")
Back to Journals » The Application of Clinical Genetics » Volume 14
Preimplantation Genetic Diagnosis for DEB by Detecting a Novel Family-Specific COL7A1 Mutation in Vietnam
Authors Trieutien S , Vu Van T, Tran Ngoc Thao M , Trinh The S , Tran Van K, Nguyen Thanh T, Tran Van T , Nguyen Thi H
Received 13 October 2021
Accepted for publication 3 December 2021
Published 9 December 2021 Volume 2021:14 Pages 467—472
DOI https://doi.org/10.2147/TACG.S344107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Sang Trieutien,1,* Tam Vu Van,2,3,* My Tran Ngoc Thao,4 Son Trinh The,5 Khoa Tran Van,1 Tung Nguyen Thanh,5 Tuan Tran Van,5 Hanh Nguyen Thi6
1Department of Biology and Genetics, Vietnam Military Medical University, Hanoi, 12108, Vietnam; 2Director Office, Hai Phong Hospital of Obstetrics and Gynecology, Haiphong, 40000, Vietnam; 3Obstetrics and Gynecology Department, Haiphong University of Medicine and Pharmacy, Haiphong, 40000, Vietnam; 4Département de formation Biologie moléculaire et cellulaire, Sorbonne University, Paris, 75006, France; 5Military Institute of Clinical Embryology and Histology, Vietnam Military Medical University, Hanoi, 12108, Vietnam; 6Faculty of Biology, VNU University of Science, Vietnam National University, Hanoi, 12108, Vietnam
*These authors contributed equally to this work
Correspondence: Son Trinh The
Military Institute of Clinical Embryology and Histology, Vietnam Military Medical University, Hanoi, 12108, Vietnam
Email [email protected]
Background: Epidermolysis bullosa (EB) is a disorder characterized by the appearance of blisters, erosions and wounds in response to minimal trauma. The disease manifests with noticeable symptoms ranging from mild to severe, classified into four major types: epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), dystrophic epidermolysis bullosa (DEB) and Kindler syndrome. Preimplantation genetic diagnosis for the disease remains the only available option for families at risk for the recurrence of the disorder without having to terminate an ongoing pregnancy.
Materials and Methods: A novel COL7A1 mutation was used to design primers for the polymerase chain reaction (PCR) to amplify the segment spanning the mutation in the family and their in-vitro fertilization (IVF) embryos. Then, the PCR products were sequenced with Sanger sequencing to detect the alteration in the allele, and some embryos would go through NGS-based preimplantation screening for chromosomal abnormalities.
Results: The established protocol for EB detected mutant allele in 6/9 embryos (66.6%), while the remaining 3 embryos (33.4%) appeared to not carry any mutation. Only one among 3 embryos was recommended to be transferred into the mother’s uterus.
Conclusion: The established preimplantation genetic diagnosis procedure is helpful to families affected by epidermolysis bullosa caused by COL7A1 mutations but wish to have healthy children.
