")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Preparation and evaluation of RGD and TAT co-modified docetaxel-loaded liposome
Received 21 August 2017
Accepted for publication 5 October 2017
Published 6 December 2017 Volume 2017:11 Pages 3481—3489
DOI https://doi.org/10.2147/DDDT.S149620
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ren Zhu,1,2 Ye Tian1,3
1Department of Radiotherapy and Oncology, The Second Affiliated Hospital of Soochow University, Suzhou, 2Department of Oncology, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, 3Institute of Radiotherapy and Oncology, Soochow University, Suzhou, People’s Republic of China
Abstract: The aim of this study is to develop a novel RGD and TAT co-modified docetaxel (DTX)-loaded liposome (LP) by the emulsification-solvent evaporation method. The prepared LPs were found to be in the size of 100 nm–110 nm. The transmission electron microscope photomicrographs were smooth, sub-spherical in shape, and aggregated to form small clusters. The DTX cumulative release from TAT and RGD co-modified LPs was significantly higher than that from other LPs due to decreased diffusion distance. Results of cell uptake showed that surface modification could indicate when cell internalization was changed and more drugs entered the cells successfully. Surprisingly, TAT and RGD co-modified DTX-LPs demonstrated a superior antiproliferative effect on A549 cells with a possible mechanism that suppressed the multidrug resistance phenomenon and exhibited a clear synergistic effect. In antitumor study, our results indicated that the form of TAT and RGD co-modified LPs had a better antitumor effect in vivo than the other formulations.
Keywords: RGD, TAT, liposome, cell uptake, cytotoxicity, antitumor
Introduction
Lung cancer is the leading cause of cancer-related death worldwide with a staggering 45% of total cancer death in People’s Republic of China alone.1 The survival rate of this cancer is much lower than that of other common cancers, such as breast and prostate cancers. Although chemotherapy has been the primary treatment for more than 30 years, its success was limited by severe dose-limiting side effects.2,3 In addition, intrinsic or acquired multidrug resistance (MDR) is a major obstacle to chemotherapy.4
Docetaxel (DTX) belongs to microtubule depolymerizing agents, which also include paclitaxel (PTX). The structure of DTX is similar to that of PTX. Clinically, DTX is used to treat many types of cancers such as breast cancer, ovarian cancer, lung cancer, and prostate cancer by inhibiting mitotic progression or cell apoptosis.5 However, poor water solubility (6–7 μg/mL) remains a critical limitation of DTX. Taxotere (commercially marketed brand) contains dehydrated alcohol (0.395 mg/mL) to improve the solubility of DTX.6 In addition, MDR of DTX decreases the accumulation of drug within the cell and increases the repairing mechanism for DNA damage, leading to therapeutic efficiency of a single agent.
In recent years, various drug delivery systems have been developed, which can provide targeted delivery of anticancer drugs to tumor tissues. Numerous types of nanoparticles (NPs) have been developed to deliver DTX to the target site, including superparamagnetic iron oxide NPs,7 targeting peptide-conjugated polylactic acid NPs,8 polydopamine-modified TPGS-PLA NPs,9 cholic acid-functionalized block copolymers consisting of PLGA and vitamin E TPGS,10 folic acid-functionalized SLN,11 Solutol nanodroplets,12 chitosan NPs,13 and DTX nanocrystals.14
Cell-penetrating peptides (CPPs), such as RKKRRQRRRC (transactivated transcription [TAT]), are short peptides that could promote cellular uptake of a variety of molecular products (from small chemical ones to nanosized particles).15 Studies have shown that CPPs play an important role in delivering molecules across body barriers, such as blood–brain barrier, blood–testis barrier, and placental barrier.16 Recently, it has been reported that CPP conjugates enhance antitumor effect of PTX in drug-resistant lung cancer.17,18 This suggests that CPPs are effective promoter of barrier penetration. However, the effect of CPP-mediated DTX carrier systems on promoting permeability has been less reported to date.
In recent years, targeted therapy of angiogenesis has become an important topic in cancer research. Integrin receptors, especially ανβ3, are often highly expressed on certain types of tumor cells and vascular endothelial tumor cells, but not in the normal vessels.19
RGD is a short peptide containing arginine, glycine, and aspartic acid, which is widespread in vivo. RGD peptides act as recognition sites for integrins and their ligands. They have adhesion between mediated cells, extracellular matrix, and cells. Exogenous RGD peptide competitively inhibits the binding of ligands to integrins, thereby inhibiting angiogenesis and migration of tumor cells. Meanwhile, tumors can be targeted and labeled, and anticancer drugs can be accurately delivered.20–22
The aim of this study is to prepare TAT and RGD co-modified DTX liposomes (LPs) by the emulsification-solvent evaporation method. The drug delivery system was characterized by physical properties, such as morphology and particle size, as well as in vitro release. In addition, the cytotoxicity and cellular uptake were investigated and compared on A549 cells.
