Back to Journals » The Application of Clinical Genetics » Volume 15
Prenatal Sonographic Features of Rare Chromosome 13 Aberrations
Authors Moczulska H ![]() , Pietrusinski M, Serafin M, Skoczylas B, Sieroszewski P
, Pietrusinski M, Serafin M, Skoczylas B, Sieroszewski P ![]() , Borowiec M
, Borowiec M
Received 4 May 2022
Accepted for publication 5 September 2022
Published 3 October 2022 Volume 2022:15 Pages 145—151
DOI https://doi.org/10.2147/TACG.S370163
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Hanna Moczulska,1 Michal Pietrusinski,1 Marcin Serafin,1 Beata Skoczylas,1 Piotr Sieroszewski,2 Maciej Borowiec1
1Department of Clinical Genetics, Medical University of Lodz, Lodz, Poland; 2Department of Fetal Medicine and Gynecology, Medical University of Lodz, Lodz, Poland
Correspondence: Hanna Moczulska, Department of Clinical Genetics, Medical University of Lodz, Lodz, Poland, Tel +48-42-201-44-92, Email [email protected]
Abstarct:
Objective: Trisomy 13 is one of the most common chromosome aberrations diagnosed in the prenatal period, and is associated with some specific dysmorphic features. Rare chromosome 13 aberrations other than trisomy 13 may cause other fetal abnormalities. The aim of the study was to analyze cases with those rare chromosome 13 aberrations.
Methods: We analyzed all prenatal tests performed in the Department of Clinical Genetics of the Medical University of Lodz from 2016 to 2021 to find all chromosome 13 aberrations.
Results: The most common aberration of chromosome 13 was a simple trisomy 13 (n = 16). We found five rare chromosome 13 aberrations other than simple chromosome 13 trisomy: mosaic trisomy 13 mos 47,XX,+13[11]/46,XX[10], mosaic monosomy 13 mos 46,XY,-13,+mar[9]/46,XY[31], duplication 13q21.1-q31, deletion 13q34 and deletion 13q31.1-q34. The deletion 13q31.1-q34 occurred in monochorionic diamniotic twin pregnancy.
Conclusion: Rare aberrations accounted for 24% of all chromosome 13 aberrations. Cases with mosaic monosomy of chromosome 13 and microdeletion 13q had similar abnormalities of the external genitalia and facial dysmorphia. The case with duplication 13q was very similar to the clinical features of chromosome 13 trisomy. Mosaic trisomy 13 can occur without any accompanying anatomical defects.
Keywords: prenatal testing, karyotype, chromosome 13, amniocentesis
Introduction
Chromosome 13 trisomy, also called Patau syndrome, is one of the most common chromosomal abnormalities with the frequency of one in 5000 total births.1 The simple chromosome 13 trisomy is frequently observed. The Robertsonian translocation is less frequent than the simple trisomy 13. There are no phenotypic differences between the simple chromosome 13 trisomy and the Robertsonian translocation. The following symptoms were observed in cases with chromosome 13 trisomy: cleft lip or palate, low-set ears, micrognathia, clenched hands, polydactyly, skeletal abnormalities, microcephaly, congenital heart defects, brain or spinal cord abnormalities, and cryptorchidism.2 Rare aberrations of chromosome 13 may have features other than observed in chromosome 13 trisomy.3–6Screening and diagnostic tests are used to diagnose chromosomal abnormalities in the fetus. Screening tests calculate the risk of the most common trisomies, including trisomy 13 in the fetus. Screening tests do not take into account rare chromosome aberrations. High risk in screening tests is an indication for diagnostic tests such as karyotype or aCGH. Diagnostic tests identify rare chromosome 13 aberrations but require an invasive procedure for collecting material for testing, such as chorionic villus sampling or amniocentesis.
The aim of the study was to analyze cases with rare chromosome 13 aberrations other than trisomy 13.
