Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Preparation and physical characterization of calcium sulfate cement/silica-based mesoporous material composites for controlled release of BMP-2
Authors Tan H, Yang S, Dai P, Li W, Yue B
Received 31 March 2015
Accepted for publication 18 May 2015
Published 7 July 2015 Volume 2015:10(1) Pages 4341—4350
DOI https://doi.org/10.2147/IJN.S85763
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Honglue Tan,1 Shengbing Yang,2 Pengyi Dai,1 Wuyin Li,1 Bing Yue2
1Luoyang Orthopedics and Traumatology Institution, Luoyang Orthopedic-Traumatological Hospital, Luoyang, 2Shanghai Key Laboratory of Orthopedic Implants, Department of Orthopedic Surgery, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
Abstract: As a commonly used implant material, calcium sulfate cement (CSC), has some shortcomings, including low compressive strength, weak osteoinduction capability, and rapid degradation. In this study, silica-based mesoporous materials such as SBA-15 were synthesized and combined with CSC to prepare CSC/SBA-15 composites. The properties of SBA-15 were characterized by X-ray diffraction, transmission electron microscopy, and nitrogen adsorption–desorption isotherms. SBA-15 was blended into CSC at 0, 5, 10, and 20 wt%, referred to as CSC, CSC-5S (5% mass ratio), CSC-10S (10% mass ratio), and CSC-20S (20% mass ratio), respectively. Fourier-transform infrared spectroscopy and compression tests were used to determine the structure and mechanical properties of the composites, respectively. The formation of hydroxyapatite on composite surfaces was analyzed using scanning electron microscopy and X-ray diffraction after soaking in simulated body fluid. BMP-2 was loaded into the composites by vacuum freeze-drying, and its release characteristics were detected by Bradford protein assay. The in vitro degradation of the CSC/SBA-15 composite was investigated by measuring weight loss. The results showed that the orderly, nanostructured, mesoporous SBA-15 possessed regular pore size and structure. The compressive strength of CSC/SBA-15 increased with the increase in SBA-15 mass ratio, and CSC-20S demonstrated the maximum strength. Compared to CSC, hydroxyapatite that formed on the surfaces of CSC/SBA-15 was uniform and compact. The degradation rate of CSC/SBA-15 decreased with increasing mass ratio of SBA-15. The adsorption of BMP-2 increased and released at a relatively slow rate; the release rate of BMP-2 in CSC-20S was the slowest, and presented characteristics of low doses of release. In vitro experiments demonstrated that the physical properties of pure CSC incorporated with SBA-15 could be improved significantly, which made the CSC/SBA-15 composite more suitable for bone repair and bone-tissue engineering.
Keywords: calcium sulfate cement (CSC), SBA-15, physical characterization, BMP-2
Introduction
With its biocompatible, biodegradable, and easily moldable properties, injectable calcium sulfate cement (CSC) is currently used to augment the anterior column of fractured vertebra.1–3 However, maintaining body height requires a balance between bone healing and graft reabsorption. If graft absorption occurs faster than new bone formation, the bone height will gradually collapse. One of the disadvantages of using CSC in vivo is that its resorption rate is faster than the rate of new bone formation, which leads to the development of immature bone in the bone defect.4–6 According to the literature, CSC can be completely resorbed within 4–8 weeks, while no obvious bone tissue had formed in the vertebral body defect.3,7 Furthermore, the rapid resorption of CSC significantly influences the quantity of newly formed bone, due to the occupation of the bone defect by avascular fibrous tissue,8 thus decreasing the mechanical properties of the fractured vertebral body, and ultimately promoting the loss of correction of local kyphosis and vertebral body height.3 Moreover, the osteoinduction of CSC to promote osteogenic cell attachment, proliferation, and osteogenic differentiation is poor.9 Therefore, to meet the clinical requirements for bone-defect repair, developing new bone cements based on CSC that have ideal degradability, mechanical performance, and bioactivity should be concerned.
Mesoporous materials have recently received considerable attention, due to their bioactivity to osteogenic cells, especially in the promotion of osteoblastic activity and bone-defect repair.10–13 Zhao et al14 first synthesized the silica-based mesoporous material, SBA-15, which showed great potential for new biological applications because of its uniformly distributed and adjustable nanopores, functionalized surfaces, and good biocompatibility. Mesoporous material has been reported as a drug carrier, and it has been shown that drug molecules can be distributed into mesopores with time- and quantity-controlled delivery.15,16 SBA-15 as a carrier material for drug release has attracted much attention, and it accommodates higher drug loads with a better sustained-release effect compared to other materials.15 BMP-2 has been proved to induce osteogenic differentiation of the stem cell and promote bone formation.17,18 However, in vivo osteoinduction of BMP-2 may be compromised, owing to its easy loss and degradation without a carrier.17
Based on the preceding analysis, this study synthesized mesoporous SBA-15 and blended it into CSC at different mass ratios to construct a CSC/SBA-15 composite. The composite was assumed to overcome the drawbacks of pure CSC and present characteristics of sustained release of BMP-2.
Materials and methods
Synthesis of mesoporous SBA-15
P123 (4 g; Sigma-Aldrich Co, St Louis, MO, USA) was dissolved in water (30 mL), followed by the addition of 2 M HCl (120 mL). After 3 hours of vigorous stirring, 8.5 g tetraethyl orthosilicate (Shanghai Ling Feng Chemical Reagent Co, Ltd, Shanghai, People’s Republic of China [PRC]) was dripped into the solution at room temperature and stirred continuously for 24 hours. The mixed solution was then transferred to an autoclave to react for 48 hours at 100°C. The obtained precipitate was filtered and washed three times with deionized water and anhydrous ethanol, then dried in a vacuum-drying oven (DHG-9203A; Shanghai Jing Hong Laboratory Instrument Co, Ltd, PRC) at 60°C overnight. A white product, SBA-15, was obtained after calcination in a muffle furnace at 550°C for 5 hours to remove the template.