Keywords: epidermolysis bullosa, rare dystrophic epidermolysis bullosa, EB, RDEB, skin disorder, COL7A1 gene mutation, preimplantation genetic diagnosis, PGD
Introduction
Epidermolysis bullosa (EB) constitutes a heterogeneous group of heritable mechano-bullous disorders characterized by the appearance of blisters, erosions and wounds in response to minimal trauma. The disease manifests with noticeable symptoms ranging from mild to severe on the epithelial layer or the mucous membrane. Much has been discovered and changed about the spectrum of inherited EB since it was first defined and classified, with the application of different fields of biology and techniques. Up to date, epidermolysis bullosa comprises four major types depending on the level of skin cleavage: epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), dystrophic epidermolysis bullosa (DEB) and Kindler syndrome.1,2 The molecular pathology of EB now consists of 21 genes with over 1000 reported pathogenic variants in either an autosomal dominant or autosomal recessive inheritance manner. These mutations lead to the change in the adhesion among specific attachment proteins or in the dermal-epidermal junction, which would eventually lead to the fragility of the skin.3,4
In dystrophic epidermolysis bullosa (DEB), the blistering occurs within the sub-lamina densa region within the uppermost dermis. Two main subtypes of DEB that are classified according to their mode of inheritance, dominant DEB (DDEB) and recessive DEB (RDEB), are only associated with the gene COL7A1. The gene is located within the 3p21 region, spanning about 32 kb and comprising 118 exons.5 In fact, they are due to mutations in the COL7A1 gene encoding for the homotrimer type VII collagen, which is the major component of the cutaneous basement membrane’s anchoring fibrils.1,6–8 The defects in the fibrils, such as the morphological alteration, physical reduction or absence, result in a considerable degree of clinical severity based on the different types and positions of the pathogenic variants. The phenotypic variability can highly differ from a limited tendency to blistering or nail dystrophy to severe extra-cutaneous complications, even lethal carcinomas and early mortality.8–10
Preimplantation genetic diagnosis (PGD), or in other terms, preimplantation genetic testing for monogenic disorder (PGT-M), is considered to be the earliest form of prenatal testing for couples at reproductive risks with a hereditary disease. Once a pathogenic variant is known, PGD would be performed to examine the cellular material from in-vitro fertilization (IVF) embryos for specific genetic abnormalities before pregnancy. This advanced specialized procedure offers appropriate genetic counseling to determine the genetic risk for future offspring and obviate the need to terminate an affected pregnancy. With the advent of technologies in biology and medicine, the spectrum of PGD is remarkably expanded when approximately 500 different conditions have been interrogated worldwide.11 In the case of DEB, not only are the pathogenic variants distributed over the entire gene making the process of screening COL7A1 gene time-consuming and expensive but also there have not been any effective treatments for the diseases. Thus, early prevention of DEB remains the only available option for families at risk for the recurrence of the disorder.2,10,12 Even though developing a standardized PGD procedure is work-intensive, complex and costly, this paper aimed to describe a PGD protocol for RDEB utilizing whole-genome amplification (WGA) and Sanger sequencing. Hence, incorporating these diverse techniques generated a more feasible and economical yet profoundly reliable approach for couples with family-specific pathogenic COL7A1 variants in Vietnam, producing an exact diagnosis of recessive dystrophic epidermolysis bullosa in future generations.
Materials and Methods
Patient Description
A Vietnamese nuclear family enrolled in the study, including a four-year-old son with noticeable characteristics of epidermolysis bullosa. The son was diagnosed with DEB and presented with the occurrence of blisters, scarring and erosion on his shoulder, back, limbs and dystrophic nails while his parents did not present any clinical or genetic alteration of interest. By studying the affected’s genetic information with whole-exome sequencing, it was reported that the son carried a recessive homozygous mutation c.8279G>A (p.G2760E) on the exon 111 of the gene COL7A1. This novel non-synonymous variant has never been reported in any public database, which was predicted as probably damaging/deleterious by in silico algorithms, such as SIFT, Polyphen2 and MutationTaster.13 With bidirectional Sanger sequencing on his parents’ DNA, the heterozygous c.8279G>A variant was also observed.
All people described in this research were signed written informed consent for the publication of the case details, and the protocol was approved by the Ethical Review Committee of Vietnam Military Medical University (No.1068/2019/VMMU-IRB). This study was also conducted using good clinical practice following the Declaration of Helsinki and its later amendments or comparable ethical standards.
DNA Extraction from Whole Blood
DNA was extracted from the collected blood samples by following the protocol of the G-spin™ Total DNA Extraction Kit (Lot. No. 105260354; Exp. June 2023). DNA went through a quality check process with a SpectraMax QuickDrop to measure the optical density (OD) and A260/A280 index. Hence, the DNA collected from the sample was qualified to be used in the next steps of the research. DNA samples were stored at −20°C.
Blastocyst Embryo Biopsy
There were nine embryos (number 1 to 9) cultured to the fifth day of the described couple who had done IVF at the Military Institute of Clinical Embryology and Histology (MICEH). Then, the embryos were biopsied (3–5 cells) and washed with PBS 1X and 1% PVP solution. After that, the washed embryo cells were contained in the 0.2 mL PCR tube. The embryo cells were stored at −20°C.