Materials and methods
Materials
DTX was purchased from JuSheng Biopharma Ltd., Co. (Hubei, People’s Republic of China). RGD peptide (MW =1,100 Da) was purchased from Biochempartner (Shanghai, People’s Republic of China). TAT peptide (MW =1,200 Da) was purchased from Bank Peptide Ltd (Hefei, People’s Republic of China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG1000–TAT and DSPE-PEG3500–RGD were synthesized following a previous report.23 Phosphatidylcholine from soybean and cholesterol were purchased from Shanghai Chemical Reagent Company of Chinese Medicine. The A549 cell was purchased from Bioleaf Biopharma Co., Ltd. (Shanghai, People’s Republic of China). 4,5-(dimethylthiazol-2-yl) 2,5-diphenyl-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were of analytical grade. Distilled water was used in all experiments.
Preparation of TAT-RGD-DTX-LPs
Different DTX-LPs were prepared by the emulsification-solvent evaporation method. Briefly, DTX (5 mg), phosphatidylcholine (100 mg), cholesterol (50 mg), and DSPE-PEG1000–TAT (2.5 mg) and DSPE-PEG3500–RGD (2.5 mg) were dissolved in 10 mL chloroform–methanol (4:1, v/v). Thereafter, the chloroform–methanol was removed under N2 and evaporated under vacuum for at least 2 hours to form a thin lipid film. A volume of 5 mL of phosphate-buffered saline (PBS; pH 7.4) was added to the hydration lipids. The LP suspensions were passed through a microfluidizer at 22,000 bar for 10 circles, and then extruded (10 times) through polycarbonate membranes of gradually decreasing pore size (200 and 100 nm). Finally, four DTX-LP preparations (DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, TAT, and RGD co-modified DTX-LPs) were freeze dried into powder. The final product was stored in a vacuum desiccator at room temperature.
Characterization
The LP size, size distribution, and zeta potential were determined using a dynamic light scattering NICOMP™ 380 (Particle Sizing Systems, Santa Barbara, CA, USA) equipped with a 50 mV laser at a scattering angle of 90°. The morphology of the LPs was examined using a transmission electron microscope (TEM, JEM-1400; Hitachi, Tokyo, Japan) after staining with a 2% sodium phosphotungstate solution. The entrapment efficiency (EE) of the prepared LPs was calculated by the following formula:
|

|

All measurements were performed in triplicates and the values are represented as mean ± SD (n=3).
Stability study
According to ICH guidelines, the particle size, zeta potential, EE, and drug loading (DL) of four DTX-LPs were evaluated at 2°C–8°C for 3 months for chemical and physical stability. The centrifuge tests were also carried out to assess the physical stability of the DTX-LP solution. The LP solution was centrifuged for 30 minutes at 650× g in the centrifuge tests.
In vitro drug release
In vitro release study of four DTX-LPs and free DTX was conducted using the dialysis bag method (MW =10–12 kDa). The release rate of free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs (each contained 10 mg DTX) were evaluated in vitro at 37°C±0.5°C using the basket method (100 r/minutes) in a buffer solution of pH 7.4 PBS (25 mL, containing 1% Tween 80 as solubilizer). Experiments were performed in triplicates. Aliquots (1 mL) were withdrawn at intervals of 0.25, 0.5, 1, 2, 4, 6, 8, 12, 16, and 24 hours, and the same replacement volume was supplied as the release medium. The collected samples were injected directly into the HPLC system for analysis.
Cell uptake
The cellular internalization of free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs was visualized by confocal microscopy using coumarin-6 as a fluorescent probe. A549 cells were grown in RPMI 1640 medium supplemented with 10% (v/v) FBS and 5% antibiotics (100 IU/mL of penicillin G sodium and 100 μg/mL of streptomycin sulfate). A549 cells were inoculated in a cell culture dish at an initial density of 4×105 cells per dish. Cells were then incubated with coumarin-6-adsorbed free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs (equivalent to 0.1 μg/mL of coumarin-6) for 2 hours at 37°C±0.5°C.