Characterization of SBA-15
X-ray diffraction
SBA-15 was characterized by a rotating anode X-ray polycrystalline diffractometer (D/Max 2550 VB/PC; Rigaku Corporation, Tokyo, Japan) using fixed monochromatized Cu-Ka radiation (λ =0.15406 nm) energized at 40 kV and 30 mA. The samples were measured at room temperature in a wide-scan range of 10°–80° (2θ) and small-scan range of 0.8°–10° (2θ) with a scan rate of 2°/min.
Transmission electron microscopy
Samples were ground into powder, then transferred to copper grids after ultrasonic dispersion in ethanol for 10 minutes and observed by high-resolution transmission electron microscopy (JEM-2100F; JEOL Ltd, Tokyo, Japan).
Specific surface area and pore volume
These measurements were performed with an automatic physisorption instrument (TriStar 3000 analyzer; Micromeritics Instrument Corporation, Norcross, GA, USA). Pore-structure characterization was obtained using low-temperature nitrogen adsorption. SBA-15 was activated for 4 hours at 573 K and 0.2–1.0 kPa to remove physically absorbed water, and the adsorption–desorption isotherms were measured. The specific surface area was calculated by the Brunauer–Emmett–Teller method, and the average pore size, pore volume, and pore distribution were obtained by the Barrett–Joyner–Halenda method.
Preparation of CSC/SBA-15 for mechanical test
SBA-15 was blended homogeneously into CSC (Sigma-Aldrich) at 0%, 5%, 10%, and 20% mass ratios. The powder was subsequently mixed with simulated body fluid (SBF) at a ratio of 1 mL/4 g in a bowl until the powder was fully wet. The compound was transferred to a polytetrafluoroethylene mold with a cylindrical hole of 6 mm diameter and 10 mm height, and was pressed between two metal plates for 24 hours. After the cement hardened, it was pulled out of the mold. Porous samples were prepared with the same method, but 60% NaCl with a particle size of 300–500 μm was added to the powder as the pore-forming agent, and the samples were immersed in deionized water for 72 hours to remove NaCl. According to the mass ratio of the SBA-15, four groups were included in this study: CSC (no SBA-15), CSC-5S (5% mass ratio), CSC-10S (10% mass ratio), and CSC-20S (20% mass ratio). The porous samples were tested only in the mechanical experiment, and not in the other experiments.
Fourier-transform infrared spectroscopy
The samples from the four groups were ground into powder and dried at 80°C in an oven. Samples were prepared by mixing with KBr at a 1:100 ratio under high-temperature argon lamp irradiation. The mixed powders were then transferred to stainless steel molds. Fourier-transform infrared spectroscopy (FT-IR) analysis (Nicolet 5700; Thermo Fisher Scientific, Waltham, MA, USA) was conducted with a resolution of 0.09 cm−1 and a scan range of 4,000–400 cm−1.
Mechanical experiments with CSC/SBA-15
Compressive strength and modulus were determined using a universal testing machine (HY-1080; Shanghai Hengyi Group Co Ltd, Shanghai, PRC), operating at a cross head speed of 5 mm/min. Load and deflection were recorded until the failure of each sample. Load–deformation and stress–strain curves were plotted, and the strength and modulus automatically calculated by a computer. Five samples were tested for each group.
Simulated body fluid-soaking experiment
The SBF solution was prepared according to the literature (Na+ 142.0, K+ 5.0, Mg2+ 1.5, Ca2+ 2.5, Cl− 147.8, HCO3− 4.2, HPO42− 1.0, SO42− 0.5 mol/m3).19 Three nonporous samples of each composite were soaked in 40 mL of SBF solution at a temperature of 37°C while being shaken at 100 rpm. The solution was replaced with fresh SBF every 3 days. After 7 and 14 days, the specimens were removed and dried overnight at a temperature of 60°C, then sputter-coated with gold, and the existence and morphology of the apatite formed on the surface were observed using scanning electron microscopy (JSM-6310LV; JEOL). In order to determine the apatite formation, wide-angle X-ray diffraction (XRD) was performed on an X-ray diffractometer (D/Max-2550 V) using fixed monochromatized Cu-Ka radiation (λ =0.15406 nm) energized at 40 kV and 40 mA. The samples were measured at room temperature in a scan range of 10°–80° (2θ) with a scan rate of 0.02°/min.
Adsorption and release characteristics of BMP-2 from CSC/SBA-15
Adsorption of BMP-2
Dried aqueous BMP-2 (Shanghai Rebone Biomaterials Co, Ltd, Shanghai, PRC) was dissolved in a diluted solution of acetic acid (2%) at a concentration of 0.5 mg/mL. The porous cylindrical samples were soaked in the BMP-2 solution. The optical density of the BMP-2 solution was measured every 5 minutes until it reached a constant value. The adsorption of BMP-2 was calculated from the residual BMP-2 in solution according to the Bradford protein assay.
Adsorption and release characteristics of BMP-2
A total of 100 μL BMP-2 was dripped into each CSC/SBA-15 composite by pipette to ensure each sample contained approximately 50 μg of BMP-2. The samples were placed in a vacuum desiccator for 30 minutes to assist BMP-2 adsorption. The samples loaded with BMP-2 were freeze-dried overnight at −40°C and stored at 4°C for further study. The release solution was prepared as follows: 7.996 g NaCl, 0.353 g NaHCO3, 0.224 g KCl, 0.228 g K2HPO4·3H2O, 0.305 g MgCl2·6H2O, 0.045 g HCl, 0.278 g CaCl2, 0.071 g Na2SO4, and 6.057 g CNH2(CH2OH)3. The pH of the solution (1 L) was adjusted to 7.25 and stored at 37°C. The CSC/SBA-15 loaded with 50 μg BMP-2 was placed into a 10 mL centrifuge tube containing 3 mL of release solution. It was then shaken at a speed of 50 rpm. The supernatant was removed to measure the release quantity of BMP-2 by Bradford protein assay after 0, 2, 6, 14, 22, 32, 40, 48, 58, 68, 78, 88, 98, 108, 118, 128, 138, 148, and 158 hours. The release properties were investigated by the protein-release curve, which was drawn according to the percentage of released BMP-2 at different times.
