Back to Journals » Vascular Health and Risk Management » Volume 11
Preeclampsia: long-term consequences for vascular health
Authors Amaral L, Cunningham Jr MW, Cornelius D, LaMarca B
Received 28 February 2015
Accepted for publication 29 April 2015
Published 15 July 2015 Volume 2015:11 Pages 403—415
DOI https://doi.org/10.2147/VHRM.S64798
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Daniel Duprez
Lorena M Amaral, Mark W Cunningham Jr, Denise C Cornelius, Babbette LaMarca
Department of Pharmacology, University of Mississippi Medical Center, Jackson, MS, USA
Abstract: Preeclampsia (PE) is a pregnancy-specific syndrome and one of the leading causes of preterm birth, neonatal and maternal morbidity and mortality. This disease is characterized by new onset hypertension usually in the third trimester of pregnancy and is sometimes associated with proteinuria, although proteinuria is not a requirement for the diagnosis of PE. In developing countries, women have a higher risk of death due to PE than more affluent countries and one of the most frequent causes of death is high blood pressure and stroke. Although PE only affects approximately 2%–8% of pregnancies worldwide it is associated with severe complications such as eclampsia, hemorrhagic stroke, hemolysis, elevated liver enzymes and low platelets (HELLP syndrome), renal failure and pulmonary edema. Importantly, there is no “cure” for the disease except for early delivery of the baby and placenta, leaving PE a health care risk for babies born from PE moms. In addition, PE is linked to the development of cardiovascular disease and stroke in women after reproductive age, leaving PE a risk factor for long-term health in women. This review will highlight factors implicated in the pathophysiology of PE that may contribute to long-term effects in women with preeclamptic pregnancies.
Keywords: preeclampsia, endothelial dysfunction, AT1-AA, CD4+ T helper cells
Introduction
The underlying pathophysiology of preeclampsia (PE) is not completely understood, but it is currently believed that the initiating event in PE is reduced placental perfusion, which develops from shallow cytotrophoblast migration toward the uterine spiral arterioles which leads to inappropriate vascular remodeling and a hypoperfused placenta.1 This placenta becomes ischemic as the pregnancy continues which leads to the release of factors that cause maternal vascular endothelial dysfunction.1–8 Endothelial dysfunction results in generalized vasoconstriction, reduced blood to multiple organs and it has been a major phenotype of PE.1,4,5,9–13 Furthermore, pre-existing conditions such as poor nutrition, diabetes, and obesity, are all risk factors for PE, and could exacerbate the maternal response to factors released from the ischemic placenta.9 While 800 women die from pregnancy complications around the world every day and 3 million of preterm births reported each year are related to PE,14,15 there is no effective treatment for this pregnancy disease except for early delivery of the fetus.
The maternal cardiovascular and renal system goes through several important adaptations during a normal pregnancy. The cardiac output, heart rate, and stroke volume increases during pregnancy due to an increase in plasma volume expansion and systemic vascular vasodilation during pregnancy.16 A lack of these changes during pregnancy is often associated with pregnancy complications and an increased risk of developing cardiovascular events (such as myocardial infarctions, venous thromboembolisms, and stroke) in the mother later in life.17 Multiple clinical studies of women with PE show an increased risk of developing cardiovascular diseases later in life.18 For example a Norwegian study from 1967 to 1992, with over 600,000 pregnant women, showed that women with PE have an 8-fold increase in death resulting from a cardiovascular event versus women who have a normal pregnancy.18 A study of ~30,000 women in the state of Washington from 1967 to 1998 showed that women with mild and severe PE had a 2-fold and 3-fold greater risk of cardiovascular events later in life respectively.18 Furthermore, a 30 year follow-up study of ~14,000 women in California noted that women with PE have a 2-fold increase in death due to cardiovascular events, and women who had the onset of PE before 34 weeks of gestation had a 9-fold increase of cardiovascular events later in life.18 Finally, a Taiwanese study of 1 million women shows that women with PE have a 12-fold increase in having a cardiovascular event.18 Thus all of these clinical studies taken together show that women with PE have an increased risk of having a cardiovascular event later in life no matter the location, thereby indicating the importance of the common mediators of this disease shared among these women to help identify the link between PE and later cardiovascular events.
Renal insufficiency is a common thread that could contribute to long-term pathology of previously preeclamptic women
Despite an increase in cardiac output during pregnancy, there is a decrease in maternal blood pressure due to a decrease in total peripheral resistance caused by maternal vasodilation during pregnancy. In addition, in the kidneys, the glomerular filtration rate (GFR) and renal blood flow increase due to a decrease in renal vascular resistance.16 Women with PE often have altered renal function, glomerular endotheliosis, and proteinuria.19 Early studies in the 1970s by Gibson, showed that the lack of rise in GFR during early pregnancy is associated with women who have an increased risk of unexplained stillbirths, abortions, or small for gestational age babies.20 A population study consisting of pregnant Norwegian patients from 1967 to 1991, showed that 477 patients out of 570,433 pregnant women developed end-stage renal disease (ESRD).21 Out of those 477 women who developed ESRD, the authors found that women who had been pregnant one or more times and developed PE during the first pregnancy are four times more likely to develop ESRD.21 They also showed that as the number of reoccurring preeclamptic pregnancies in the mother increased, the greater her risk of developing ESRD.21
Importantly, there is a decrease in GFR and proteinuria in preeclamptic patients.19 Several studies show that podocyturia, which is the urinary excretion of podocytes, are elevated in women with PE.22–24 The amount of podocyturia, podocytes in the urine, can be determined by quantifying podocalyxin, podocin, nephrin, and synaptopodin protein structures in the urine.22–24 A more recent study by Garovic et al,22 showed that podocyturia had a greater predictive value for the diagnosis of PE versus other angiogenic factors, such as soluble fms-like tyrosine kinase-1 (sFLT-1), endoglin, and placental growth factor (PlGF). It is important to note that the loss of podocytes, which have a very limited regenerative capacity, from the glomerulus may lead to an increase in proteinuria and glomerulosclerosis due to a disruption of the glomerular filtration barrier. Therefore, podocyturia may serve as a sensitive biomarker for the development of PE and the degree of renal injury damage observed in PE.