Subsequently, the cells were washed several times with PBS, and fixed with 4% paraformaldehyde for 10 minutes before being observed under a confocal microscope. For DTX uptake quantitative estimates, the density of cells inoculated on 24-well plates was 3×104 cells. When reaching a confluence of 70%–80%, the cells were incubated with coumarin-6-adsorbed free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs (equivalent to 0.1 μg/mL of coumarin-6). After 2 hours of culture, the cells were washed several times with cold PBS and then dissolved by addition of Triton X-100 (0.1%). Fluorescence intensities were measured by a multimode microplate reader at an excitation wavelength of 440 nm and an emission wavelength of 520 nm.
Cytotoxicity study
The cytotoxicity of blank LPs, free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs was evaluated in A549 cells by MTT assay. Briefly, A549 cells were seeded in a 96-well plate at a density of 3–4×103 cells per well. After 12 hours, different DTX formulations (DTX concentrations ranging from 0.002 to 20 μg/mL [equivalent to 0.001616–16.16 mol/L]) were added, and plates were incubated for 24, 48, and 72 hours. DTX standard solution was prepared with DTX dissolved in ethanol ranging from 0.05 to 2 mg/mL and then diluted 100 times with distill water. Measurements were taken using a microplate reader.
In vivo antitumor study
The antitumor effect of DTX-LPs was evaluated in vivo using BALB/c nude mice (age: 6 weeks and weight: 20–22 g) that had been inoculated subcutaneously with 2×109 human lung adenocarcinoma (A549) cells.
The treatments were started on the day when the tumor volume reached 100–150 mm3, which was set as day 0. On day 0, the mice were randomly divided into 6 groups (blank LPs, free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs) (n=5).
Samples of drugs at a dose of 10 mg/kg were injected intravenously via the tail vein every 3 days for 3 administrations in total. Tumor size and body weight were measured three times per week during the study.
On day 10, the mice were sacrificed and the resected tumor weighed. The test drug administration procedure was as follows: weighing and first injection on day 0; weighing and second injection on day 3; weighing and third injection on day 9; and euthanasia on day 10.
Tumor volume (V) was calculated by the following formula: (W2 × L)/2, where W was the widest tumor measured and L was the longest tumor dimension. Relative tumor volume (R) was calculated by the following formula: Vi/V0, where V0 is the tumor volume at day 0 and Vi is the tumor volume at the measurement point. Antitumor activity was estimated by the relative tumor inhibitory rate (%), which was calculated by the formula: (1-[R(treatment group)/R(negative control group)]) ×100%.
Data analysis
The data are shown as the mean ± SD. Statistical comparisons were made using Student’s t-tests and one-way analysis of variance. A P-value of <0.05 was considered statistically significant.
Results and discussion
Characterization of LPs
The characterization of four DTX-LPs is shown in Table 1. All batches of LPs were found to be in the size of 100–110 nm and the larger particle size of TAT-RGD-DTX-LPs as compared to others could be caused by the anchoring of TAT or RGD molecule at the surface of LPs. This could be the reason that an increment in size of LPs was observed. The TEM photomicrographs are shown in Figure 1. It was observed from these photomicrographs that all samples of particles were smooth, sub-spherical in shape, and aggregated to form small clusters. The zeta potential of LPs is commonly used to characterize the surface charge property of particles. The zeta potential, EE, and DL are also listed in Table 1. A relatively higher percentage of drug entrapment was observed in coated LPs as compared to their conventional counterparts, which could be due to minimum repulsion between drug and polymer.