Methods
To select chromosome 13 aberrations, we retrospectively analyzed 1573 invasive tests performed from January 2016 to December 2021 in the Department of Clinical Genetics of the Medical University of Lodz. One case was diagnosed before 2016 (case 4). All patients were Caucasian. The study was conducted in a tertiary referral center. Prenatal genetic tests were ordered in high-risk pregnancies diagnosed in the Department of Clinical Genetics and in the Department of Fetal Medicine and Gynecology of the Medical University of Lodz. All tests were conducted as part of a prenatal screening program reimbursed by the national health care system. The criteria for inclusion in the prenatal testing program in Poland are the age above 35 years, the occurrence of a chromosomal aberration in the previous fetus or child, structural chromosomal aberration in a pregnant woman or in the father of a fetus, a significantly increased risk of having a child with a monogenic or multifactorial disease, abnormal fetal ultrasound or biochemical tests that indicate increased the risk of chromosomal abnormalities or fetal abnormalities.
Each patient has a detailed fetal ultrasound examination of the first and second trimesters. All scans were performed by certified sonographers (certificates of the Foetal Medicine Foundation and the Polish Society of Gynaecologists and Obstetricians). If a chromosomal aberration was suspected in the fetus, invasive diagnostics with aCGH microarray examination or karyotype assessment were performed.
Fetal material was collected by amniocentesis or chorionic villus sampling (case 1). DNA was isolated from direct amniocyte and chorionic villus samples. The amniotic fluid samples were cultured for conventional cytogenetic analysis according to standard protocols. The chromosomes obtained in the metaphase were subjected to Giemsa staining after trypsin treatment and analyzed on the Cytovision karyotyping platform. Chromosomal microarray was performed using Agilent, GenetiSure Pre-Screen Kit 8x60K with a resolution of approximately 0.50 Mb. Suspected pathogenic areas were analyzed using ClinGen, ISCA, DGV, Decipher, Ensemble, OMIM, RareChromo, or PubMed databases.
The patient’s age, obstetric interview, and pregnancy age are presented in Table 1.
 |
Table 1 Cases with Chromosome 13 Aberration Other than Trisomy 13 |
Results
We selected all chromosome 13 aberrations (n=21), which constituted 1.3% of the studied group (21/1573). The most common aberration was trisomy 13 n = 16. We found five other aberrations of chromosome 13: mosaic trisomy of chromosome 13 mos 47,XX,+13[11]/46,XX[10], mosaic monosomy of chromosome 13 mos 46,XY,-13,+mar[9]/46,XY[31], duplication 13q21.1-q31, deletion 13q34 and deletion 13q31.1-q34. The deletion 13q31.1-q34 occurred in monochorionic diamniotic twin pregnancy. Rare aberrations accounted for 24% of all chromosome 13 aberrations. Table 1 presents the phenotype of cases with chromosome 13 aberration other than trisomy 13. The first case was diagnosed by chorionic villus sampling, and duplication of 13q21.1q34(56622219_115083342) was found. Foetal congenital defects were an indication for invasive diagnostics. The risk in the combined test was low, and the foetal heart rate was normal. The foetal sex was female. The 13q21.1q34 region was 58.5 Mb long and contained 111 genes. The patient did not report for further examinations. The remaining cases were diagnosed with amniocentesis. In the second case (twin pregnancy), deletion 13q31.1q34(100484940_115092648) was found. The deletion size was 30.4 Mb long, and the region contained 111 genes; the fetal sex was male. The presence of the deletion was confirmed in the karyotype. The same aberration was found in both foetuses; twins’ phenotypes were very similar (Table 1). Parents’ karyotypes were normal. The pregnancy was terminated. A post-mortem examination was not performed. The macroscopic evaluation confirmed the presence of dysmorphia and abnormal structure of the external genitalia (hypospadias with bifid scrotum). In the third case, a potentially pathogenic copy number variation (CNV) of 2.17 Mb was found: deletion 13q34(112925399_115097664). The indication for amniocentesis was increased risk in the combined test (risk for trisomy 21 1:147). The second-trimester sonography revealed right sited choroid plexus cyst. The parents’ results were normal (karyotype and aCGH). The fourth and fifth cases consisted of the mosaic karyotype. Fourth case: normal male karyotype with monosomy of chromosome 13 and marker chromosome. The fifth case: normal female karyotype with trisomy 13. The phenotypes in cases fourth and fifth were highly different. The fourth case showed lethal defects (e.g. encephalocele and common arterial trunk), the pregnancy continued, and the child died after delivery. The only abnormality in a fifth case was high risk in the combined test; no abnormalities were found in sonography. Unfortunately, follow-up is not available.