Endothelial dysfunction is a common thread that could contribute to long-term pathology of previously preeclamptic women
Nitric oxide (NO)
Endothelial dysfunction is characterized by an increase in ET-1, a potent vasoconstrictor, secretion from endothelial cells and a lack of appropriate vascular responses to endothelial mediated vasodilators. While the mechanisms responsible for systemic maternal vascular dysfunction are unknown, mediators of endothelial dysfunction such as decreased NO have been shown to play a role in the development of hypertension in preeclamptic women.25–30 Previous studies have shown that inhibition of NO synthase in pregnant rats is associated with intrauterine growth retardation (IUGR)31 and L-arginine supplementation prevents fetal growth restriction in animal models of IUGR.32 In addition, the supplementation with L-arginine in IUGR pregnant women improves vasorelaxation indicating this pathway could be an important role in the pathophysiology of IUGR in human pregnancies.33
NO, originally identified as the endothelium derived relaxing factor, is an important vasodilator synthetized in response to mechanical and chemical stimuli from L-arginine by a family of calcium–calmodulin-dependent enzymes, called nitric oxide synthases (NOS).34 NO induces vascular smooth muscle relaxation via soluble guanylate cyclase (sGC)/cyclic guanosine monophosphate (cGMP)-dependent and independent mechanisms. In addition, NO inhibits leukocytes adhesion, and has antithrombotic and anti-apoptotic effects.35
NO derived from endothelial nitric oxide synthase (eNOS) is an important mediator of vascular homeostasis.35 An up-regulation of eNOS, resulting in increased NO production has been shown to contribute to increases in uteroplacental blood flow via changes in vascular tone.36 In addition, there is evidence that genetic eNOS polymorphisms may affect the susceptibility to hypertensive disorders of pregnancy.37,38
Alterations during normal pregnancy such as increased blood volume is accommodated within the cardiovascular system by systemic vasodilatation associated with NO production,39 thereby suggesting that NO deficiency could be playing an important role in hypertensive disorders of pregnancy. In fact, several studies in human and animal models have shown that impaired vascular relaxation in PE has been attributed to reduced bioavailability of NO produced via eNOS.14,40,41 Previous studies have demonstrated elevated eNOS expression during PE,42 however both increased arginase expression (regulator of NOS)43 and elevated levels of a natural NOS inhibitor, asymmetric dimethylarginine44 were found which in turn, could impair the balance between vasodilator and vasoconstrictor effects on the vascular smooth muscle thereby contributing to rise in blood pressure in PE. Furthermore, Mutlu-Turkoglu et al have published that circulating NOx levels were decreased in preeclamptic women compared to normal pregnant women before delivery. After 24 hours, post-delivery, these levels did not change in the normal pregnant group, however, nitrate-nitrite (NOx) levels increased in the preeclamptic group supporting the idea that alterations in NO regulation may play a key role in PE.45
Experimental animal models of placental ischemia including reduced uterine perfusion pressure (RUPP) have been shown to exhibit impaired vasorelaxation in conduit vessels and activity of NO has been investigated.28,39,46,47 Moreover, previous investigators have demonstrated a loss or absence of NO-mediated vasorelaxation in RUPP plasma-treated vessels.48 In our recent study we have shown that circulating NOx levels and vascular eNOS expression were decreased in RUPP rats.49
Although extensive research is needed to further investigate the benefits of NO, drugs that target components of NO pathway could increase NO bioavailability contributing to improve the maternal outcomes such as hypertension and endothelial dysfunction. Furthermore, markers of endothelial dysfunction such as ET-1, soluble vascular adhesion molecule and IL-8, or endothelial leukocyte adhesion molecule 1 (ELAM-1), which could be elevated weeks prior the clinical manifestation of PE.50 Indeed, detection of endothelial dysfunction or vasoconstriction may serve as predictor of this disease and may be helpful in order to understand the pathways associated with PE. Peripheral arterial tone and other methods which rely on flow mediated dilatation have been used to assess the endothelial function during pregnancy and PE.51 Although the peripheral arterial tone did not identify women who will develop PE, this technique could be associated with a relative peripheral vasoconstriction in preeclamptic women after delivery and might be used to detect abnormalities that persisted after pregnancy. Furthermore, peripheral pulse pressure waveforms determined by method of applanation tonometry demonstrate the changes in radial artery pulse waveform, and its correlation with central aortic pressure and pulse wave during pregnancy. In fact, tonometry has showed elevated augmentation index in preeclamptic women compared with normal pregnant women.52 Non-invasive measurements of vasoconstriction during pregnancy in future studies may help predict PE and the risks of cardiovascular disease post-delivery.
sFlt-1
Abnormalities in the placenta are associated with increased anti-angiogenic factors such as sFlt-1 or soluble vascular endothelial growth factor receptor 1 which contribute to decreased renal function and hypertension during pregnancy. Preeclamptic women have increased circulating sFlt-1 levels and placental sFlt-1 mRNA compared to women who have normal pregnancies.42 Furthermore, the effects of pro-angiogenic factors VEGF and PlGF which are important to maintain the vascular endothelium are antagonized by sFlt-1 which is a possible cause of endothelial dysfunction during this disease.53 Previous studies have shown that decreased levels of VEGF and PIGF were correlated with increased levels of sFlt-1 during PE.42 In addition, VEGF is important to induce and maintain the integrity of fenestrated endothelium in various tissues, including the renal glomerulus, and VEGF blockade by sFlt-1 may be a cause for renal damage and a decrease in renal function.
The role for sFlt-1 in the pathogenesis of this hypertensive disorder during pregnancy has been supported by animal data. When administered to pregnant and non-pregnant rats using an adenoviral vector, sFlt-1 induces PE-like syndrome resulting in the classical signs such as hypertension, proteinuria, and glomerular endotheliosis.42 Recent studies by Murphy et al demonstrate that infusion of sFlt-1 into normal pregnant rats caused hypertension which was associated with endothelial dysfunction characterized by increased ET-1 and decreased NO.54 Importantly, L-arginine improved hypertension and ET-1 expression.28 These data further support the hypothesis that increased NO availability may be an innovative pathway to target improved treatment of PE.
Importantly, a recent paper has shown that sFlt-1 levels normalized after 2 months post-delivery in PE-exposed compared with control mice. In the same study, an enhanced vascular response to injury in vessels exposed to PE was observed after delivery.55 Additionally, women who develop PE did not show differences in serum sFlt-1 levels at 12 weeks postpartum compared with their third trimester.56 Therefore, high sFlt-1 levels during and after PE could play a persistent role in endothelial dysfunction and damage after PE. Furthermore, altered expression of angiogenesis-proteins was presented in women who had prior PE more than 1 year post-delivery.57 Although PE is resolved by delivery of the placenta, and altered angiogenesis and vascular damage have been associated with long-term cardiovascular risk, larger studies are necessary to verify whether postpartum measures of sFlt-1 levels have a role in predicting future cardiovascular disease.
Chronic inflammation is a common thread that could contribute to long-term pathology of previously preeclamptic women
During normal pregnancy, the fetus must overcome immune rejection by the maternal immune system. Therefore, tolerance for the semi-allogenic fetal antigens must be established, particularly at the maternal-fetal interface, while still maintaining immune protection against pathogens. At the maternal-fetal interface, leukocytes make up 30%–40% of the decidual cells and are comprised of mainly NK cells, CD14+ myelomonocytic cells, and T lymphocytes.58 Normal pregnancy proceeds with mild inflammation, however, women with PE exhibit chronic immune activation and have an exaggerated innate inflammatory response. Multiple studies implicate a number of immune factors in mediating hypertension and endothelial dysfunction during pregnancy.59–69 Clinical studies have demonstrated an increase in pro-inflammatory cytokine production in PE compared to normal pregnancy, and these data are also observed in animal models of PE.5,46,70–76 T lymphocytes are instrumental to immunological memory and could play a role in the long-term sequelae of PE such as stroke or other cardiovascular events.
CD4+ T helper cells
CD4+ T-cells are a heterogeneous group of cells made up of several different subsets to include TH1, TH2, TH17, and CD4+ TReg cells. Clinical studies have shown that the populations of all subsets of CD4+ T-cells are not increased, but rather the TH1 and the TH17 subsets are increased while the TH2 and TReg subsets are decreased in women with PE compared to women with normal pregnancies.77–86 These alterations in the CD4+ populations of cells are described as a CD4+ T-cell imbalance, and recent studies have partially determined how changes in these specific populations contribute to pathophysiology during PE.
TH1 cells produce IL-2 and IFN-γ, are pro-inflammatory, and are involved in cellular immunity, while TH2 cells produce IL-4, IL-5, and IL-13, are anti-inflammatory, and are involved in humoral immunity. Normal pregnancy is associated with a predominant TH2 profile and suppressed TH1-type immunity, whereas an increased ratio of TH1:TH2 cells is observed in PE.77,80,86,87 Zenclussen et al performed adoptive transfer of activated BALB/c TH1-like splenocytes into allogeneically pregnant BALB/c female mice during late gestation.76 This resulted in the development of a PE-like model presenting with increased blood pressure, glomerulonephritis, and proteinuria. Furthermore, in response to adoptive transfer of activated TH1-like cells, uterine cytokine production and fetal reabsorptions were also increased. However, adoptive transfer of these cells into non-pregnant mice did not elicit any pathological response.76 This study demonstrates that increased activation of the TH1 population of cells causes pathophysiological symptoms exclusively during pregnancy.
In our studies with the RUPP rat model of PE, the T-cell profile is similar to that seen in PE patients.66,88 RUPP rats have an increase in CD4+ T helper cells characterized by increased TH17 and decreased TRegs. We demonstrate that adoptive transfer of RUPP CD4+ T-cells into normal pregnant rats increases blood pressure and reduces renal function.64 Furthermore, RUPP CD4+ T-cells cause placental and renal oxidative stress and increased ET-1.64,66,67,88 We believe one mechanism stimulated by T-cells is to facilitate B-cell production of AT1-AA, as discussed in greater detail below and seen in Figure 1.