Degradation of CSC/SBA-15 composite in vitro
CSC/SBA-15 composite was used for degradation testing, prepared according to the aforementioned method, and samples with diameter of 6 mm and height of 10 mm. The total weight of each sample was 0.5 g. Sample degradation was measured by the weight-loss method. Five samples of each group were put into a polythene bottle containing 50 mL Tris-HCl. The solutions were shaken at a speed of 100 rpm for 15 days, and the samples were removed daily to dry to a constant weight. Weight loss was calculated by the following formula:
|
|
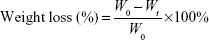
where W0 is the original weight and Wt is the weight of a sample immersed for time t.
Statistical analysis
All experiments were performed in triplicate. The results are reported as means ± standard deviation. The results were tested using one-way analysis of variance with Tukey’s multiple-comparison tests. The differences observed between samples were considered to be significant at P<0.05 (SAS Institute Inc, Cary, NC, USA).
Results
Characterization of ordered mesoporous SBA-15
TEM
Figures 1A1 and A2 show the ordered mesoporous material SBA-15. It can be seen that the pore sizes are uniform. The high-magnification image (Figure 1A2) indicates the long-range orderly mesopores of the synthesized SBA-15 throughout the sphere. Figures 1A3 and A4 show the sample without the addition of the pore-forming reagent P123. It was observed that the appearance was completely random, and there was no mesoporous structure under high magnification.
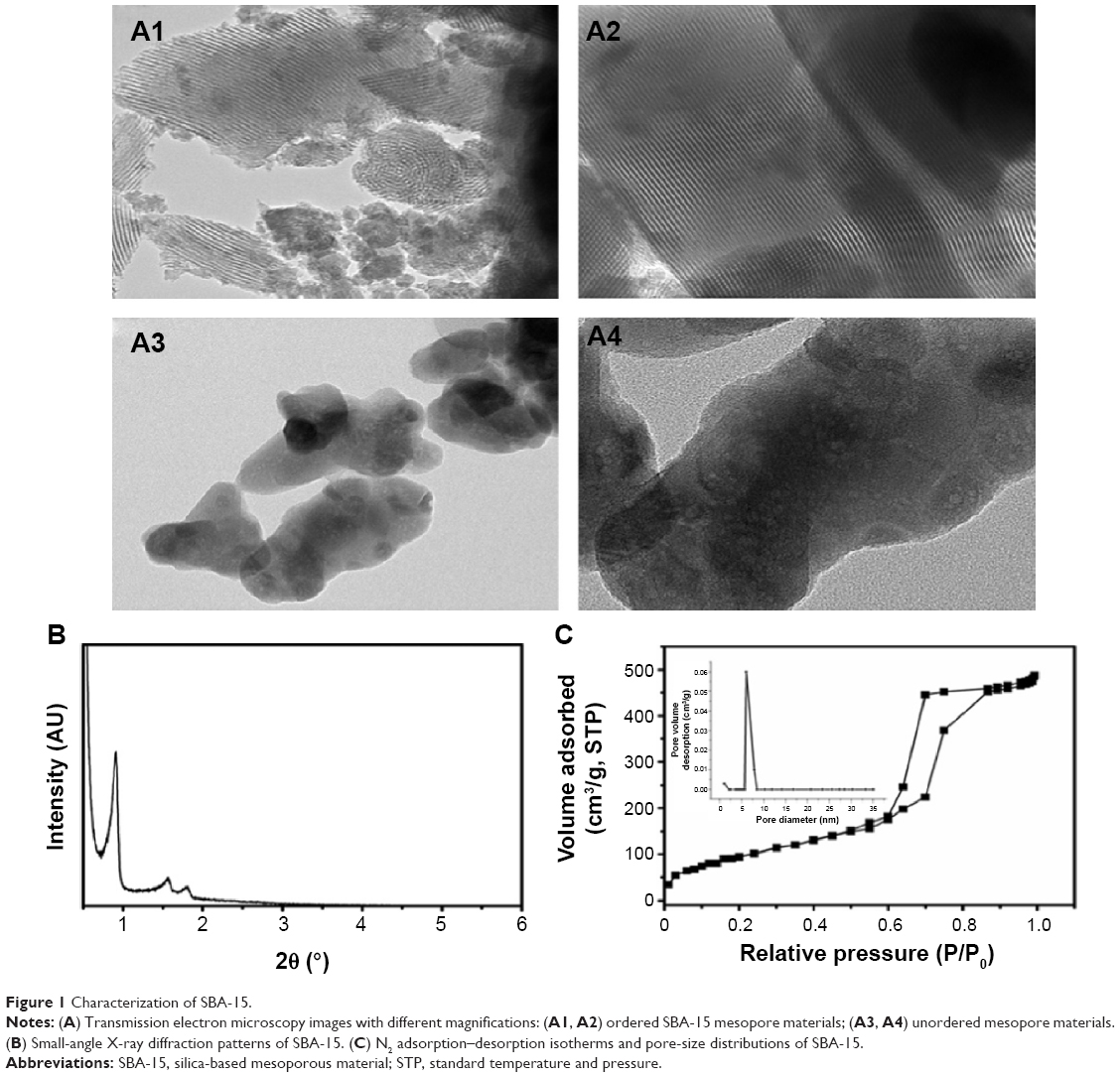 | Figure 1 Characterization of SBA-15. |
Small-angle XRD
Three characteristic diffraction peaks were observed (Figure 1B). There was a strong diffraction peak at 2θ =0.9°, corresponding to the (100) crystal plane, and two weak diffraction peaks between 1° and 2° that were representative of the (110) and (200) crystal planes. The results indicated that SBA-15 possessed excellent order, with a 2-D hexagonal mesoporous structure.