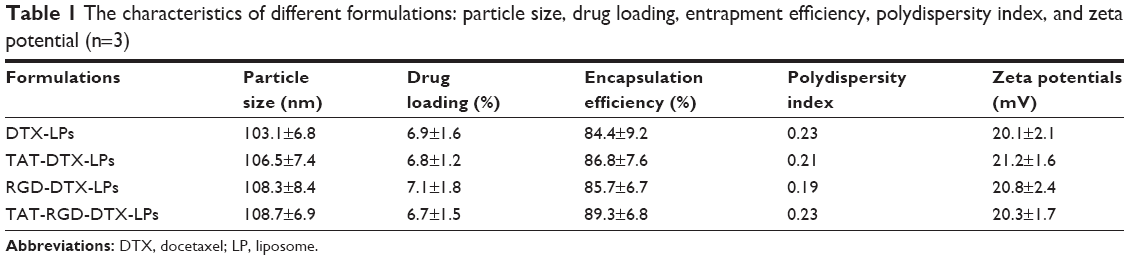 | Table 1 The characteristics of different formulations: particle size, drug loading, entrapment efficiency, polydispersity index, and zeta potential (n=3) |
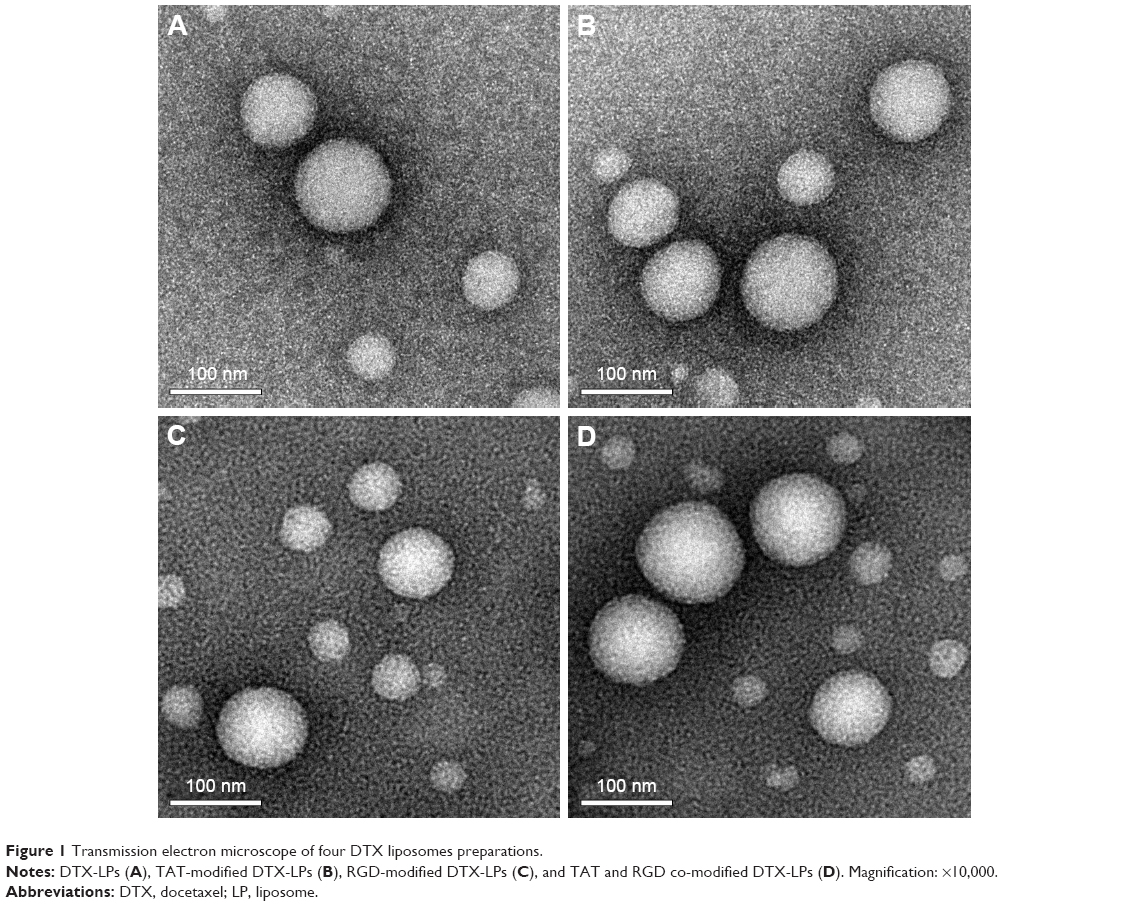 | Figure 1 Transmission electron microscope of four DTX liposomes preparations. |
Stability studies
Following the ICH guidelines, the stability of LPs was assessed. After 3 months of storage at 2°C–8°C, lyophilized LPs appeared as stable as dried cakes as they showed no collapse or contraction (Figure 2). No significant change in clarity and phase separation was observed in the tests of centrifugation. DTX in the solution formulations showed no degradation (Table 1).