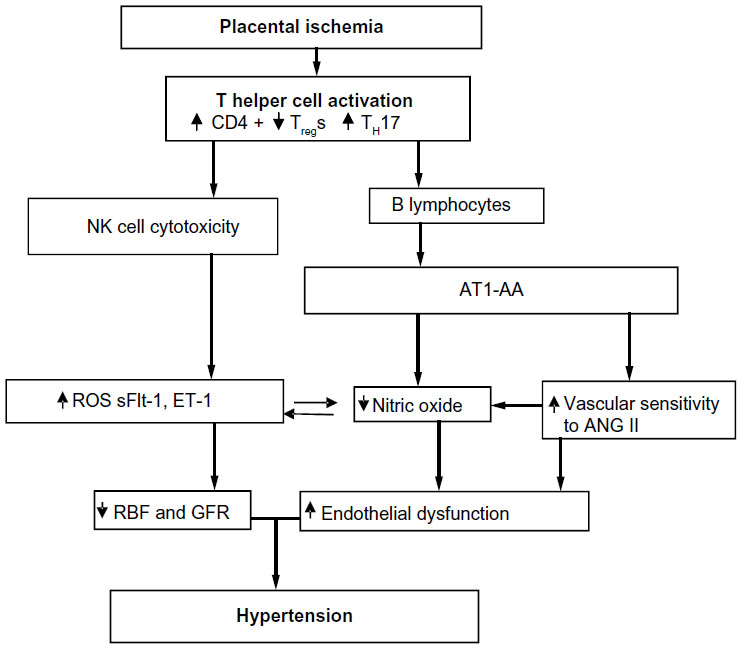 | Figure 1 Placental ischemia is a stimulus for chronic inflammation that leads to vasoactive factors that could play a role in later CVD in previously preeclamptic women. |
A number of clinical studies have reported that the population of TH17 cells is significantly increased in PE compared to normal pregnancy.66,79,83,89 Recent preclinical studies have begun to elucidate a role for TH17s and IL-17 to cause much of the pathophysiology observed in PE.66,90–92 TH17 cells are distinguished from other populations of CD4+ T-cells by their secretion of IL-17, surface expression of the IL-23 receptor, and intracellular expression of the lineage-specific transcription factor retinoic acid receptor-related organ receptor (RORγ).93
Studies have demonstrated that the increased inflammation observed during PE persists. Vitoratos et al demonstrated that women with PE remained under inflammatory stress up to 12–14 weeks postpartum.94 A later study by Kvehaugen et al showed evidence of chronic systemic inflammation and persistent endothelial dysfunction 5–8 years postpartum in women and their offspring after PE.95 These data suggest that persistent inflammation, postpartum, may play a role in future cardiovascular disease (CVD) risk in women with PE. Furthermore, a preclinical study by Pruthi et al demonstrated that exposure to experimental PE led to increased vascular damage after injury compared with normal pregnancy in mice.55 It is possible that the vascular dysfunction observed in preeclamptic women postpartum is due to the persistent inflammation after delivery. However, more studies are still needed to determine this definitively. Additionally, studies to determine if inflammatory CD4+ helper T-cell populations remain elevated postpartum in preeclamptic women should be performed.
A number of clinical studies demonstrate that in contrast to the increased population of inflammatory cells, such as TH17s, a decrease in the number of CD4+ TRegs occurs in preeclamptic women compared with their normal pregnant counterparts.83,96–99 CD4+ TRegs are classically identified by surface expression of CD4 and CD25, as well as intracellular expression of the transcription factor forkhead box P3 (FoxP3).100,101 TRegs are responsible for maintenance of maternal immune tolerance during pregnancy.102–105 Maternal immune tolerance is dependent on TRegs and uterine NK cells recognizing and accepting the fetal antigens and facilitating placental growth. Breakdown in maternal-fetal tolerance is thought to be a mediating factor in PE development and pathology. Previous studies have shown that depletion of TRegs prior to conception or in early pregnancy resulted in implantation failure and increased resorptions.102,105 Therefore, the importance of TRegs in the maintenance of early pregnancy has previously been established. However, TReg maintenance of proper immune function may be important in mid to late pregnancy, as well.
Because of the roles of the over-stimulated immune response in causing the pathophysiology of PE, proper immune regulation by TRegs may be important to blunt the inflammatory mediators leading to vasoactive pathways that can cause hypertension and PE symptoms. CD4+ TRegs may inhibit endogenous activation of the pro-inflammatory TH1 and TH17 cells leading to decreased inflammation and oxidative stress. Without T-cell activation, inflammatory cytokine production would be inhibited, resulting in fewer inflammatory cells and less production of reactive oxygen species.
To date, no studies have investigated the possibility of a full long-term autoimmune response after PE involving CD4+ T-cells or B-cells. Furthermore, very few studies have investigated the risk of PE leading to other autoimmune diseases later in life. To date, two such studies have determined a slightly increased risk for rheumatoid arthritis (RA) in women who were previously diagnosed with PE. A national cohort study in Denmark examined the possible impact of live births, pregnancy loss, and pregnancy complications on the risk for the development of RA later in life. These studies in Danish women found the rate ratio of RA to be 1.42 in women previously diagnosed with PE compared to women with normal pregnancies, while a recent update reported the hazard ratio of RA in women diagnosed with PE to be 1.96 compared to 1.18 in women diagnosed with gestational hypertension, without PE.106,107 Although these hazard ratios are relatively small, the dysregulation of the preeclamptic woman’s immune system during pregnancy that leads to PE may be related to the subsequent risk for RA. Similarly there remains a need for studies to investigate an association between PE and increased risk for other autoimmune disorders, especially those that disproportionally affect women and are associated with cardiovascular disease, such as systemic lupus erythematosus and psoriasis.
NK cells
NK cells make up 10% of lymphocytes in human peripheral blood and are identified by their expression of CD56 in the absence of CD3. NK cells can differentiate into two distinct subsets, type 1 (NK1) or type 2 (NK2). The NK1 subset is characterized by its release of IFN-γ and potent cytolytic activity upon activation.58,108 IL-2 and IL-12 signaling promote differentiation of NK cells to the NK1 subset and are secreted by CD4+ T helper cells. CD4+ TRegs can inhibit IL-2 mediated differentiation by decreasing availability of the cytokine.109,110 It has also been demonstrated that IL-17 may enhance cytolytic activity of NK cells, suggesting that TH17 cells may play a role in mediating differentiation into the NK1 population subset.111 PE is associated with a shift in the NK cell population from NK2 to NK1 (type 1 shift). A prominence of NK1 cells secreting tumor necrosis factor alpha (TNF-α) has previously been shown in preeclamptic women.112 NK-mediated killing of fetal trophoblasts can contribute to the shallow trophoblast invasion that results in insufficient spiral artery remodeling. Thereby promoting development of the ischemic placenta, leading to the release of vasoactive factors mediating maternal vascular dysfunction. Recent studies in mice suggest a role for cytotoxic NK cells in Angiotensin II (ANGII) mediated vascular dysfunction and artherosclerosis.113–115 Additionally, a study in humans demonstrated an association with NK cells and increased plaque rupture.113,116 The role of the cytolytic NK1 cell population in the development and pathophysiology of PE and other cardiovascular disease is poorly understood and warrants further investigation.