N2 adsorption–desorption isotherm
Figure 1C shows the N2 adsorption–desorption isotherm and the pore-size distribution of SBA-15. The isotherms of the mesoporous material could be described as Langmuir isotherms. The isotherms exhibited an abrupt change over the range of P/P0 =0.6–0.8, which was due to capillary condensation of adsorbed N2 molecules in mesoporous pores with narrow size distribution at low temperature and the rapid increase of N2 adsorption. As the relative pressure increased to 0.9–1.0 kPa, the isotherm exhibited another abrupt change, indicating the presence of regular and cylindrical mesopores with uniform aperture height. The pore-size distribution is shown in the inset photograph in Figure 1C. This plot shows that the pores are distributed regularly and uniformly. The specific surface area was 750 m2/g, pore size was 5.8 nm, and pore volume was 1.2 mL/g. These values demonstrated that SBA-15 was a mesoporous material.
FT-IR spectra of CSC/SBA-15
FT-IR spectra of CSC/SBA-15 are shown in Figure 2. There were three absorption peaks located at 462.1, 808.6, and 1,085.1 cm−1 that were related to the bending vibration of the Si–O band and the asymmetric and symmetric stretching vibrations of the Si–O–Si band, respectively. The peak located approximatley 972.2 cm−1 belonged to the stretching vibration of Si–OH at the surface of SiO2. The absorption peak located at 1,626.9 cm−1 was attributed to the bending mode of water molecules due to the absorbed H2O when the materials were exposed to air. The broad peak approximatley 3,402.7 cm−1 was the absorbance band of free hydroxyls. Compared to CSC, the chemical composition of CSC/SBA-15 was unchanged, implying that the composite was a physical mixture.
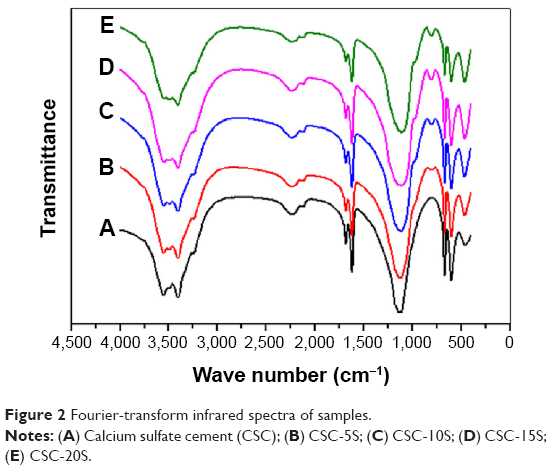 | Figure 2 Fourier-transform infrared spectra of samples. |
Mechanical properties of CSC/SBA-15
Figure 3A shows the stress–strain curve of CSC/SBA-15. The maximum mechanical loading of the composite increased with increasing SBA-15 mass ratio. Figures 3B1 and B2 show that the compressive strength of CSC/SBA-15 also increased with increasing SBA-15 quantity. The average compressive strengths of porous CSC, CSC-5S, CSC-10S, and CSC-20S were 7.2±2.1 MPa, 9.8±2.3 MPa, 15.81±4.4 MPa, and 22.72±6.56 MPa, respectively; a statistical difference existed in both groups (P<0.05) (Figure 3B1). Figure 3B2 shows that the mean compressive strengths of nonporous CSC, CSC-5S, CSC-10S, and CSC-20S were 18.82±2.54 MPa, 21.5±2.87 MPa, 29.71±4.29 MPa, and 47.72±9.56 MPa, respectively. Compared to CSC and CSC-5S, the strengths of CSC-10S and CSC-20S had obvious increases (P<0.05). There was a significant difference between CSC-10S and CSC-20S (P<0.01).