Whole Genome Amplification for Embryos’ Genome
The DNA from the biopsied embryos was amplified with REPLI-g® Single Cell Kit (Lot. No. 169023130; Exp. May 2022) and then diluted with nuclease-free water. The concentration and purification were calculated with a SpectraMax QuickDrop. Therefore, the amplified DNA collected from the embryonic cells was qualified to be used in the research. DNA samples were stored at −20°C.
Polymerase Chain Reaction (PCR) Analysis
First, primers were designed to amplify the segment targeting exon 111 and spanning the detected mutation c.8279G>A. Afterward, a PCR was performed by using the designed primers and DNA collected from the whole family and DNA amplified from biopsied embryonic cells to confirm the inheritance of mutation. PCR products were then electrophoresed on 2% agarose gel on multiSUB Choice, Wide Midi Horizontal Electrophoresis System (Cleaver Scientific, SKU: MSCHOICE10) to check for the appropriate desired products.
Sanger Sequencing and the Next-Generation Sequencing Analysis
The amplified PCR products showing the accurate bands on electrophoresis results would be sequenced by Sanger sequencing to scan for the c.8279G>A mutation. The process was carried out on the ABI 3500 Genetic Analyzer according to the manufacturer’s instructions. Next, embryos that do not carry any mutated alleles would go through preimplantation genetic aneuploidy testing (PGT-A) based on Thermo Fisher Scientific Ion Proton™ semiconductor system, using the clinical testing kits to screen for chromosomal abnormalities. Then, the obtained next-generation sequencing data would be analyzed automatically with the manufacturer’s software.
Results
Preimplantation Genetic Diagnosis Program for DEB
The PCR reaction utilizing the appropriate primers, components and the thermal cycle was performed in triplicate. After each reaction round, the products went under electrophoresis on 2% agarose gel and were observed under UV light. The obtained results were consistent in all reactions. The gel electrophoresis was then analyzed, resulting in the successful amplification of all desired gene segments in all samples and embryos in comparison to the positive control that was the DEB homozygotes. The products’ bands appeared bright and clear at a position corresponding to the standard scale size of around 250 base pairs, proving that the amplified segment was consistent with the initial expectation of 241 base pairs. From here, the PCR products would be purified and sequenced via the Sanger sequencing method to detect the c.8279G>A variant in these cells.
Sanger Sequencing and NGS Results
Sanger sequencing was carried out using forward and reverse primers to amplify the segment spanning exon 111 from both ends in order to ensure the accurate interpretation of the results. The Sanger sequencing’s electropherograms were annotated by SnapGene software and summarized in Table 1.
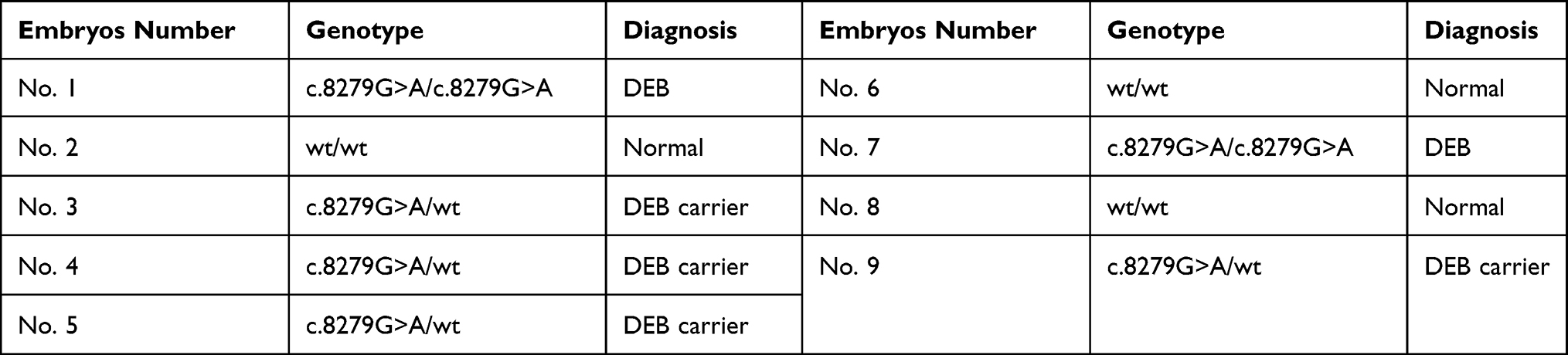 |
Table 1 Genotypes of Nine IVF Embryos by Sanger Sequencing |
From the information provided by Sanger sequencing, among nine biopsied embryos, two of which (22.2%) were DEB homozygotes, while four (44.4%) were DEB carriers. In fact, these embryos inherited at least one pathogenic allele from either the mother or the father, whose genotypes were determined to be heterozygous according to Sanger sequencing. Although the embryos that carried one mutant allele for DEB might not develop any clinical symptoms later in life, these embryos, along with the mutant homozygotes, were advised not to be transferred to completely prevent the inheritance of the pathogenic variant. The remaining embryos number 2, 6 and 8 were recommended to carry out PGT-A to screen for any chromosomal abnormalities to reduce the risk of miscarriage and to increase the possible pregnancy and live birth rate.