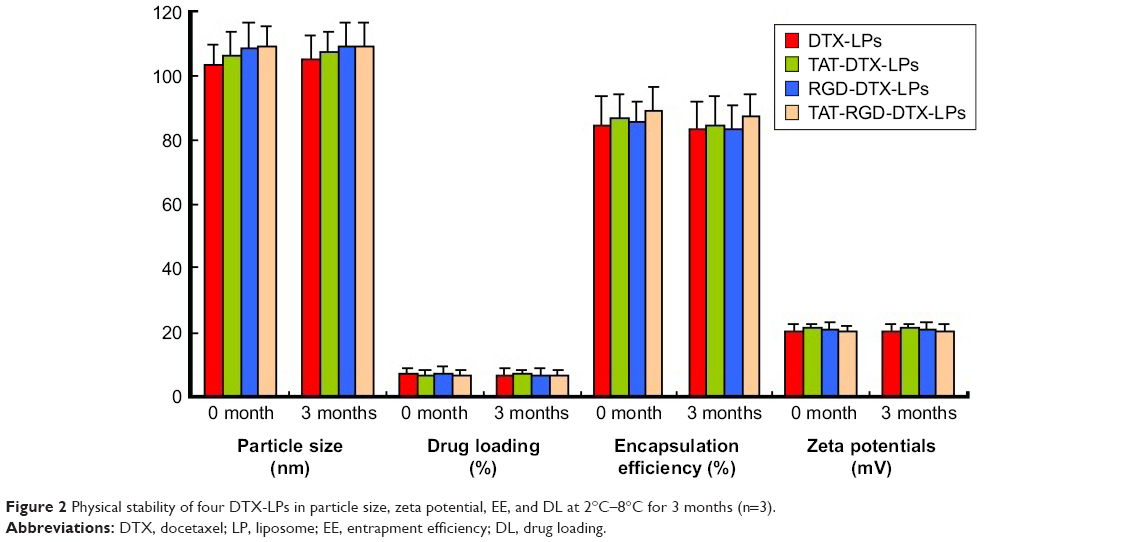 | Figure 2 Physical stability of four DTX-LPs in particle size, zeta potential, EE, and DL at 2°C–8°C for 3 months (n=3). |
In vitro drug release
In vitro drug release of free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs was evaluated in PBS containing Tween 80 (1% w/v) for 24 hours (Figure 3). During the in vitro release experiments, insoluble drugs are difficult to maintain a good sinking state. In the present study, the sink conditions were maintained by the addition of Tween 80 and the same fresh buffer volume was frequently replaced during the experiment. Free DTX showed poor drug release because of its poor water solubility. Only 5% of DTX release was observed at 24 hours. The DTX cumulative release from TAT and RGD co-modified LPs was significantly higher than that from others preparations. However, there was no significant difference in DTX release between TAT- and RGD-modified LPs. In previous studies, surface-modified drug systems showed faster release than free drugs.24 Drug LPs have smaller size than free drugs. They have an increased dissolution rate due to the decrease in diffusion distance.25
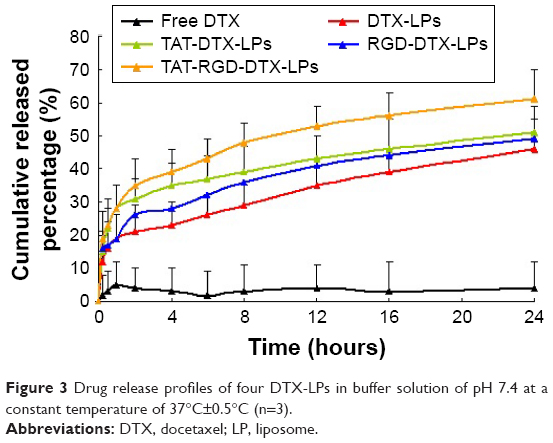 | Figure 3 Drug release profiles of four DTX-LPs in buffer solution of pH 7.4 at a constant temperature of 37°C±0.5°C (n=3). |
Cell uptake
Confocal microscopy was employed to characterize the cellular internalization of free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs in A549 cells. As shown in Figure 4, a strong green fluorescence was observed in the cytoplasmic region after incubation of TAT and RGD co-modified DTX-LPs for 2 hours. Cell internalization in A549 cells was higher for TAT and RGD co-modified DTX-LPs than others. Results showed that depending on surface modification could indicate when cell internalization was changed and more drugs entered the cells successfully. In quantitative cell uptake studies, coumarin-6 on the five formulations was quantified by recovering the drug LPs from cells and measuring their fluorescence (normalized to per mg of the total cellular protein contents). Quantitative results were in agreement with the confocal images (data not shown).