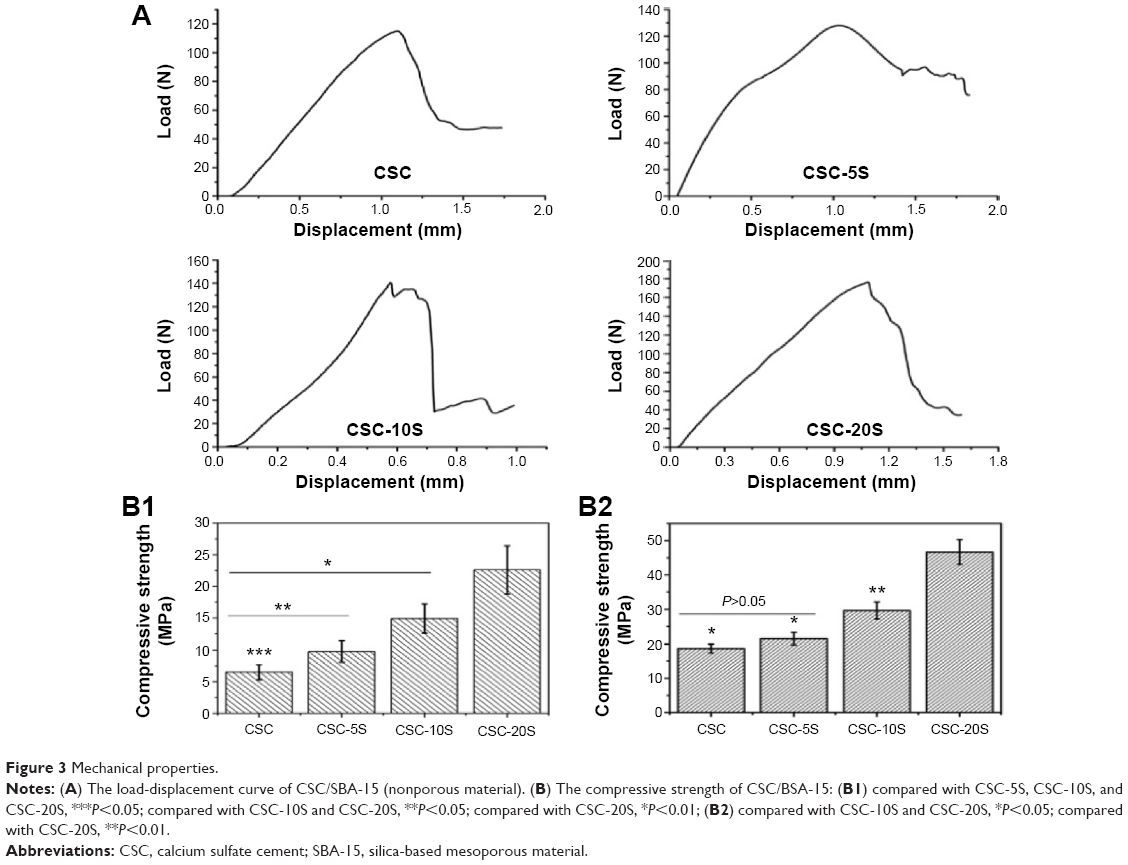 | Figure 3 Mechanical properties. |
Formation of hydroxyapatite after SBF soaking
Figure 4 shows no obvious formation of hydroxyapatite (HA) on the surface of CSC after soaking in SBF for 7 days. However, spherical HA particles formed on the surfaces of CSC-5S, CSC-10S, and CSC-20S. The spherical particles aggregated to form layered HA on the surfaces of CSC-10S and CSC-20S.
 | Figure 4 Scanning electron microscopy after 7-day immersion in simulated body fluid. |
Figure 5 shows that HA formed on the surface of the compound material after soaking in SBF for 14 days. Compared to CSC, the HA crystals were more uniform and compact on the surfaces of CSC-5S, CSC-10S, and CSC-20S. The HA on the CSC-20S surface was thick and coral-like.
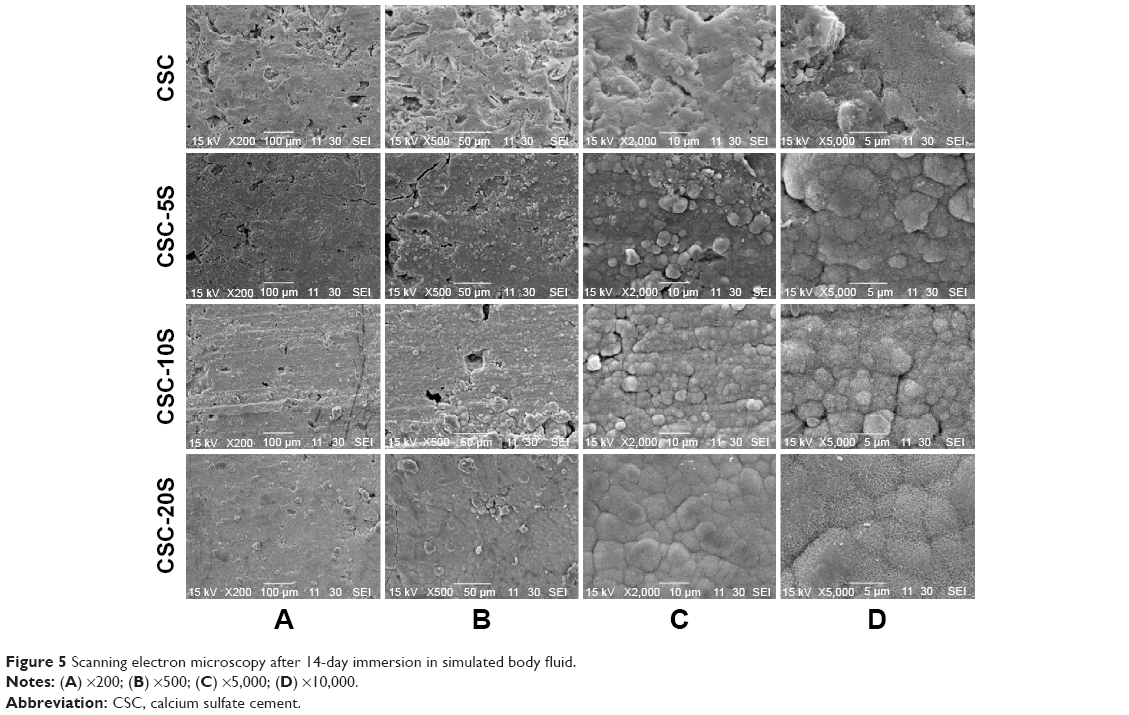 | Figure 5 Scanning electron microscopy after 14-day immersion in simulated body fluid. |
XRD of SBF soaking
Figure 6 shows an XRD pattern of compound surfaces after 14-day immersion in SBF. The diffraction peaks (2θ =25.9°, 32.2°, and 49.6°) corresponding to HA were present in all compounds. The diffraction peaks increased in intensity with the increasing mass percentage of SBA-15. The results indicated that the degradation rate on the surface of compound material reduced after the addition of SBA-15, which benefited the deposition and formation of HA.