Based on the obtained NGS results of the Ion Proton™ system, embryos number 2 and 6 observed a trisomy pattern in chromosomes 15 and 21, respectively. Embryo number 8 was found not to carry any aneuploidies; therefore, it could be concluded that this embryo should be selected for transfer.
Discussion
The variant c.8279G>A (p.G2760E) that was first found in the son was reported to be a novel variant by Whole Exome Sequencing. This substitution mutation of the amino acid glycine by glutamic acid happens on the collagenous encoding domain, which might affect the protein’s stability and its triple helix structure.13 Since the variant was discovered, the genetic information of both parents was thoroughly studied to exclude the possibility of a de novo mutation, mainly when his mother and father did not express any clinical symptoms regarding dystrophic epidermolysis bullosa. Indeed, the pathogenic variant was identified in both of them with one copy of the mutated allele, consequently transmitted to their firstborn, leading to his current state. The disease leaves significant psychological and physical morbidity for the patients’ families since the couples constantly fear the recurrence of RDEB in the future offspring, especially when unfamiliar with genetic services. Therefore, the need to promote the advantages of PGD or PGT-M for the affected families is growing tremendously to help the ones in need.
PGT-M is a robust and highly specialized procedure involving the testing of genetic material in IVF human embryos. It begins with the stimulation of the ovaries to collect oocytes, then fertilization by sperm injection (ICSI). Then, the embryos are biopsied at different stages of development, either by a single blastomere at the cleavage stage or multiple trophectoderm (TE) at the blastocyst stage. However, a blastocyst stage biopsy is becoming preferable in terms of PGD approaches across the globe. The reason behind this trend is that more cells can be removed at this stage, which means there would be higher DNA yield to carry out more accurate diagnosis yet does not affect the blastocyst viability if appropriately performed by experienced hands.11,14–17 Accordingly, much effort has been made to establish a feasible and widely applicable preimplantation genetic diagnosis procedure using advanced biopsy and genetic analysis techniques. In literature, various studies are focusing on the early detection of different types of epidermolysis bullosa. All the designed tests had to reflect the complexity of the disease’s molecular pathology, which comprises mutations spanning almost every exon of the gene COL7A1. In each case, the discovered pathogenic variants are rarely recurrent yet specific to individual families, sometimes novel.18–20
Moreover, some other researches also focused on polymorphic markers within and flanking the genes responsible for other EB types, such as junctional epidermolysis bullosa.21,22 Therefore, in EB, where mutation screening in preimplantation genetic diagnosis is not convenient and economical, an approach utilizing a straightforward analysis tool for a specific mutation is more applicable and practical. In our research, based on discovering the mutation of exon 111, a set of primers were designed for both conventional PCR programs and direct sequencing of the PCR products. Since one pair of primers was needed to detect the novel family-specific variant, the labor for carrying out the diagnosis has decreased immensely. At the same time, the test’s accuracy is strengthened, and it can be applied to other couples in the same family.