Neutrophils
Neutrophils are activated in response to the placental hypoxia and inflammation that occurs during PE. These cells can also be recruited and activated by increased IL-17.89 These cells may contribute to the increased vascular resistance and fetal morbidity of PE through production of oxidative stress and the release of neutrophil extracellular traps (NETs).89,117 NETs are extracellular structures released from neutrophils that kill extracellular bacteria and fungi, but are implicated in pathogenesis of inflammatory disorders and autoimmunity.89,117 Oxidative stress-induced inflammation and vascular damage and dysfunction may be attributable in part to the activation of neutrophils and subsequent release of NETs in response to hypoxia and IL-17 signaling during PE and requires further study.89,117
AT1-AA
AT1-AAs are elevated in women with PE.118,119 AT1-AAs bind with a high affinity to the 7 amino acid sequence on the second extracellular loop of the AT1R. Binding of the AT1-AA to the AT1R, increases AT1 receptor activity, intracellular calcium levels, and activation of intracellular mitogen activated protein kinase/ extracellular signal regulated kinases (MAPK/ERK) pathways.120–122
AT1-AAs are hypothesized to be generated from an immunological loss of self-tolerance toward the AT1R, which results in the accumulation of antibodies against the AT1R. AT1-AAs are also elevated in normotensive pregnancies with uterine growth-restricted fetuses, kidney transplant recipients, and patients with systemic sclerosis, vasculopathy, tissue fibrosis, hypertension, renovascular disease, and pregnant women with hemolysis, elevated liver enzymes and low platelets (HELLP syndrome).123–127
In 1999, Wallukat et al was the first to identify AT1-AAs in women with PE and not in healthy pregnant women or in women with pre-existing essential hypertension and pregnancy.118 In this study, the AT1-AAs were isolated by affinity column antibody purification techniques and used to stimulate cultured neonatal rat cardiomyocytes. However, in the presence of losartan, an AT1R blocker, purified AT1-AAs from preeclamptic patients were not able to stimulate cultured cardiomyocytes. Thus, this study was the first to identify that the actions of the AT1-AA on cardiomyocytes are facilitated through AT1-AA binding to the AT1R.
In vitro experiments with the AT1-AA on vascular smooth muscle cells and trophoblast cells increased reactive oxygen species production, nicotinamide adenine dinucleotide phosphate oxidase components, and activation of nuclear factor kappa-light-chain-enhancer of activated B cells.128 Human coronary artery vascular smooth muscle cells incubated with AT1-AAs increased tissue factor levels, which are elevated in the placenta of preeclamptic patients.122 AT1-AAs administered to human trophoblast cells, increased the secretion of plasminogen activator inhibitor-1 and decreased trophoblast invasiveness.129 Together these effects on the trophoblast cells suggest a mechanism of poor placental placentation, a possible cause of PE. Human mesangial cells incubated with the AT1-AA increased plasminogen activator inhibitor-1 and IL-6 secretions.130 AT1-AAs cultured with cardiomyocytes, increased the beating rate of cardiomyocytes and apoptosis of cardiomyocytes via a TNF-α pathway.131,132 Furthermore, we have shown that human umbilical vein endothelial cells (HUVECs) incubated 6 hours with ANGII (10−7 M) and AT1-AAs drastically increased ET-1 protein expression.133 This ~100 fold increase in ET-1 expression was absent in HUVECs incubated with ANGII or AT1-AAs alone, which suggest a possible enhancement of ANGII signaling when AT1-AAs are present.133 Therefore the increase in ET-1 expression from HUVECs may play a significant role in hypertension and enhanced ANGII sensitivity during PE. The results from the HUVEC study further emphasize the importance of ET-1 expression in PE, where there is increased AT1-AA production and hypertension. In an earlier study we showed that blockade of ETa receptor in rats administered the AT1-AA and ANGII displayed a decrease in blood pressure.63 Thus this in vitro HUVEC data taken along with the in vivo data strongly supports the notion that ET-1 is a major mediator of hypertension during pregnancy.
Animal models of PE, such as the RUPP model, the transgenic human angiotensinogen and renin gene pregnant rat model, the adoptive transfer of CD4+ T lymphocytes from placental ischemic rats to normal pregnant rats, and the cytokine (TNF-α, IL-6, and IL-17) administration during pregnancy model, display elevations in AT1-AAs during pregnancy.64,90,119,134–138 Isolated human and rodent AT1-AAs administered to rats during pregnancy increase several circulating factors associated with PE, such as sFlt-1, sEng, oxidative stress, ET-1, and endothelial microparticles (EMPs).63,134,136,139,140 EMPs are small endothelial cell debris fragments that are elevated in women with PE and serve as a marker of endothelial cell dysfunction. The mechanism by which AT1-AA increases EMPs is through the p38 MAP kinase signaling transduction pathway.120 Furthermore AT1-AAs administered to pregnant rats on day 12 or 13 of pregnancy exhibited a marked increase in blood pressure on day 19 of pregnancy.63,134,136,140 The rats also display and increase in placental oxidative stress, and ET-1 expression in the kidney cortex, aorta, and placenta.63,90,134,140
The mechanism of increased ANGII sensitivity in PE is unknown. However, we hypothesize that AT1-AAs play an important role in increasing ANGII sensitivity in PE. Studies from our lab have shown that acute ANGII along with AT1-AA infusion during a rat pregnancy increased the blood pressure ~40 mmHg above AT1-AA administered alone.133,134 In addition, chronic ANGII and AT-AAs together increase blood pressure, oxidative stress, and ET-1 secretion and increases the renal artery resistive index above ANGII or AT1-AA administered separately.133,134
One mechanism by which the AT1-AA can increase ANGII sensitivity may be by altering the binding affinity of ANGII to the AT1R. Preliminary studies from our lab have shown that AT1-AAs increase (~15-fold) the binding affinity of ANGII to the AT1Rs on HUVECs incubated with fluorescent labeled ANGII and human AT1-AAs isolated from preeclamptic patients for 1 hour. This increase in ANGII binding was also correlated with an increase in ET-1 secretion. Another proposed mechanism by which AT1-AAs increase ANGII affinity to AT1R is via increasing the dimerization of the AT1 receptors.141 Multiple studies have shown an increase in AT1R responsiveness to ANGII, when the AT1R heterodimerizes with the vasodepressor bradykinin receptor (B2).141–143 AbdAlla et al showed that women with PE have increased B2 protein amounts and AT1/B2 heterodimerization in platelets and the omental vessels of preeclamptic patients.141–143 Therefore it can be postulated, that the process by which the AT1-AAs increase ANGII sensitivity in PE is mediated by AT1R receptor dimerization, however, more studies are needed to verify this hypothesis.
Studies by Hubel et al show that ~18% of postpartum preeclamptic women have elevated circulating AT1-AAs 1 year after delivery.144 These women with elevated AT1-AAs have elevated sFlt-1 levels, decreased VEGF, and increased insulin sensitivity.144 The correlation between increased circulating AT1-AAs and these variables may suggest a mechanism by which women with PE have an increased risk of cardiovascular events later in life. Of caution, this study only consisted of 64 women (35 controls and 29 preeclamptic women), in which ~18% (five out of 29) of postpartum preeclamptic women had elevated circulating AT1-AAs thereby indicating the importance of studies determining the significance of AT1-AAs with larger populations of postpartum preeclamptic women.
Rats administered AT1-AA during pregnancy displayed no differences in cardiac function as compared with normal pregnant rats 16 weeks postpartum in a study conducted by Wang et al.145 However in this study, there were several morphological changes in cardiomyocytes, increased collagen content, and changes in the myocardium structure of rats administered the AT1-AA during pregnancy.145 These changes were prevented when losartan was administered along with AT1-AA during pregnancy.145 These data taken together suggest that the AT1-AA may play a pivotal role in the development or pathogenesis of cardiovascular dysfunction and CVDs in the mother.