Discussion
We found that mosaic monosomy of chromosome 13 and microdeletion 13q are characterized by similar abnormalities of the external genitalia and facial dysmorphia. The 13q duplication phenotype is similar to the clinical features of trisomy 13.
Partial trisomy 13q is a rare chromosomal abnormality with variable phenotypic expression. Machado et al reported prenatal diagnosis of partial trisomy 13q (arr 13q14qterx3) in the fetus with partial agenesis of the cerebellum vermis, agenesis of the corpus callosum, hydrops, and polyhydramnios. In newborn evaluation, they revealed a short neck, low-set ears, nasal bridge hypoplasia, camptodactyly, clinodactyly, and thin umbilical cord. Additionally, an autopsy showed agenesis of the diaphragm and severe pulmonary hypoplasia.7 We presented a case with duplication 13q21.1-q34 (58,5 Mb), abnormal features like bilateral cleft lip and palate, polydactyly in the hands and feet, a bright spot in the left ventricle, and the two-vessel umbilical cord were recognised (Figure 1). Jotterand et al compared a partial chromosome 13 trisomy with a complete trisomy 13 and found common features, e.g. polydactyly, microphthalmia, low set, and/or malformed ears.8 Congenital heart defect and cleft lip or/and palate were rarely recognised in partial trisomy chromosome 13.8 Similar conclusions were made by Schinzel et al.9 Contrary to these observations, our case with duplication 13q presented a bilateral cleft lip and palate.
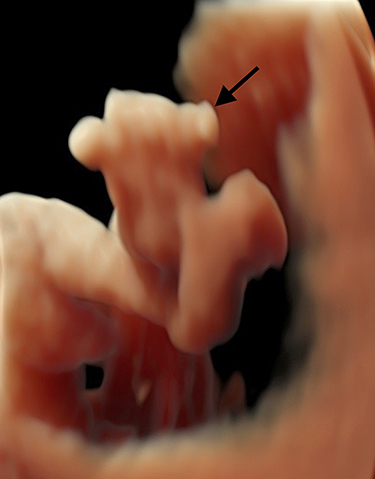 |
Figure 1 Postaxial polydactyly of the left foot in the fetus with 13q21.1-q34 duplication (case number 1 in Table 1). |
Deletions on the long arm of chromosome 13 have been described in patients with mental retardation, facial anomalies (flattened nasal bridge, hypertelorism, microcephaly, and micrognathia), and severe malformations in the central nervous system, distal limbs, genitourinary tract, and congenital heart defects.2 We diagnosed twin monozygotic pregnancy with a large deletion (30,4 Mb) in region 13q31.1q34. Ultrasound examination at the 12th week of pregnancy revealed the increased nuchal translucency in both fetuses and an increased risk of Down syndrome in the combined test. In the second trimester, we recognised ventriculomegaly, agenesis of the corpus callosum, hypospadias with the bifid scrotum, increased nuchal fold, flat profile, and shortening of the nasal bones in both twins with the 13q31.1-q34 deletion (Figures 2 and 3). Additionally, one of the twins had a right aortic arch. The second twin had normal heart anatomy, but there was an effusion to a pericardial sac. Wang et al observed that congenital heart disease, anorectal/genitourinary, and gastrointestinal tract malformations are the prominent anomalies observed in 13q deletion syndrome.10 Vervecken et al presented a prenatal case with the same deletion as our case (deletion 13q31.1-q34), but the phenotypic features were similar rather to those of trisomy 13. Abnormal cerebral anatomy, with symptoms of holoprosencephaly, was diagnosed at the 11th week of gestation. A follow-up ultrasound at the 17th week of pregnancy showed a unilateral facial cleft, holoprosencephaly with Dandy-Walker malformation, and abnormal positioning of feet and knees. On autopsy, dysmorphic facial features including unilateral cleft lip and palate, low-set implantation of the ears, hypertelorism, and neck edema were diagnosed. Vervecken et al reported also upper and lower limb abnormalities, e.g. the right thumb was absent, the left was only rudimentary.11 In our study group, we also found a smaller 13q deletion (2,17 Mb) covering the 13q34 region. The aberration was classified as potentially pathogenic copy number variation. The indication for invasive diagnosis was a high risk of trisomy 21 in the combined test. In the second trimester, the only abnormality was a right-sided choroid plexus cyst. Due to the potential pathogenic nature of the lesion and the almost normal ultrasound image of the fetus, the prognosis was very difficult to determine.