Apart from developing the general idea behind this PGD procedure, some common technical obstacles had to be overcome regarding single-cell assay. These problems include insufficient DNA template, amplification failure, or allele dropout.23–25 However, using a non-PCR-based amplification technique called multiple displacement amplification (MDA), adequate DNA was generated uniformly with high molecular weight and large fragments. The high processivity and excellent proofreading activity exploited from Phi29 DNA polymerase pose a greater possibility of developing a robust and non-bias amplifying procedure for limited DNA sources.26,27 Moreover, incorporating one more round of DNA targeting the exon with the mutation with specifically designed primers would diminish the amplification failure for further analysis. Nonetheless, a small fraction of erroneous diagnoses occur, mainly caused by ADO as directly working on the embryos’ genetic materials is not practical; hence, this raises a problematic issue regarding preimplantation testing for in vitro fertilization processes. The false-negative result, especially in recessive Mendelian disorders, has always been the main concern for IVF practitioners, genetic specialists, and patients and should be avoided at all costs.
Overall, our research focused on developing a contemporary procedure to balance between giving out the best results in genetic consultations and ensuring the highest vitality of in vitro fertilized embryos. Blastocyst stage TE biopsy, multiple displacement amplification, then performing both PGT-M to detect epidermolysis bullosa and PGT-A for chromosomal abnormalities make up for each other’s shortcomings when being applied individually. The utilization of Sanger sequencing – an exact sequencing technique – enables a fast and reliable approach to studying each nucleotide of the pathogenic variant’s exon. With its improvements in methodology and automation, this is irrefutably suitable for examining single nucleotide replacement in a short segment. In addition, by coordinating NGS-based PGT-A on amplified DNA from human embryos, embryonic mosaicism can be identified in whole chromosomes as well as segmental regions along with aneuploidy and structural rearrangements. Thanks to the advent of bioinformatics tools, PGT-A technology has been widely adopted by various assisted reproduction centers. It has been reported to increase rates of positive clinical outcomes compared to transfers without PGT-A in some studies. According to Sacchi et al, the live birth rate per transferred embryo was significantly higher in the PGT-A group (40.3%) than in the control group (11.0%). Moreover, much higher aneuploidy in the control group was witnessed in cytogenetic analysis of amniocentesis products compared to PGT-A (0% versus 17.9% and 19.9%).28 Furthermore, the application of PGT-A on elective single embryo transfer (eSET) has tremendously improved implantation rates despite only one embryo being transferred since it considerably decreased the complications from multiple pregnancies.29–31 Although NGS-based PGT-A should be offered to patient groups with a high incidence of embryonic aneuploidy, evidence demonstrates that it likewise benefits young and good-prognosis patients.32,33 It not only reduced the risks of miscarriage but also time to pregnancy. Additionally, it helped increase the live births rate and enabled confident single embryo transfer, even for patients younger than 35 years old.34
Conclusion
The established PGD procedure is helpful to families affected by epidermolysis bullosa caused by COL7A1 mutations. With proper counseling towards the hereditary disease, couples at reproductive risk for recurrence of EB would be in an active state to make plans for their future offspring without the inheritance of the pathogenic allele. Incorporating advances in molecular techniques provide patients and their families with a robust, precise, and cost-effective mutation detection process that is practical among genetic centers worldwide.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fine J-D, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–1126. doi:10.1016/j.jaad.2014.01.903
2. Järvikallio A, Pulkkinen L, Uitto J. Molecular basis of dystrophic epidermolysis bullosa: mutations in the type VII collagen gene (COL7A1). Hum Mutat. 1997;10(5):338–347. doi:10.1002/(SICI)1098-1004(1997)10:5<338::AID-HUMU2>3.0.CO;2-B
3. Vahidnezhad H, Youssefian L, Saeidian AH, Uitto J. Phenotypic spectrum of epidermolysis bullosa: the paradigm of syndromic versus non-syndromic skin fragility disorders. J Investig Dermatol. 2019;139(3):522–527. doi:10.1016/j.jid.2018.10.017
4. Has C, Fischer J. Inherited epidermolysis bullosa: new diagnostics and new clinical phenotypes. Exp Dermatol. 2019;28(10):1146–1152. doi:10.1111/exd.13668
5. Christiano AM, Hoffman GG, Chung-Honet LC, et al. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics. 1994;21(1):169–179. doi:10.1006/geno.1994.1239
6. Uitto J. The molecular basis of the dystrophic forms of epidermolysis bullosa. In: Epidermolysis Bullosa Clinical, Epidemiologic, and Laboratory Advances and the Findings of the National Epidermolysis Bullosa Registry; 1999.