PE has long-term effects on the offspring
Not only does PE have a long-term effect on the cardiovascular system of the mother later in life, but it also has an effect on the cardiovascular system of the offspring later in life. PE is one the major contributors to preterm and low birth weight babies. It has been known for several years, especially with the development of the Barker hypothesis, that fetal adaptions during pregnancy, such as malnutrition caused by hypertension and/or placental ischemia during pregnancy, increase the offspring’s risk of developing hypertension, stroke, diabetes, and CVDs later in life.146,147 Furthermore, several recent meta-analysis studies have shown that for each 1 kg increase in birthweight, there is a 9%–15% decrease in cardiovascular mortality and 2–4 mmHg decrease in systolic blood pressure in adulthood.147 The offspring of women with PE have elevated blood pressure, body mass index, and increased triglycerides and cholesterol content in both adolescence and adulthood.147,148 A meta-analysis study conducted by Davis et al in 2011, examined over 18 cohorts of ~45,000 women with normal and hypertensive pregnancies. In this study they discovered an increase of 2.39 mmHg in systolic pressure, 1.35 mmHg in diastolic pressure, and an 0.32 kg/m2 increase in body mass index in the offspring (4–30 years old) of preeclamptic mothers versus the offspring of normal pregnant women.148
Conclusion
The clinical symptoms of PE can be resolved after delivery of the placenta, however women and their offspring affected by PE have double the risk for subsequent cardiovascular complications such as heart disease, stroke, and venous thromboembolism over the 5–15 years after delivery and these women have greater risks of dying from cerebrovascular disease after pregnancy than women who had a healthy pregnancy.149–151 A more recent study examined the incidence of long-term atherosclerotic morbidity in preeclamptic women and the risk is greater for patients with severe and recurrent episodes of PE.152 Moreover, endothelial dysfunction is a common feature of pregnancies with PE, atherosclerosis, and cardiovascular disease and thus, endothelial dysfunction could serve as an underlying mechanism in the development of cardiovascular disease in preeclamptic women or their offspring. Furthermore, PE women exhibit chronic inflammation indicative of immunological memory with the activation of CD4+ T helper cells and the secretion of specialized IgG in the form of the AT1-AA. The prevalence of AT1-AA, immunological memory with NK or T-cells and the sequela of endothelial dysfunction could all mediate the development of CVD or cardiovascular events later in life among PE women. In addition, because of this prolonged chronic inflammation, PE women may be at risk for autoimmune diseases later in life. In recent years, a number of small studies or cases have been published that suggest use of the monoclonal antibody, rituximab, for the treatment of lymphoma or autoimmune disease is safe during pregnancy.153–159 However, these same groups performed follow-up studies that identified the drug within the nasal cavity of newborns born to pregnant macaques treated during pregnancy. Therefore, this avenue of treatment may prove to be deleterious for the immune development during the first few months of life of the newborn. With the development of immune suppression therapies proving successful for the treatment of cancer and lymphomas, if we can prove they are safe for both mother and infant ante and postpartum, should we consider the use of such therapies for PE women?
Acknowledgment
This research was supported by National Institutes of Health grants HL105324, HL124715, HL51971, HL78147, and HD067541.
Disclosure
The authors have no conflicts of interest to disclose.
References
Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension. 2005;46(6):1243–1249. | |
Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;294(2):H541–H550. | |
Granger JP, Alexander BT, Bennett WA, Khalil RA. Pathophysiology of pregnancy-induced hypertension. Am J Hypertens. 2001;14(6 Pt 2):178S–185S. | |
Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of preeclampsia: linking placental ischemia/hypoxia with microvascular dysfunction. Microcirculation. 2002;9(3):147–160. | |
Noris M, Perico N, Remuzzi G. Mechanisms of disease: Pre-eclampsia. Nat Clin Pract Nephrol. 2005;1(2):98–114. | |
Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–1594. | |
Sankaralingam S, Arenas IA, Lalu MM, Davidge ST. Preeclampsia: current understanding of the molecular basis of vascular dysfunction. Expert Rev Mol Med. 2006;8(3):1–20. | |
Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005; 365(9461):785–799. | |
Brennan LJ, Morton JS, Davidge ST. Vascular dysfunction in preeclampsia. Microcirculation. 2014;21(1):4–14. | |
Goulopoulou S, Davidge ST. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol Med. 2015;21(2):88–97. | |
Sandrim VC, Palei AC, Metzger IF, Gomes VA, Cavalli RC, Tanus-Santos JE. Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble fms-like tyrosine kinase-1 and soluble endogline in preeclampsia. Hypertension. 2008;52(2):402–407. | |
Murphy SR, LaMarca BB, Parrish M, Cockrell K, Granger JP. Control of soluble fms-like tyrosine-1 (sFlt-1) production response to placental ischemia/hypoxia: role of tumor necrosis factor-alpha. Am J Physiol Regul Integr Comp Physiol. 2013;304(2):R130–R135. | |
Wang A, Rana S, Karumanchi SA. Preeclampsia: the role of angiogenic factors in its pathogenesis. Physiology (Bethesda). 2009;24: 147–158. | |
Serrano NC, Casas JP, Diaz LA, et al. Endothelial NO synthase genotype and risk of preeclampsia: a multicenter case-control study. Hypertension. 2004;44(5):702–707. | |
Kinney MV, Lawn JE, Howson CP, Belizan J. 15 Million preterm births annually: what has changed this year? Reprod Health. 2012;9:28. | |
Chapman AB, Abraham WT, Zamudio S, et al. Temporal relationships between hormonal and hemodynamic changes in early human pregnancy. Kidney Int. 1998;54(6):2056–2063. | |
Garovic VD, Hayman SR. Hypertension in pregnancy: an emerging risk factor for cardiovascular disease. Nat Clin Pract Nephrol. 2007;3(11):613–622. | |
Ahmed R, Dunford J, Mehran R, Robson S, Kunadian V. Pre-eclampsia and future cardiovascular risk among women: a review. J Am Coll Cardiol. 2014;63(18):1815–1822. | |
Muller-Deile J, Schiffer M. Preeclampsia from a renal point of view: Insides into disease models, biomarkers and therapy. World J Nephrol. 2014;3(4):169–181. | |
Gibson HM. Plasma volume and glomerular filtration rate in pregnancy and their relation to differences in fetal growth. J Obstet Gynaecol Br Commonw. 1973;80(12):1067–1074. | |
Vikse BE, Irgens LM, Leivestad T, Skjaerven R, Iversen BM. Preeclampsia and the risk of end-stage renal disease. N Engl J Med. 2008;359(8):800–809. | |
Garovic VD, Wagner SJ, Turner ST, et al. Urinary podocyte excretion as a marker for preeclampsia. Am J Obstet Gynecol. 2007;196(4):320. e1–e7. | |
Craici IM, Wagner SJ, Bailey KR, et al. Podocyturia predates proteinuria and clinical features of preeclampsia: longitudinal prospective study. Hypertension. 2013;61(6):1289–1296. | |
Aita K, Etoh M, Hamada H, et al. Acute and transient podocyte loss and proteinuria in preeclampsia. Nephron Clin Pract. 2009; 112(2):c65–c70. | |
Matsubara K, Matsubara Y, Hyodo S, Katayama T, Ito M. Role of nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. J Obstet Gynaecol Res. 2010;36(2):239–247. | |
Roberts JM, Von Versen-Hoeynck F. Maternal fetal/placental interactions and abnormal pregnancy outcomes. Hypertension. 2007;49(1):15–16. | |