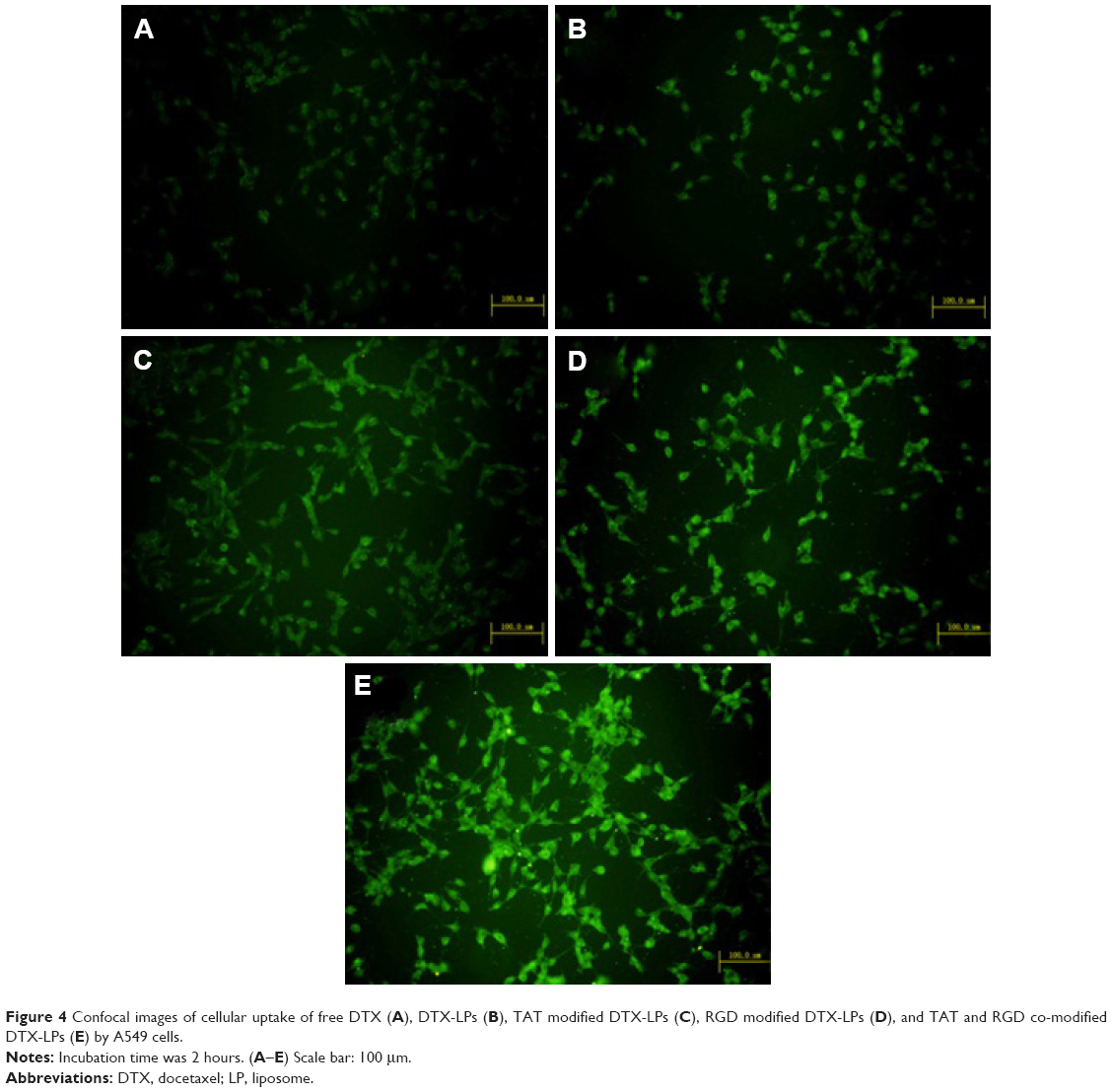 | Figure 4 Confocal images of cellular uptake of free DTX (A), DTX-LPs (B), TAT modified DTX-LPs (C), RGD modified DTX-LPs (D), and TAT and RGD co-modified DTX-LPs (E) by A549 cells. |
Cytotoxicity study
Cell inhibition effects of different DTX formulations on A549 cells were evaluated by MTT assay (Figure 5A–C). It can be seen that free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs exhibited a time-dependent and dose-dependent cytotoxicity on the cell lines tested. Although DTX is a powerful antitumor drug, it cannot completely inhibit cell proliferation. Similarly, LP conjugation with DTX did not result in superior effect. On the other hand, TAT or RGD conjugation with DTX significantly decreased cell proliferation and improved therapeutic efficiency. Surprisingly, TAT and RGD co-modified DTX-LPs showed a superior antiproliferative effect on A549 cells with a possible mechanism that suppressed the MDR phenomenon and exhibited a clear synergistic effect. The half maximal inhibitory concentration (IC50) values of all formulations are listed in Table 2. It was indicated that IC50 value was reduced to varied extents depending on incubation times, and TAT and RGD co-modified DTX-LPs have strong targeting ability, which can improve the cellular uptake as well as cytotoxicity of DTX.