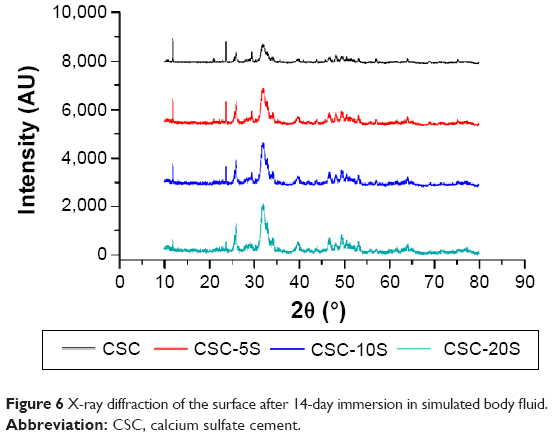 | Figure 6 X-ray diffraction of the surface after 14-day immersion in simulated body fluid. |
BMP-2 adsorption and release
The adsorption and release of BMP-2 in the porous CSC/SBA-15 composite are shown in Figure 7. Figure 7A indicates that the adsorption of BMP-2 increased with increasing mass percentage of SBA-15, which might have been due to the large specific surface area of SBA-15 and the increased adsorption of BMP-2 after SBA-15 addition. BMP-2 loading in CSC, CSC-5S, CSC-10S, and CSC-20S was 18±0.9 μg, 19.3±0.97 μg, 24.7±1.1 μg, and 36.7±1.63 μg, respectively. Compared to CSC, CSC-5S, and CSC-10S, there was a statistical difference for BMP-2 loading in CSC-20S (P<0.05). Figure 7B shows the cumulative quantity of BMP-2 that was released from CSC/SBA-15. The release rate of BMP-2 slowed with increasing mass percentage of SBA-15. After 24 hours, the released quantities from CSC, CSC-5S, CSC-10S, and CSC-20S were 82.3%, 50.8%, 43.9%, and 34.6% of the total drug incorporated into the composite, respectively. Unlike the rapid initial burst release of BMP-2 from CSC, the CSC/SBA-15 showed relatively slow release. After 164 hours, the released quantities of CSC, CSC-5S, CSC-10S, and CSC-20S were 93.6%, 89.7%, 77.9%, and 69.6%, respectively, and the BMP-2 in CSC-20S still showed constant low-dose release.
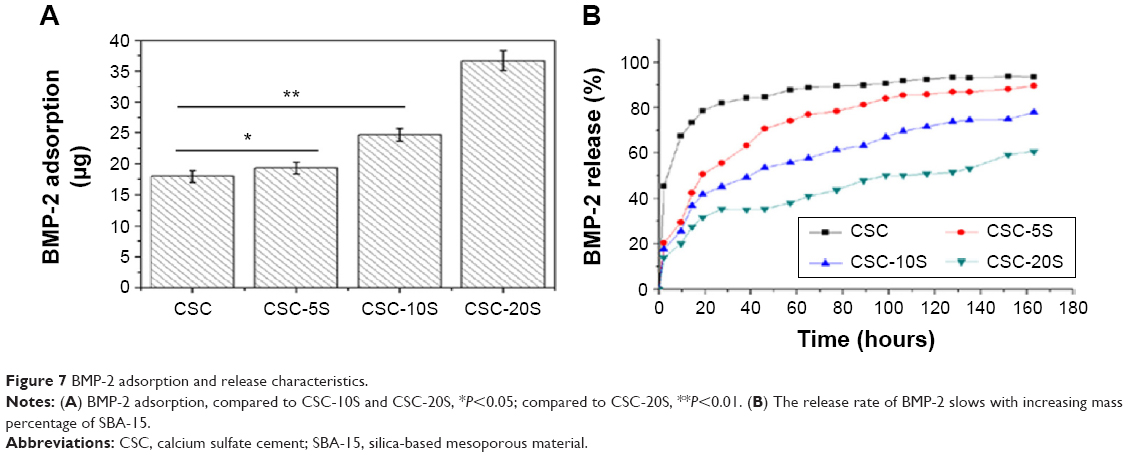 | Figure 7 BMP-2 adsorption and release characteristics. |
Degradation characteristics of CSC/SBA-15
Figure 8A shows the weight loss of CSC/SBA-15 with different mass percentages in Tris-HCl solution. Compared to CSC, the weight loss of CSC-5S, CSC-10S, and CSC-20S was slower. After 15 days of immersion, the weights of CSC-10S and CSC-20S (0.37±0.03 and 0.38±0.09 g, respectively) were higher than that of CSC and CSC-5S (0.27±0.09 and 0.31±0.02 g, respectively; P<0.05). The degradation rates are shown in Figure 8B. It was obvious that the degradation rates of CSC and CSC-5S were fast, while CSC-10S and CSC-20S had constant and slow degradation rates. After 15 days, the degradation rates of CSC and CSC-5S, which were 49.5%±0.92% and 38.9%±0.56% g, respectively, higher than those of CSC-10S (26.4%±0.28%) and CSC-20S (21.3%±1.12%) (P<0.05). Meanwhile, there was a significant difference in degradation rates between CSC-10S and CSC-20S.