Shaamash AH, Elsnosy ED, Makhlouf AM, Zakhari MM, Ibrahim OA, HM EL-d. Maternal and fetal serum nitric oxide (NO) concentrations in normal pregnancy, pre-eclampsia and eclampsia. Int J Gynaecol Obstet. 2000;68(3):207–214. | |
Murphy SR, LaMarca B, Cockrell K, Arany M, Granger JP. L-arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2012;302(2):R259–R263. | |
Sandrim VC, Montenegro MF, Palei AC, et al. Increased circulating cell-free hemoglobin levels reduce nitric oxide bioavailability in preeclampsia. Free Radic Biol Med. 2010;49(3):493–500. | |
Pimentel AM, Pereira NR, Costa CA, et al. L-arginine-nitric oxide pathway and oxidative stress in plasma and platelets of patients with pre-eclampsia. Hypertens Res. 2013;36(9):783–788. | |
Molnar M, Suto T, Toth T, Hertelendy F. Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am J Obstet Gynecol. 1994;170(5 Pt 1):1458–1466. | |
Vosatka RJ, Hassoun PM, Harvey-Wilkes KB. Dietary L-arginine prevents fetal growth restriction in rats. Am J Obstet Gynecol. 1998;178(2):242–246. | |
Neri I, Mazza V, Galassi MC, Volpe A, Facchinetti F. Effects of L-arginine on utero-placental circulation in growth-retarded fetuses. Acta Obstet Gynecol Scand. 1996;75(3):208–212. | |
Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329(27):2002–2012. | |
Moncada S, Higgs EA. The discovery of nitric oxide and its role in vascular biology. Br J Pharmacol. 2006;147 Suppl 1: S193–S201. | |
Nelson SH, Steinsland OS, Wang Y, Yallampalli C, Dong YL, Sanchez JM. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ Res. 2000;87(5):406–411. | |
Sandrim VC, Palei AC, Cavalli RC, et al. eNOS haplotypes associated with gestational hypertension or preeclampsia. Pharmacogenomics J. 2008;9(10):1467–1473. | |
Sandrim VC, Palei AC, Luizon MR, Izidoro-Toledo TC, Cavalli RC, Tanus-Santos JE. eNOS haplotypes affect the responsiveness to antihypertensive therapy in preeclampsia but not in gestational hypertension. Pharmacogenomics J. 2010;10(1):40–45. | |
Noris M, Todeschini M, Cassis P, et al. L-arginine depletion in preeclampsia orients nitric oxide synthase toward oxidant species. Hypertension. 2004;43(3):614–622. | |
Eleuterio NM, Palei AC, Rangel Machado JS, Tanus-Santos JE, Cavalli RC, Sandrim VC. Relationship between adiponectin and nitrite in healthy and preeclampsia pregnancies. Clin Chim Acta. 2013;423: 112–115. | |
Sandrim VC, Palei AC, Metzger IF, Cavalli RC, Duarte G, Tanus- Santos JE. Interethnic differences in ADMA concentrations and negative association with nitric oxide formation in preeclampsia. Clin Chim Acta. 2010;411(19–20):1457–1460. | |
Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003; 111(5):649–658. | |
Sankaralingam S, Xu H, Davidge ST. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc Res. 2010;85(1):194–203. | |
Alpoim PN, Godoi LC, Freitas LG, Gomes KB, Dusse LM. Assessment of L-arginine asymmetric 1 dimethyl (ADMA) in early-onset and late-onset (severe) preeclampsia. Nitric Oxide. 2013;33:81–82. | |
Mutlu-Turkoglu U, Aykac-Toker G, Ibrahimoglu L, Ademoglu E, Uysal M. Plasma nitric oxide metabolites and lipid peroxide levels in preeclamptic pregnant women before and after delivery. Gynecol Obstet Invest. 1999;48(4):247–250. | |
Alexander BT, Kassab SE, Miller MT, et al. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension. 2001;37(4):1191–1195. | |
Crews JK, Herrington JN, Granger JP, Khalil RA. Decreased endothelium-dependent vascular relaxation during reduction of uterine perfusion pressure in pregnant rat. Hypertension. 2000; 35(1 Pt 2):367–372. | |
Walsh SK, English FA, Johns EJ, Kenny LC. Plasma-mediated vascular dysfunction in the reduced uterine perfusion pressure model of preeclampsia: a microvascular characterization. Hypertension. 2009;54(2):345–351. | |
Amaral LM, Cornelius DC, Harmon A, Moseley J, Martin JN Jr, LaMarca B. 17-hydroxyprogesterone caproate significantly improves clinical characteristics of preeclampsia in the reduced uterine perfusion pressure rat model. Hypertension. 2015;65(1):225–231. | |
Lamarca B. Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol. 2012;64(4):309–320. | |
Carty DM, Anderson LA, Duncan CN, et al. Peripheral arterial tone: assessment of microcirculatory function in pregnancy. J Hypertens. 2012;30(1):117–123. | |
Spasojevic M, Smith SA, Morris JM, Gallery ED. Peripheral arterial pulse wave analysis in women with pre-eclampsia and gestational hypertension. BJOG. 2005;112(11):1475–1478. | |
Clark DE, Smith SK, He Y, et al. A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol Reprod. 1998;59(6):1540–1548. | |
Murphy SR, LaMarca BB, Cockrell K, Granger JP. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension. 2010;55(2):394–398. | |
Pruthi D, Khankin EV, Blanton RM, et al. Exposure to experimental preeclampsia in mice enhances the vascular response to future injury. Hypertension. 2015;65(4):863–870. | |
Noori M, Donald AE, Angelakopoulou A, Hingorani AD, Williams DJ. Prospective study of placental angiogenic factors and maternal vascular function before and after preeclampsia and gestational hypertension. Circulation. 2010;122(5):478–487. | |
Wolf M, Hubel CA, Lam C, et al. Preeclampsia and future cardiovascular disease: potential role of altered angiogenesis and insulin resistance. J Clin Endocrinol Metab. 2004;89(12):6239–6243. | |
Vacca P, Moretta L, Moretta A, Mingari MC. Origin, phenotype and function of human natural killer cells in pregnancy. Trends Immunol. 2011;32(11):517–523. | |
Cid MC, Kleinman HK, Grant DS, Schnaper HW, Fauci AS, Hoffman GS. Estradiol enhances leukocyte binding to tumor necrosis factor (TNF)-stimulated endothelial cells via an increase in TNF-induced adhesion molecules E-selectin, intercellular adhesion molecule type 1, and vascular cell adhesion molecule type 1. J Clin Invest. 1994;93(1):17–25. | |
Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of hypertension during preeclampsia linking placental ischemia with endothelial dysfunction. Hypertension. 2001;38(3 Pt 2):718–722. | |
Keiser SD, Veillon EW, Parrish MR, et al. Effects of 17-hydroxyprogesterone on tumor necrosis factor-alpha-induced hypertension during pregnancy. Am J Hypertens. 2009;22(10):1120–1125. | |
LaMarca BB, Cockrell K, Sullivan E, Bennett W, Granger JP. Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats. Hypertension. 2005;46(1):82–86. | |
LaMarca B, Parrish M, Ray LF, et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension. 2009;54(4):905–909. | |
Novotny SR, Wallace K, Heath J, et al. Activating autoantibodies to the angiotensin II type I receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. Am J Physiol Regul Integr Comp Physiol. 2012;302(10):R1197–R1201. | |
Taylor RN, de Groot CJ, Cho YK, Lim KH. Circulating factors as markers and mediators of endothelial cell dysfunction in preeclampsia. Semin Reprod Endocrinol. 1998;16(1):17–31. | |
Wallace K, Richards S, Dhillon P, et al. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension. 2011;57(5):949–955. | |
Wallace K, Novotny S, Heath J, et al. Hypertension in response to CD4(+) T cells from reduced uterine perfusion pregnant rats is associated with activation of the endothelin-1 system. Am J Physiol Regul Integr Comp Physiol. 2012;303(2):R144–R149. | |
Wu CF, Huang FD, Sui RF, Sun JX. Preeclampsia serum upregulates CD40/CD40L expression and induces apoptosis in human umbilical cord endothelial cells. Reprod Biol Endocrinol. 2012;10:28. | |
Zhou CC, Irani RA, Dai Y, et al. Autoantibody-mediated IL-6-dependent endothelin-1 elevation underlies pathogenesis in a mouse model of preeclampsia. J Immunol. 2011;186(10):6024–6034. | |
Benyo DF, Miles TM, Conrad KP. Hypoxia stimulates cytokine production by villous explants from the human placenta. J Clin Endocrinol Metab. 1997;82(5):1582–1588. | |