7. Uitto J, Pulkkinen L. The genodermatoses: candidate diseases for gene therapy. Hum Gene Ther. 2000;11(16):2267–2275. doi:10.1089/104303400750035807
8. Mariath LM, Santin JT, Frantz JA, Doriqui MJR, Schuler‐Faccini L, Kiszewski AE. Genotype‐phenotype correlations on epidermolysis bullosa with congenital absence of skin: a comprehensive review. Clin Genet. 2021;99(1):29–41. doi:10.1111/cge.13792
9. Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype–genotype correlations in the dystrophic subtypes. J Med Genet. 2007;44(3):181–192. doi:10.1136/jmg.2006.045302
10. Kern JS, Kohlhase J, Bruckner-Tuderman L, Has C. Expanding the COL7A1 mutation database: novel and recurrent mutations and unusual genotype–phenotype constellations in 41 patients with dystrophic epidermolysis bullosa. J Investig Dermatol. 2006;126(5):1006–1012. doi:10.1038/sj.jid.5700219
11. Simpson JL, Kuliev A, Rechitsky S. Overview of preimplantation genetic diagnosis (PGD): historical perspective and future direction. In: Prenatal Diagnosis; 2019:23–43.
12. Uitto J, Pulkkinen L, Ringpfeil F. Progress in molecular genetics of heritable skin diseases: the paradigms of epidermolysis bullosa and pseudoxanthoma elasticum.
13. Ma THT, Luong TLA, Hoang TL, et al. Novel and very rare causative variants in the COL7A1 gene of Vietnamese patients with recessive dystrophic epidermolysis bullosa revealed by whole‐exome sequencing. Mol Genet Genomic Med. 2021;9(8):e1748. doi:10.1002/mgg3.1748
14. Coonen E, Van Montfoort A, Carvalho F, et al. ESHRE PGT Consortium data collection XVI–XVIII: cycles from 2013 to 2015. Hum Reprod Open. 2020;2020(4):hoaa043. doi:10.1093/hropen/hoaa043
15. Sciorio R, Tramontano L, Catt J. Preimplantation genetic diagnosis (PGD) and genetic testing for aneuploidy (PGT-A): status and future challenges. Gynecol Endocrinol. 2020;36(1):6–11. doi:10.1080/09513590.2019.1641194
16. Committee EPCS, Carvalho F, Coonen E, et al. ESHRE PGT Consortium good practice recommendations for the organisation of PGT. Hum Reprod Open. 2020;2020(3):hoaa021. doi:10.1093/hropen/hoaa021
17. Viotti M. Preimplantation genetic testing for chromosomal abnormalities: aneuploidy, mosaicism, and structural rearrangements. Genes. 2020;11(6):602. doi:10.3390/genes11060602
18. Fassihi H, Renwick P, Black C, McGrath J. Single cell PCR amplification of microsatellites flanking the COL7A1 gene and suitability for preimplantation genetic diagnosis of Hallopeau–Siemens recessive dystrophic epidermolysis bullosa. J Dermatol Sci. 2006;42(3):241–248. doi:10.1016/j.jdermsci.2006.01.005
19. Ozge A, Safak H, Ebru H, et al. First successful preimplantation genetic diagnosis of epidermolysis bullosa with pyloric atresia: case study of a novel c. 4505-4508insACTC mutation. J Assist Reprod Genet. 2012;29(4):347–352. doi:10.1007/s10815-012-9728-8
20. Vendrell X, Bautista-Llácer R, Alberola TM, et al. Pregnancy after PGD for recessive dystrophic epidermolysis bullosa inversa: genetics and preimplantation genetics. J Assist Reprod Genet. 2011;28(9):825–832. doi:10.1007/s10815-011-9601-1
21. Cserhalmi‐Friedman P, Tang Y, Adler A, Krey L, Grifo J, Christiano A. Preimplantation genetic diagnosis in two families at risk for recurrence of Herlitz junctional epidermolysis bullosa. Exp Dermatol. 2000;9(4):290–297.