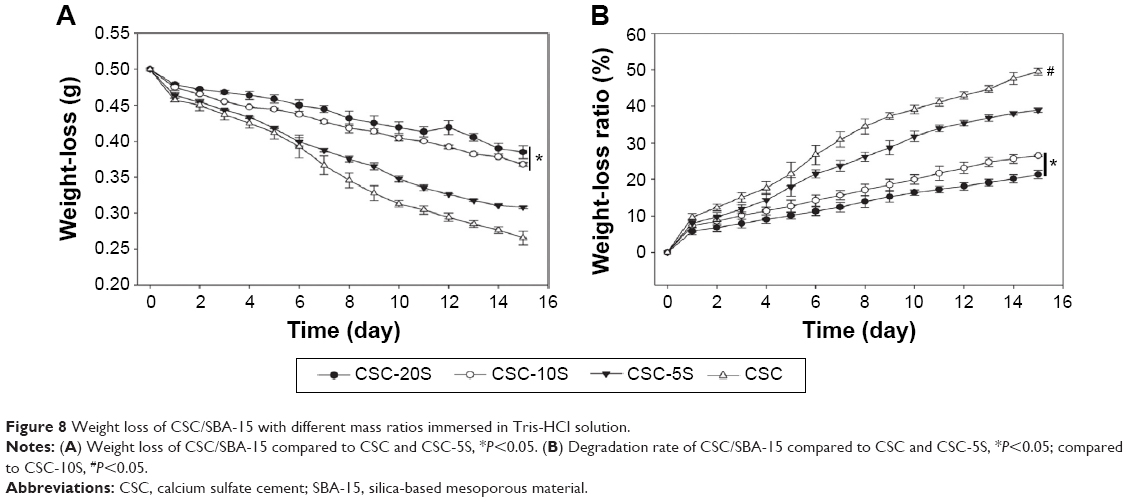 | Figure 8 Weight loss of CSC/SBA-15 with different mass ratios immersed in Tris-HCl solution. |
Discussion
This study adopted tetraethyl orthosilicate as the precursor of mesoporous silica, and SBA-15 was synthesized with regular pores by hydrolysis, condensation, adsorption, and deposition.20 The results indicated that SBA-15 possessed good long-range-order pore structures and uniform pore-size distribution. The specific surface area was 750 m2/g, the pore size was 5.8 nm, and the pore volume was 1.2 mL/g. Therefore, these materials are designated as mesoporous materials that can be used as drug-carrier systems for sustained release.
Biomaterials should have a certain mechanical strength for bone repair. However, the compressive strength of CSC did not meet the biomechanical requirements for augmentation of the bone defects in weight-bearing areas. Therefore, in order to improve the biomechanical property of CSC, calcium phosphate or HA have been mixed into CSC to prepare compound materials, and the clinical results were satisfactory.5,6,21 This study showed that the compressive strength of CSC/SBA-15 was obviously improved compared to pure CSC in the preparation of both porous and nonporous materials. The compressive strengths of these compounds increased with increasing mass percentage of SBA-15 into CSC, which was related to the filling of SBA-15 particles in the gaps between calcium sulfate particles, thus decreasing the effect of liquid scour, and then reduced microcracking in the CSC composite.
One requirement for osteointegration between the bone graft and bone tissue is the formation of bone-like HA on the surface of the bone-grafting material, which can be reflected by the formation of HA on the graft surface in SBF in vitro.19 It was found in this study that there was no obvious HA formation on the surface of CSC after 7 days of immersion. The spherical particles were formed on the surfaces of CSC-5S, CSC-10S, and CSC-20S, and grew to form layered HA in CSC-10S and CSC-20S. With the immersion time prolonged, there were large quantities of HA that formed on the surface of CSC, but it was less uniform and compact than that of CSC-5S, CSC-10S, and CSC-20S. In addition, the HA on the surface of CSC-20S was thick and coral-like, indicating that the bioactivity of CSC was obviously improved after recombination with SBA-15. The collapsibility of CSC in water was reduced by the addition of SBA-15, which also benefited the deposition of HA.
CSC, which does not have a mesoporous structure, shows obvious burst releases of drugs. This study showed that the adsorption of BMP-2 in CSC was reduced, and the release quantity achieved 80% within 24 hours. The molecular weight of the BMP-2 was ~25 kDa, and the volume was 7×3.5×2.5 nm3, as reported previously.22 The mesopore size of SBA-15 was 5.8 nm, and the specific surface area was 750 m2/g. Therefore, SBA-15 was able to adsorb a large amount of protein with high molecular weight within the pores and on the surface, and meanwhile possessed slow-release effects. In this study, the adsorption quantities of BMP-2 in CSC/SBA-15 increased with increasing mass percentage of SBA-15, while the release rate of BMP-2 was relatively slow. After 169 hours, the release quantities of CSC-10S and CSC-20S were 69.6% and 61.3% of the original loading amount, respectively. SBA-15 possessed a mesoporous structure and high specific surface area, and it was able to effectively adsorb the BMP-2 protein. Meanwhile, there was abundant Si–OH in SBA-15, which was able to form hydrogen bonds with –OH and –NH2 of BMP-2. Therefore, BMP-2 loading was increased by the addition of SBA-15, which was able also to effectively reduce the BMP-2 release.
The degradation performance of material in a solution can be investigated by weight-loss measurements. After the same soaking time, greater weight loss corresponded to faster degradation rate, which indicated better degradation performance. The weight-loss curves of CSC/SBA-15 with different mass percentages immersed in Tris-HCl solution after 15 days showed that the weight loss of each composition increased with time, signifying degradation. However, the degradation rates of CSC and CSC-5S were fast, while those of CSC-10S and CSC-20S were constant and slow. Therefore, the degradation rate of CSC was able to be effectively reduced by the addition of SBA-15. This was because the material became more compact as SBA-15 particles entered the gaps between CSC particles, which reduced the access of liquid into the interior of the particle and avoided the washing effect. Therefore, the composite was found to be more stable, and did not easily collapse or break in the solution. In addition, mesoporous SBA-15 had a degradation time of approximately 3 months.23 However, as for CSC, it easily collapsed or broke, owing to poor stability of the surfaces under washing with water. The present in vitro degradation experiment was investigated by immersing the material in Tris-HCl solution, which differed from the real degradation in vivo. Because the in vivo situation was more complex, the materials exhibited different degradable behavior, due to the effects of endocytosis and tissue metabolism. However, the degradation performances of materials could still be detailed by the solution-immersion method in vitro.