Conrad KP, Benyo DF. Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol. 1997;37(3):240–249. | |
Roberts JM, Pearson G, Cutler J, Lindheimer M; Pregnancy NWGoRoHD. Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertension. 2003;41(3):437–445. | |
Roberts JM, Lain KY. Recent Insights into the pathogenesis of pre-eclampsia. Placenta. 2002;23(5):359–372. | |
Lamarca B. The role of immune activation in contributing to vascular dysfunction and the pathophysiology of hypertension during preeclampsia. Minerva Ginecol. 2010;62(2):105–120. | |
LaMarca BB, Bennett WA, Alexander BT, Cockrell K, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of tumor necrosis factor-alpha. Hypertension. 2005; 46(4):1022–1025. | |
Zenclussen AC, Fest S, Joachim R, Klapp BF, Arck PC. Introducing a mouse model for pre-eclampsia: adoptive transfer of activated Th1 cells leads to pre-eclampsia-like symptoms exclusively in pregnant mice. Eur J Immunol. 2004;34(2):377–387. | |
Saito S, Umekage H, Sakamoto Y, et al. Increased T-helper-1-type immunity and decreased T-helper-2-type immunity in patients with preeclampsia. Am J Reprod Immunol. 1999;41(5):297–306. | |
Saito S, Sakai M, Sasaki Y, Tanebe K, Tsuda H, Michimata T. Quantitative analysis of peripheral blood Th0, Th1, Th2 and the Th1:Th2 cell ratio during normal human pregnancy and preeclampsia. Clin Exp Immunol. 1999;117(3):550–555. | |
Toldi G, Rigo J Jr, Stenczer B, Vasarhelyi B, Molvarec A. Increased prevalence of IL-17-producing peripheral blood lymphocytes in pre-eclampsia. Am J Reprod Immunol. 2011;66(3):223–229. | |
Saito S, Nakashima A, Shima T, Ito M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am J Reprod Immunol. 2010;63(6):601–610. | |
Saito S. Th17 cells and regulatory T cells: new light on pathophysiology of preeclampsia. Immunol Cell Biol. 2010;88(6):615–617. | |
Jianjun Z, Yali H, Zhiqun W, Mingming Z, Xia Z. Imbalance of T-cell transcription factors contributes to the Th1 type immunity predominant in pre-eclampsia. Am J Reprod Immunol. 2010;63(1):38–45. | |
Darmochwal-Kolarz D, Kludka-Sternik M, Tabarkiewicz J, et al. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J Reprod Immunol. 2012;93(2):75–81. | |
El-Kabarity RH, Naguib AH. Serum levels of IL-18, IL-12 and TH-1/TH-2 ratio in patients with pre-eclampsia. Egypt J Immunol. 2011;18(1):1–8. | |
Dong M, He J, Wang Z, Xie X, Wang H. Placental imbalance of Th1- and Th2-type cytokines in preeclampsia. Acta Obstet Gynecol Scand. 2005;84(8):788–793. | |
Saito S, Sakai M. Th1/Th2 balance in preeclampsia. J Reprod Immunol. 2003;59(2):161–173. | |
Borzychowski AM, Croy BA, Chan WL, Redman CW, Sargent IL. Changes in systemic type 1 and type 2 immunity in normal pregnancy and pre-eclampsia may be mediated by natural killer cells. Eur J Immunol. 2005;35(10):3054–3063. | |
Wallace K, Cornelius DC, Scott J, et al. CD4+ T cells are important mediators of oxidative stress that cause hypertension in response to placental ischemia. Hypertension. 2014;64(5):1151–1158. | |
Laresgoiti-Servitje E. A leading role for the immune system in the pathophysiology of preeclampsia. J Leukoc Biol. 2013;94(2):247–257. | |
Dhillion P, Wallace K, Herse F, et al. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2012;303(4):R353–R358. | |
Cornelius DC, Wallace K, Kipprono L, Dhillion P, Moseley J, Lamarca B. Endothelin-1 is not a Mechanism of IL-17 Induced Hypertension during Pregnancy. Med J Obstet Gynecol. 2013;1(1).pii:1006. | |
Cornelius DC, Hogg JP, Scott J, et al. Administration of interleukin-17 soluble receptor C suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension. 2013;62(6):1068–1073. | |
Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181(1):8–18. | |
Vitoratos N, Economou E, Iavazzo C, Panoulis K, Creatsas G. Maternal serum levels of TNF-alpha and IL-6 long after delivery in preeclamptic and normotensive pregnant women. Mediators Inflamm. 2010;2010:908649. | |
Kvehaugen AS, Dechend R, Ramstad HB, Troisi R, Fugelseth D, Staff AC. Endothelial function and circulating biomarkers are disturbed in women and children after preeclampsia. Hypertension. 2011;58(1):63–69. | |
Toldi G, Saito S, Shima T, et al. The frequency of peripheral blood CD4+ CD25high FoxP3+ and CD4+ CD25− FoxP3+ regulatory T cells in normal pregnancy and pre-eclampsia. Am J Reprod Immunol. 2012;68(2):175–180. | |
Tilburgs T, Roelen DL, van der Mast BJ, et al. Differential distribution of CD4(+)CD25(bright) and CD8(+)CD28(−) T-cells in decidua and maternal blood during human pregnancy. Placenta. 2006;27 Suppl A: S47–S53. | |
Santner-Nanan B, Peek MJ, Khanam R, et al. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J Immunol. 2009;183(11):7023–7030. | |
Prins JR, Boelens HM, Heimweg J, et al. Preeclampsia is associated with lower percentages of regulatory T cells in maternal blood. Hypertens Pregnancy. 2009;28(3):300–311. | |
Geiger TL, Tauro S. Nature and nurture in Foxp3(+) regulatory T cell development, stability, and function. Hum Iimmunol. 2012;73(3):232–239. | |
Peterson RA. Regulatory T-cells: diverse phenotypes integral to immune homeostasis and suppression. Toxicol Pathol. 2012;40(2):186–204. | |
Shima T, Sasaki Y, Itoh M, et al. Regulatory T cells are necessary for implantation and maintenance of early pregnancy but not late pregnancy in allogeneic mice. J Reprod Immunol. 2010;85(2):121–129. | |
Bettini M, Vignali DA. Regulatory T cells and inhibitory cytokines in autoimmunity. Curr Opin Immunol. 2009;21(6):612–618. | |
Arck PC, Hecher K. Fetomaternal immune cross-talk and its consequences for maternal and offspring’s health. Nat Med. 2013;19(5):548–556. | |
Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. 2004;5(3):266–271. | |
Jorgensen KT, Pedersen BV, Jacobsen S, Biggar RJ, Frisch M. National cohort study of reproductive risk factors for rheumatoid arthritis in Denmark: a role for hyperemesis, gestational hypertension and pre-eclampsia? Ann Rheum Dis. 2010;69(2):358–363. | |
Jorgensen KT, Harpsoe MC, Jacobsen S, Jess T, Frisch M. Increased risk of rheumatoid arthritis in women with pregnancy complications and poor self-rated health: a study within the Danish National Birth Cohort. Rheumatology (Oxford). 2014;53(8):1513–1519. | |
Peritt D, Robertson S, Gri G, Showe L, Aste-Amezaga M, Trinchieri G. Cutting Edge: Differentiation of Human NK Cells into NK1 and NK2 Subsets. The Journal of Immunology. 1998;161(11):5821–5824. | |
Kerdiles Y, Ugolini S, Vivier E. T cell regulation of natural killer cells. J Exp Med. 2013;210(6):1065–1068. | |
Katsumoto T, Kimura M, Yamashita M, et al. STAT6-dependent differentiation and production of IL-5 and IL-13 in murine NK2 cells. J Immunol. 2004;173(8):4967–4975. | |
Al Omar S, Flanagan BF, Almehmadi M, Christmas SE. The effects of IL-17 upon human natural killer cells. Cytokine. 2013;62(1):123–130. | |
Fukui A, Yokota M, Funamizu A, et al. Changes of NK cells in preeclampsia. Am J Reprod Immunol. 2012;67(4):278–286. | |
Hedrick CC. Lymphocytes in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(2):253–257. | |
Selathurai A, Deswaerte V, Kanellakis P, et al. Natural killer (NK) cells augment atherosclerosis by cytotoxic-dependent mechanisms. Cardiovasc Res. 2014;102(1):128–137. | |
Kossmann S, Schwenk M, Hausding M, et al. Angiotensin II-induced vascular dysfunction depends on interferon-gamma-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol. 2013;33(6):1313–1319. | |
Martinez-Rodriguez JE, Munne-Collado J, Rasal R, et al. Expansion of the NKG2C+ natural killer-cell subset is associated with high-risk carotid atherosclerotic plaques in seropositive patients for human cytomegalovirus. Arterioscler Thromb Vasc Biol. 2013;33(11):2653–2659. | |