22. Fassihi H, Liu L, Renwick P, Braude P, McGrath J. Development and successful clinical application of preimplantation genetic haplotyping for Herlitz junctional epidermolysis bullosa. Br J Dermatol. 2010;162(6):1330–1336.
23. Thornhill AR, Snow K. Molecular diagnostics in preimplantation genetic diagnosis. J Mol Diagn. 2002;4(1):11–29. doi:10.1016/S1525-1578(10)60676-9
24. Thornhill AR, deDie-Smulders C, Geraedts JP, et al. ESHRE PGD Consortium ‘Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS)’. Hum Reprod. 2005;20(1):35–48. doi:10.1093/humrep/deh579
25. Harton G, De Rycke M, Fiorentino F, et al. ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum Reprod. 2011;26(1):33–40. doi:10.1093/humrep/deq231
26. Spits C, Le Caignec C, De Rycke M, et al. Optimization and evaluation of single‐cell whole‐genome multiple displacement amplification. Hum Mutat. 2006;27(5):496–503. doi:10.1002/humu.20324
27. Ling J, Zhuang G, Tazon-Vega B, et al. Evaluation of genome coverage and fidelity of multiple displacement amplification from single cells by SNP array. Mol Hum Reprod. 2009;15(11):739–747. doi:10.1093/molehr/gap066
28. Sacchi L, Albani E, Cesana A, et al. Preimplantation genetic testing for aneuploidy improves clinical, gestational, and neonatal outcomes in advanced maternal age patients without compromising cumulative live-birth rate. J Assist Reprod Genet. 2019;36(12):2493–2504. doi:10.1007/s10815-019-01609-4
29. Forman EJ, Hong KH, Franasiak JM, Scott RT
30. Penzias A, Bendikson K, Butts S, et al. The use of preimplantation genetic testing for aneuploidy (PGT-A): a committee opinion. Fertil Steril. 2018;109(3):429–436. doi:10.1016/j.fertnstert.2018.01.002
31. Zakaria M, Ennaji M, Wassym SR, et al. PGT-A for embryos for transfer and to improve clinical outcomes in terms of embryo implantation, and recommendations embryo biopsy for at blastocyst stage. J Basic Clin Reprod Sci. 2020;9(5). doi:10.4103/2278-960X.1945146
32. Franasiak JM, Forman EJ, Hong KH, et al. The nature of aneuploidy with increasing age of the female partner: a review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil Steril. 2014;101(3):656–663. e1. doi:10.1016/j.fertnstert.2013.11.004
33. Rubio C, Rodrigo L, Garcia-Pascual C, et al. Clinical application of embryo aneuploidy testing by next-generation sequencing. Biol Reprod. 2019;101(6):1083–1090. doi:10.1093/biolre/ioz019
34. Anderson R, Whitney J, Schiewe M. Clinical benefits of preimplantation genetic testing for aneuploidy (PGT-A) for all in vitro fertilization treatment cycles. Eur J Med Genet. 2020;63(2):103731. doi:10.1016/j.ejmg.2019.103731
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.