Conclusion
Mesoporous SBA-15 was blended into CSC at different mass percentages to prepare a CSC/SBA-15 composite. The compressive strength of the composite increased with increasing mass percentage of SBA-15, and the CSC/SBA-15 with 20% mass ratio had maximum strength. HA on the surface of CSC/SBA-15 was uniform and compact after SBF soaking, and CSC-20S was thick and coral-like. The adsorption of BMP-2 in CSC/SBA-15 increased with increasing SBA-15 content; meanwhile, the release was slow and the degradation rate of the composite was reduced, which made the CSC/SBA-15 composite more suitable for bone repair and bone-tissue engineering.
Acknowledgment
This research was supported by the Henan Basic and Advanced Technology Research Program (2015), PRC.
Disclosure
The authors report no conflicts of interest in this work.
References
Liao JC, Fan KF, Keorochana G, Chen WJ, Chen LH. Transpedicular grafting after short-segment pedicle instrumentation for thoracolumbar burst fracture: calcium sulfate cement versus autogenous iliac bone graft. Spine (Phila Pa 1976). 2010;35(15):1482–1488. | ||
Liao JC, Fan KF, Chen WJ, Chen HL. Posterior instrumentation with transpedicular calcium sulphate graft for thoracolumbar burst fracture. Int Orthop. 2009;33(6):1669–1675. | ||
Bu BX, Wang MJ, Liu WF, Wang YS, Tan HL. Short-segment posterior instrumentation combined with calcium sulfate cement vertebroplasty for thoracolumbar compression fractures: radiographic outcomes including nonunion and other complications. Orthop Traumatol Surg Res. 2015;101(2):227–233. | ||
Walsh WR, Morberg P, Yu Y, et al. Response of a calcium sulfate bone graft substitute in a confined cancellous defect. Clin Orthop Relat Res. 2003;(406):228–236. | ||
Hu GF, Xiao LW, Fu H, Bi D, Ma H, Tong P. Study on injectable and degradable cement of calcium sulphate and calcium phosphate for bone repair. J Mater Sci Mater Med. 2010;21(2):627–634. | ||
Vallet-Regí M, González-Calbet JM. Calcium phosphates as substitution of bone tissues. Prog Solid State Chem. 2004;32(1–2):1–31. | ||
Parikh SN. Bone graft substitutes in modern orthopedics. Orthopedics. 2002;25(11):1301–1309. | ||
Hing KA, Wilson LF, Buckland T. Comparative performance of three ceramic bone substitutes. Spine J. 2007;7(4):475–490. | ||
Yang GJ, Lin M, Zhang L, Gou ZR. Progress of calcium sulfate and inorganic composites for bone defect repair. J Inorgan Mater. 2013;28(8):795–803. | ||
Trejo CG, Lozano D, Manzano M, et al. The osteoinductive properties of mesoporous silicate coated with osteostatin in a rabbit femur cavity defect model. Biomaterials. 2010;31(33):8564–8573. | ||
Wu CT, Fan W, Gelinsky M, et al. Bioactive SrO-SiO2 glass with well-ordered mesopores: characterization, physiochemistry and biological properties. Acta Biomater. 2011;7(4):1797–1806. | ||
Wei GF, Yan XX, Yi J, et al. Synthesis and in vitro bioactivity of mesoporous bioactive glasses with tunable macropores. Microporous Mesoporous Mater. 2011;143(1):157–165. | ||
Zhou HJ, Wu XH, Wei J, et al. Stimulated osteoblastic proliferation by mesoporous silica xerogel with high specific surface area. J Mater Sci Mater Med. 2011;22(3):731–739. | ||
Zhao DY, Feng JL, Huo QS, et al. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science. 1998;279(5350):548–552. | ||
Song SW, Hidajat K, Kawi S. Functionalized SBA-15 materials as carriers for controlled drug delivery: influence of surface properties on matrix-drug interactions. Langmuir. 2005;21(21):9568–9575. | ||
Horcajada P, Rámila A, Férey G, Vallet-Regí M. Influence of superficial organic modification of MCM-41 matrices on drug delivery rate. Solid State Sci. 2006;8(10):1243–1249. | ||
Yang HS, La WG, Cho YM, Shin W, Yeo GD, Kim BS. Comparison between heparin-conjugated fibrin and collagen sponge as bone morphogenetic protein-2 carriers for bone regeneration. Exp Mol Med. 2012;44(5):350–355. | ||
Cui X, Zhang BX, Wang Y, Gao YL. Effects of chitosan-coated pressed calcium sulfate pellet combined with recombinant human bone morphogenetic protein 2 on restoration of segmental bone defect. J Craniofac Surg. 2008;19(2):459–465. | ||
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27(15):2907–2915. | ||
Bi ZC, Shi Y, Yan WH, Yu JY. Adsorption of cetyl trimethylammonium bromide on surface of silicon dioxide. Chem Reagents. 1997;19(6):331–333. | ||
Thomas MV, Puleo DA. Calcium sulfate: properties and clinical applications. J Biomed Mater Res B Appl Biomater. 2009;88(2):597–610. | ||
Laub M, Jennissen HP, Seul T, Schmachtenberg E. Molecular modelling of bone morphogenetic protein-2 (BMP-2) by 3D-rapid prototyping. Materwiss Werksttech. 2001;32(12):926–930. | ||
Xia LG, Zeng DL, Sun XJ, et al. Engineering of bone using rhBMP-2-loaded mesoporous silica bioglass and bone marrow stromal cells for oromaxillofacial bone regeneration. Microporous Mesoporous Mater. 2013;173:155–165. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.