Gupta AK, Hasler P, Holzgreve W, Hahn S. Neutrophil NETs: a novel contributor to preeclampsia-associated placental hypoxia? Semin Immunopathol. 2007;29(2):163–167. | |
Wallukat G, Homuth V, Fischer T, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103(7):945–952. | |
Herse F, LaMarca B. Angiotensin II type 1 receptor autoantibody (AT1-AA)-mediated pregnancy hypertension. Am J Reprod Immunol. 2013;69(4):413–418. | |
Yang S, Zhong Q, Qiu Z, et al. Angiotensin II receptor type 1 autoantibodies promote endothelial microparticles formation through activating p38 MAPK pathway. J Hypertens. 2014;32(4):762–770. | |
Thway TM, Shlykov SG, Day MC, et al. Antibodies from preeclamptic patients stimulate increased intracellular Ca2+ mobilization through angiotensin receptor activation. Circulation. 2004;110(12):1612–1619. | |
Dechend R, Homuth V, Wallukat G, et al. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101(20):2382–2387. | |
Dragun D, Muller DN, Brasen JH, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med. 2005;352(6):558–569. | |
Fu ML, Herlitz H, Schulze W, et al. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens. 2000;18(7):945–953. | |
Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against AT1-receptor and alpha1-adrenergic receptor in patients with hypertension. Hypertens Res. 2002;25(4):641–646. | |
Fischer T, Wallukat G, Schneider MP, Schlembach D, Munz W, Homuth V. HELLP syndrome in the 18th week of gestation in association with elevated angiotensin AT(1)-receptor autoantibodies. Eur J Obstet Gynecol Reprod Biol. 2001;97(2):255–257. | |
Zhang L, Cui L, Miao GB, Zhao WS, Wang SY, Liu XL. [Study of autoantibodies against the G-protein-coupled beta 2- and alpha 1-adrenergic and AT1 receptors in patients with primary hypertension]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2002;24(4):367–369. Chinese. | |
Dechend R, Viedt C, Muller DN, et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation. 2003;107(12):1632–1639. | |
Xia Y, Wen H, Bobst S, Day MC, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human trophoblast cells. J Soc Gynecol Investig. 2003;10(2):82–93. | |
Bobst SM, Day MC, Gilstrap LC 3rd, Xia Y, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human mesangial cells and induce interleukin-6 and plasminogen activator inhibitor-1 secretion. Am J Hypertens. 2005;18(3):330–336. | |
Chai W, Zhang W, Jin Z, Feng Y, Kuang Y, Zhi J. Angiotensin II type I receptor agonistic autoantibody-induced apoptosis in neonatal rat cardiomyocytes is dependent on the generation of tumor necrosis factor-alpha. Acta Biochim Biophys Sin (Shanghai). 2012;44(12):984–990. | |
Jin Z, Wang J, Zhang W, Zhang G, Jiao X, Zhi J. Changes in cardiac structure and function in rats immunized by angiotensin type 1 receptor peptides. Acta Biochim Biophys Sin (Shanghai). 2011; 43(12):970–976. | |
Wenzel K, Rajakumar A, Haase H, et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension. 2011;58(1):77–84. | |
Brewer J, Liu R, Lu Y, et al. Endothelin-1, oxidative stress, and endogenous angiotensin II: mechanisms of angiotensin II type I receptor autoantibody-enhanced renal and blood pressure response during pregnancy. Hypertension. 2013;62(5):886–892. | |
LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension. 2008;52(6):1168–1172. | |
Li J, LaMarca B, Reckelhoff JF. A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. Am J Physiol Heart Circ Physiol. 2012;303(1):H1–H8. | |
Dechend R, Gratze P, Wallukat G, et al. Agonistic autoantibodies to the AT1 receptor in a transgenic rat model of preeclampsia. Hypertension. 2005;45(4):742–746. | |
Lamarca B, Brewer J, Wallace K. IL-6-induced pathophysiology during pre-eclampsia: potential therapeutic role for magnesium sulfate? Int J Interferon Cytokine Mediator Res. 2011;2011(3):59–64. | |
Parrish MR, Murphy SR, Rutland S, et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am J Hypertens. 2010;23(8):911–916. | |
Parrish MR, Wallace K, Tam Tam KB, et al. Hypertension in response to AT1-AA: role of reactive oxygen species in pregnancy-induced hypertension. Am J Hypertens. 2011;24(7):835–840. | |
Xia Y, Kellems RE. Angiotensin receptor agonistic autoantibodies and hypertension: preeclampsia and beyond. Circ Res. 2013;113(1):78–87. | |
Quitterer U, Lother H, Abdalla S. AT1 receptor heterodimers and angiotensin II responsiveness in preeclampsia. Semin Nephrol. 2004;24(2):115–119. | |
AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med. 2001;7(9):1003–1009. | |
Hubel CA, Wallukat G, Wolf M, et al. Agonistic angiotensin II type 1 receptor autoantibodies in postpartum women with a history of preeclampsia. Hypertension. 2007;49(3):612–617. | |
Wang HP, Zhang WH, Wang XF, et al. Exposure to AT1 receptor autoantibodies during pregnancy increases susceptibility of the maternal heart to postpartum ischemia-reperfusion injury in rats. Int J Mol Sci. 2014;15(7):11495–11509. | |
Barker DJ. Fetal origins of coronary heart disease. Br Heart J. 1993; 69(3):195–196. | |
Yeung EH, Robledo C, Boghossian N, Zhang C, Mendola P. Developmental Origins of Cardiovascular Disease. Curr Epidemiol Rep.2014;1(1):9–16. | |
Davis EF, Lazdam M, Lewandowski AJ, et al. Cardiovascular risk factors in children and young adults born to preeclamptic pregnancies: a systematic review. Pediatrics. 2012;129(6):e1552–e1561. | |
Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation. 2011;123(24):2856–2869. | |
Fraser A, Nelson SM, Macdonald-Wallis C, et al. Associations of pregnancy complications with calculated cardiovascular disease risk and cardiovascular risk factors in middle age: the Avon Longitudinal Study of Parents and Children. Circulation. 2012;125(11):1367–1380. | |
Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women – 2011 update: a guideline from the American Heart Association. J Am Coll Cardiol. 2011;57(12):1404–1423. | |
Kessous R, Shoham-Vardi I, Pariente G, Sergienko R, Sheiner E. Long-term maternal atherosclerotic morbidity in women with pre-eclampsia. Heart. 2015;101(6):442–446. | |
Lee EJ, Ahn KH, Hong SC, Lee EH, Park Y, Kim BS. Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) chemotherapy for diffuse large B-cell lymphoma in pregnancy may be associated with preterm birth. Obstet Gynecol Sci. 2014;57(6):526–529. | |
Mandal PK, Dolai TK, Bagchi B, Ghosh MK, Bose S, Bhattacharyya M. B cell suppression in newborn following treatment of pregnant diffuse large B-cell lymphoma patient with rituximab containing regimen. Indian J Pediatr. 2014;81(10):1092–1094. | |
Sangle SR, Lutalo PM, Davies RJ, Khamashta MA, D’Cruz DP. B-cell depletion therapy and pregnancy outcome in severe, refractory systemic autoimmune diseases. J Autoimmun. Jun 2013;43:55–59. | |
Ojeda-Uribe M, Afif N, Dahan E, et al. Exposure to abatacept or rituximab in the first trimester of pregnancy in three women with autoimmune diseases. Clin Rheumatol. 2013;32(5):695–700. | |
Ringelstein M, Harmel J, Distelmaier F, et al. Neuromyelitis optica and pregnancy during therapeutic B cell depletion: infant exposure to anti-AQP4 antibody and prevention of rebound relapses with low-dose rituximab postpartum. Mult Scler. 2013;19(11):1544–1547. | |
Sarno MA, Mancari R, Azim HA Jr, Colombo N, Peccatori FA. Are monoclonal antibodies a safe treatment for cancer during pregnancy? Immunotherapy. 2013;5(7):733–741. | |
Burnette BL, Jentoft MA, Porrata LF, Boyce TG, Witzig TE. Single-agent rituximab for primary CNS lymphoma during pregnancy as a bridge to definitive management. J Clin Oncol. 2014;32(7):e14–e17. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.