 | Figure 5 In vitro cytotoxicity analysis of free DTX, DTX-LPs, TAT-modified DTX-LPs, RGD-modified DTX-LPs, and TAT and RGD co-modified DTX-LPs on A549 cell lines. |
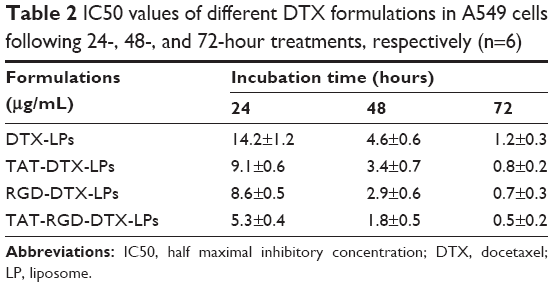 | Table 2 IC50 values of different DTX formulations in A549 cells following 24-, 48-, and 72-hour treatments, respectively (n=6) |
In vivo antitumor study
To test the antitumor activity of DTX, mice bearing human lung adenocarcinoma (A549) were treated with DTX in LPs with different formulations on three occasions over 10 days. As shown in Figure 6, all types of LPs inhibited the growth of human lung adenocarcinoma (A549) cells and reduced tumor size to a greater degree than the control group. However, compared with the control group, there was a marked reduction in tumor volume in the group treated with TAT and RGD co-modified LPs, while only a slight decrease in tumor volume was observed in the group treated with others. The average weights and volumes of the tumor are shown in Table 3. The appearance of the tumors was consistent with the statistical analysis of the tumor volume data, that is, when compared with the control group, all treatments significantly (P<0.05) inhibited tumor volume, with the greatest effect seen in TAT and RGD co-modified LPs. Similarly, the tumor weights were significantly (P<0.05) inhibited by both DTX treatments. Once again, the effect of TAT and RGD co-modified LPs was the most pronounced. Overall, our results indicate that the form of TAT and RGD co-modified LPs had a better antitumor effect in vivo than the other formulations.
 | Figure 6 Changes in tumor volume in nude mice transplanted with a human adenocarcinoma (A549) cell line on day 10. |
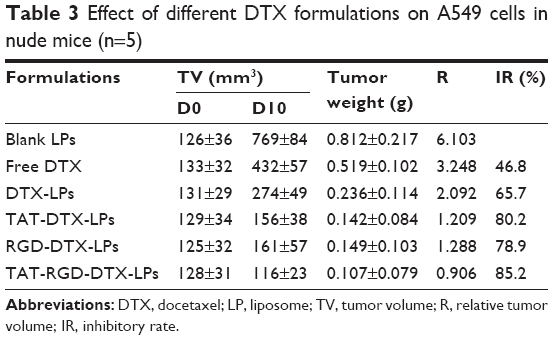 | Table 3 Effect of different DTX formulations on A549 cells in nude mice (n=5) |
Disclosure
The authors report no conflicts of interest in this work.
