopurine-Chitosan nanoparticles in rats. Saudi Pharm J. 2020;28(1):147–154. doi:10.1016/j.jsps.2019.11.018
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.