 |
Figure 2 The female appearance of the external genitalia in a fetal ultrasound examination with a deletion 13q31.1q34 (case number 2 in Table 1). |
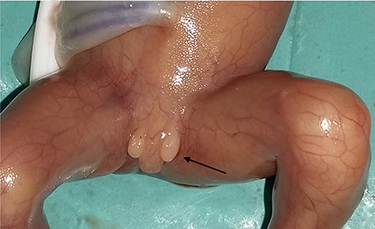 |
Figure 3 Hypospadias with the bifid scrotum in the fetus with a deletion 13q31.1q34 (case number 2 in Table 1). |
Additionally, we diagnosed mosaic monosomy of chromosome 13 with a marker chromosome (mos 46,XY,-13,+mar[9]/46,XY[31]). The origin of the marker chromosome has not been established in our case. Increased nuchal translucency and encephalocele were recognised in the first trimester. The combined test revealed a high risk of Down syndrome 1:48. In the second trimester, we diagnosed encephalocele, common arterial trunk, hypospadias with the bifid scrotum, abnormal face profile, and a broad nasal bridge (Figures 4 and 5). Chen et al presented the fetus with mosaic ring chromosome 13 with monosomy 13 (46XY,r(13)/45,XY,-13), the indication for amniocentesis was intrauterine growth restriction. The pregnancy was terminated, the fetus was delivered with facial dysmorphism of microcephaly, hypertelorism, a small mouth, low-set ears, mid-face hypoplasia, and a bulbous nose.12 A review of the literature shows that the symptoms of monosomy and partial monosomy of chromosome 13 are very similar. When comparing fetuses with 13q deletion (case 2a and 2b) with mosaic monosomy 13 (case 4), common phenotypic features were observed: abnormalities of the external genitalia - hypospadias with the bifid scrotum, abnormal face profile, and a broad nasal bridge. Similar abnormalities reported by Kataoka et al presented a fetus with mosaic ring chromosome 13 with monosomy 13 (46,XX,r(13)(p11q33)/45,XX,-13) with slightly sloping forehead, hyponasal bridge, hypertelorism, ambiguous genitalia, low-set ear, neck edema, and webbing.5
 |
Figure 4 Facial dysmorphia in the fetus with mosaic monosomy of chromosome 13 with a marker chromosome (case number 4 in Table 1). |
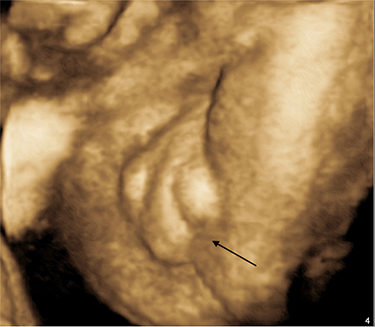 |
Figure 5 Hypospadias with the bifid scrotum in the fetus with mosaic monosomy of chromosome 13 with a marker chromosome (case number 4 in Table 1). |
We reported a case with mosaic trisomy 13 with no malformations in the first and in the second trimester. The only abnormalities in our case were tachycardia and low levels of PAPPA and BhCG in the maternal blood. The risk for Patau syndrome was increased. Amniocentesis was performed at the 15th week of pregnancy and the karyotype was mosaic mos 47,XX,+13[11]/46,XX[10]. Parents refused to follow up. Chen et al presented a case with mosaic trisomy 13 without malformations. The neonate was doing well and presented neither phenotypic abnormalities nor psychomotor disorders at two months.13 Individuals with mosaic trisomy 13 have been reported to present phenotypic variability from the normal phenotype to the abnormal phenotype of trisomy 13.13 Griffith et al showed no clear correlation between the percentage of trisomic cells and the level of intellectual function. The features of dysmorphia vary significantly from patient to patient, which makes the clinical diagnosis of trisomy 13 mosaicism difficult.14
Conclusion
Rare aberrations accounted for 24% of all chromosome 13 aberrations. Mosaic monosomy of chromosome 13 and microdeletion 13q are characterized by similar abnormalities of the external genitalia and facial dysmorphia. The 13q duplication phenotype is similar to the clinical features of chromosome 13 trisomy. Mosaic trisomy 13 can occur without any accompanying anatomical defects.