References
She J, Yang P, Hong Q, Bai C. Lung cancer in China: challenges and interventions. Chest. 2013;143(4):1117–1126. | ||
Nixon NA, Khan OF, Imam H, et al. Drug development for breast, colorectal, and non-small cell lung cancers from 1979 to 2014. Cancer. Epub 2017 Aug 17. | ||
Sheng J, Liu X, Yang Y, Ma Y, Huang Y, Zhao H. A prospective survey of depressive disorder among patients with advanced lung cancer in South China. J Clin Oncol. 2015;33(29 Suppl):216. | ||
Meschini S, Marra M, Calcabrini A, et al. Role of the lung resistance-related protein (LRP) in the drug sensitivity of cultured tumor cells. Toxicol In Vitro. 2002;16(4):389–398. | ||
Du N, Song LP, Li XS, et al. Novel pH-sensitive nanoformulated docetaxel as a potential therapeutic strategy for the treatment of cholangiocarcinoma. J Nanobiotechnology. 2015;13:17. | ||
Blumenthal GM, Scher NS, Cortazar P, et al. First FDA approval of dual anti-HER2 regimen: pertuzumab in combination with trastuzumab and docetaxel for HER2-positive metastatic breast cancer. Clin Cancer Res. 2013;19(18):4911–4916. | ||
Nagesh PKB, Johnson NR, Boya VKN, et al. PSMA targeted docetaxel-loaded superparamagnetic iron oxide nanoparticles for prostate cancer. Colloids Surf B Biointerfaces. 2016;144:8–20. | ||
Jiang Y, Yang N, Zhang H, et al. Enhanced in vivo antitumor efficacy of dual-functional peptide-modified docetaxel nanoparticles through tumor targeting and Hsp90 inhibition. J Control Release. 2016;221:26–36. | ||
Zhu D, Tao W, Zhang H, et al. Docetaxel (DTX)-loaded polydopamine-modified TPGS-PLA nanoparticles as a targeted drug delivery system for the treatment of liver cancer. Acta Biomater. 2016;30:144–154. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Pawar H, Surapaneni SK, Tikoo K, et al. Folic acid functionalized long-circulating co-encapsulated docetaxel and curcumin solid lipid nanoparticles: in vitro evaluation, pharmacokinetic and biodistribution in rats. Drug Deliv. 2016;23(4):1453–1468. | ||
Oh KS, Kim K, Yoon BD, et al. Docetaxel-loaded multilayer nanoparticles with nanodroplets for cancer therapy. Int J Nanomedicine. 2016;11:1077–1087. | ||
Jain A, Thakur K, Sharma G, Kush P, Jain UK. Fabrication, characterization and cytotoxicity studies of ionically cross-linked docetaxel loaded chitosan nanoparticles. Carbohydr Polym. 2016;137:65–74. | ||
Pawar VK, Gupta S, Singh Y, et al. Pluronic F-127 stabilised docetaxel nanocrystals improve apoptosis by mitochondrial depolarization in breast cancer cells: pharmacokinetics and toxicity assessment. J Biomed Nanotechnol. 2015;11(10):1747–1763. | ||
Trabulo S, Cardoso AL, Mano M, De Lima MC. Cell-penetrating peptides-mechanisms of cellular uptake and generation of delivery systems. Pharmaceuticals (Basel). 2010;3(4):961–993. | ||
Farkhani SM, Valizadeh A, Karami H, Mohammadi S, Sohrabi N, Badrzadeh F. Cell penetrating peptides: efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides. 2014;57:78–94. | ||
Duan Z, Chen C, Qin J, et al. Cell-penetrating peptide conjugates to enhance the antitumor effect of paclitaxel on drug-resistant lung cancer. Drug Deliv. 2017;24(1):752–764. | ||
Koutsiouki K, Angelopoulou A, Ioannou E, et al. TAT peptide-conjugated magnetic PLA-PEG nanocapsules for the targeted delivery of paclitaxel: in vitro and cell studies. AAPS PharmSciTech. 2017;18(3):769–781. | ||
Zetter BR. On target with tumor blood vessel markers. Nat Biotechnol. 1997;15(12):1243–1244. | ||
Danhier F, Le Breton A, Préat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9(11):2961–2973. | ||
Garanger E, Boturyn D, Dumy P. Tumor targeting with RGD peptide ligands – design of new molecular conjugates for imaging and therapy of cancers. Anticancer Agents Med Chem. 2007;7(5):552–558. | ||
Zitzmann S, Ehemann V, Schwab M. Arginine-glycine-aspartic acid (RGD)-peptide binds to both tumor and tumor-endothelial cells in vivo. Cancer Res. 2002;62(18):5139–5143. | ||
Mei L, Fu L, Shi K, et al. Increased tumor targeted delivery using a multistage liposome system functionalized with RGD, TAT and cleavable PEG. Int J Pharm. 2014;468(1–2):26–38. | ||
Park JJ, Meghani N, Choi JS, Lee BJ. Development and evaluation of decorated aceclofenac nanocrystals. Colloids Surf B Biointerfaces. 2016;143:206–212. | ||
Chu KR, Lee E, Jeong SH, Park ES. Effect of particle size on the dissolution behaviors of poorly water-soluble drugs. Arch Pharm Res. 2012;35(7):1187–1195. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.