Ethical Approval
The present study was approved by the Ethics and Research Committee of the Medical University of Lodz, Poland with reference no: RNN/76/10/KE. The study was performed according to the Declaration of Helsinki on research involving human subjects. Written informed consent was signed by the participants authorizing to perform genetic tests and use of the data for research and education.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Springett A, Wellesley D, Greenlees R, et al. Congenital anomalies associated with trisomy 18 or trisomy 13: a registry-based study in 16 European countries, 2000–2011. Am J Med Genet A. 2015;167:3062–3069.
2. Jones KL, Jones MC, Campo MD. Smith’s Recognizable Patterns of Human Malformation. Elsevier Health Sciences.
3. Dalal SS, Berry T, Pimentel VM. Prenatal sacrococcygeal teratoma diagnosed in a fetus with partial trisomy 13q22. Case Rep Obstet Gynecol. 2019;2019:2892869.
4. Allderdice PW, Davis JG, Miller OJ, et al. The 13q-deletion syndrome. Am J Hum Genet. 1969;21:499–512.
5. Kataoka A, Hirakawa S, Iwamoto M, Sakumura Y, Yoshinaga R, Ohba T. Prenatal diagnosis of a case of partial monosomy/monosomy 13 mosaicism: 46,XX,r(13)(p11q33)/45,XX,-13 suspected by nuchal fold translucency increasing. Kurume Med J. 2011;58:127–130.
6. Jønch AE, Larsen LG, Pouplier S, Nielsen K, Brøndum-Nielsen K, Tümer Z. Partial duplication of 13q31.3-q34 and deletion of 13q34 associated with diaphragmatic hernia as a sole malformation in a fetus. Am J Med Genet A. 2012;158A:2302–2308.
7. Machado IN, Heinrich JK, Campanhol C, Rodrigues-Peres RM, Oliveira FM, Barini R. Prenatal diagnosis of a partial trisomy 13q (q14-->qter): phenotype, cytogenetics and molecular characterization by spectral karyotyping and array comparative genomic hybridization. Genet Mol Res. 2010;9:441–448.
8. Jotterand M, Juillard E. A new case of trisomy for the distal part of 13q due to maternal translocation, t(9;13)(p21;q21). Hum Genet. 1976;33:213–222.
9. Schinzel A, Hayashi K, Schmid W. Further delineation of the clinical picture of trisomy for the distal segment of chromosome 13: report of three cases. Hum Genet. 1976;32:1–12.
10. Wang YP, Wang DJ, Niu ZB, Cui WT. Chromosome 13q deletion syndrome involving 13q31‑qter: a case report. Mol Med Rep. 2017;15:3658–3664.
11. Vervecken E, Blaumeiser B, Vanderheyden T, Hauspy J, Janssens K. Terminal deletion of chromosome 13 in a fetus with normal NIPT: the added value of invasive prenatal diagnosis in the NIPT era. Clin Case Rep. 2020;8:1461–1466.
12. Chen CP, Chen CY, Chern SR, et al. Prenatal diagnosis and molecular cytogenetic characterization of mosaicism for r(13), monosomy 13 and idic r(13) by amniocentesis. Taiwan J Obstet Gynecol. 2020;59:130–134.
13. Chen CP, Chern SR, Wu FT, Chen YY, Lee MS, Wang W. Low-level mosaic trisomy 13 at amniocentesis associated with a favorable outcome in a pregnancy. Taiwan J Obstet Gynecol. 2020;59:935–937.
14. Griffith CB, Vance GH, Weaver DD. Phenotypic variability in trisomy 13 mosaicism: two new patients and literature review. Am J Med Genet A. 2009;149:1346–1358.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.