")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Predictive value of genomics in the screening of type 2 diabetes: limitations and current status
Received 27 February 2013
Accepted for publication 14 March 2014
Published 15 May 2014 Volume 2014:4 Pages 45—57
DOI https://doi.org/10.2147/AGG.S18705
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
V Radha, V Mohan
Madras Diabetes Research Foundation, Dr Mohan’s Diabetes Specialities Centre, World Health Organization Collaborating Centre for Non-Communicable Diseases Prevention and Control, International Diabetes Federation Centre of Education, Chennai, India
Abstract: Multiple genetic variants and environmental factors interact resulting in the causation of type 2 diabetes. The advent of genome-wide association studies has accelerated the pace of discovery of genetic variants associated with type 2 diabetes. These variants could potentially be useful for the prediction, prevention, and early treatment of diabetes. Although a number of studies have been conducted on the predictive value of genetic polymorphisms, its value in the general population is unclear. Although in monogenic forms of diabetes genetic screening yields excellent predictive value, genetic profiling for polygenic type 2 diabetes currently appears to be limited in its predictive ability compared with conventional clinical risk scores. Performing a genetic profiling of strongly associated and replicated genetic variants seem to be the way forward, although such analysis is not yet successful. It is hoped that combined analyses of these genetic factors or hitherto unidentified genes would help in better genetic prediction of type 2 diabetes in the future.
Keywords: predictive value, genomics, monogenic diabetes, polygenic type 2 diabetes, genes, genetic risk variants, clinical risk factors
Introduction
Type 2 diabetes (T2D) is a metabolic disorder characterized by hyperglycemia, insulin resistance, and relative insulin deficiency. Diabetes is a leading cause of blindness, renal failure, and limb amputation, and a major risk factor for cardiovascular morbidity and mortality.1 It is possible to slow, or sometimes even reverse the disease process by early intervention, weight loss and physical activity, and the judicious use of medications.
Identification of population subgroups at particularly high risk for T2D might facilitate the targeting of prevention efforts to those who might benefit from them. This is the goal of risk prediction of a disease.
Prediction of diabetes risk for healthy individuals is commonly attempted using multivariate diabetes risk scores, and some of them are recommended in current practice guidelines for diabetes prevention2 and are also implemented in prevention programs in some Western countries.3–6 However, the predictive ability of diabetes risk scores, which have been developed in populations of varying ethnic backgrounds, differs considerably between populations.
Clinical T2D risk-prediction model
Based on the information available in routine clinical practice and gathered through questionnaires, a number of risk scores exist that help in prediction of T2D. Large studies such as the Framingham Offspring Study and the Atherosclerosis Risk in Communities Study have developed prediction models for T2D in middle-aged adults using common clinical measurements (Table 1).7–9 These models usually include measures of glycemia, adiposity, dyslipidemia, and blood pressure, in addition to age, family history of diabetes, physical activity, and, in some cases, race.10 They discriminate future cases from noncases, with a C-statistic (also called the area under the receiver operating characteristic [ROC] curve) ranging from 0.7 to 0.9. It has been possible to derive risk scores that predict T2D using basic clinical information, examples being the Finnish Diabetes Risk Score and diabetes risk calculator, and the Indian Diabetes Risk Score (IDRS; Table 1).9,11,12 Using these scores, prediction has been considered to be modest, leading to the question whether genetic risk prediction might prove to be superior. This article will focus on the genetic risk prediction for diabetes.
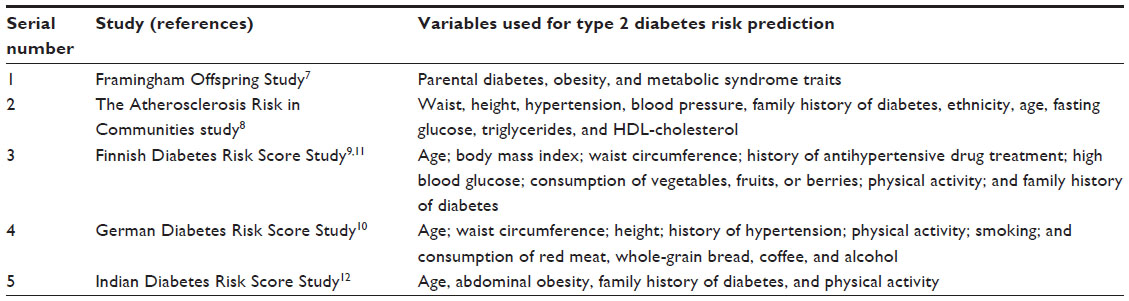 | Table 1 Type 2 diabetes risk prediction using clinical variables |
Genetic risk prediction model for T2D
Risk models that include genetic variants exclusively or genetic variants added to clinical risk factors have been assessed for their clinical predictive ability. For a genetic marker, its predictive value is its ability to predict disease. Predictive genetic tests can be used to identify persons who have a disease at the time of testing (diagnosis) or who will develop the disease in the future (prediction). Before ordering genetic tests as routine investigations, it is very important to see if they have both clinical validity and utility. For clinical validity to be proved, the discriminative accuracy of the test is a very important factor. As the name indicates, it is the extent to which a marker can discriminate between individuals who will develop the disease and those who will not. The important indicators are the sensitivity and specificity. The proportion of carriers among persons who will develop the disease is sensitivity and the proportion of noncarriers who will not develop the disease is specificity. Sensitivity is known as “true positive rate” and specificity is known as “true negative rate”. Conversely, the false positive rate is equal to one minus the specificity, and the false negative rate is equal to one minus the sensitivity.
The ability to predict disease risk by a genetic marker is its predictive value. If genetic testing improves disease prediction beyond conventional risk factors, then it is said to have good clinical utility. When a genetic marker is associated with risk of disease, carriers of a risk genotype have a higher risk of disease and noncarriers have a lower risk of disease compared with the average disease risk. A test that is useful for predicting disease in one population may not be useful in another population, since among populations disease risks and genotype frequencies are likely to vary.13 Common diseases such as T2D are caused by an interaction of several genetic and nongenetic factors, each of which conveys a minor increase in the risk of disease.14 For this reason, the genetic prediction of common diseases has proved to be more challenging.
Genetic prediction of monogenic diabetes
Monogenic forms of diabetes mellitus constitute a heterogeneous group of single-gene disorders that are characterized by impaired insulin secretion of the pancreatic β-cells. They account for up to 2%–5% of all cases of diabetes mellitus. Monogenic disorders are characterized by different modes of inheritance and different ages of disease onset.15,16 Maturity-onset diabetes of the young (MODY) and neonatal diabetes mellitus (NDM) are two main types of monogenic diabetes. In addition, there are syndromic forms of monogenic diabetes (Table 2).
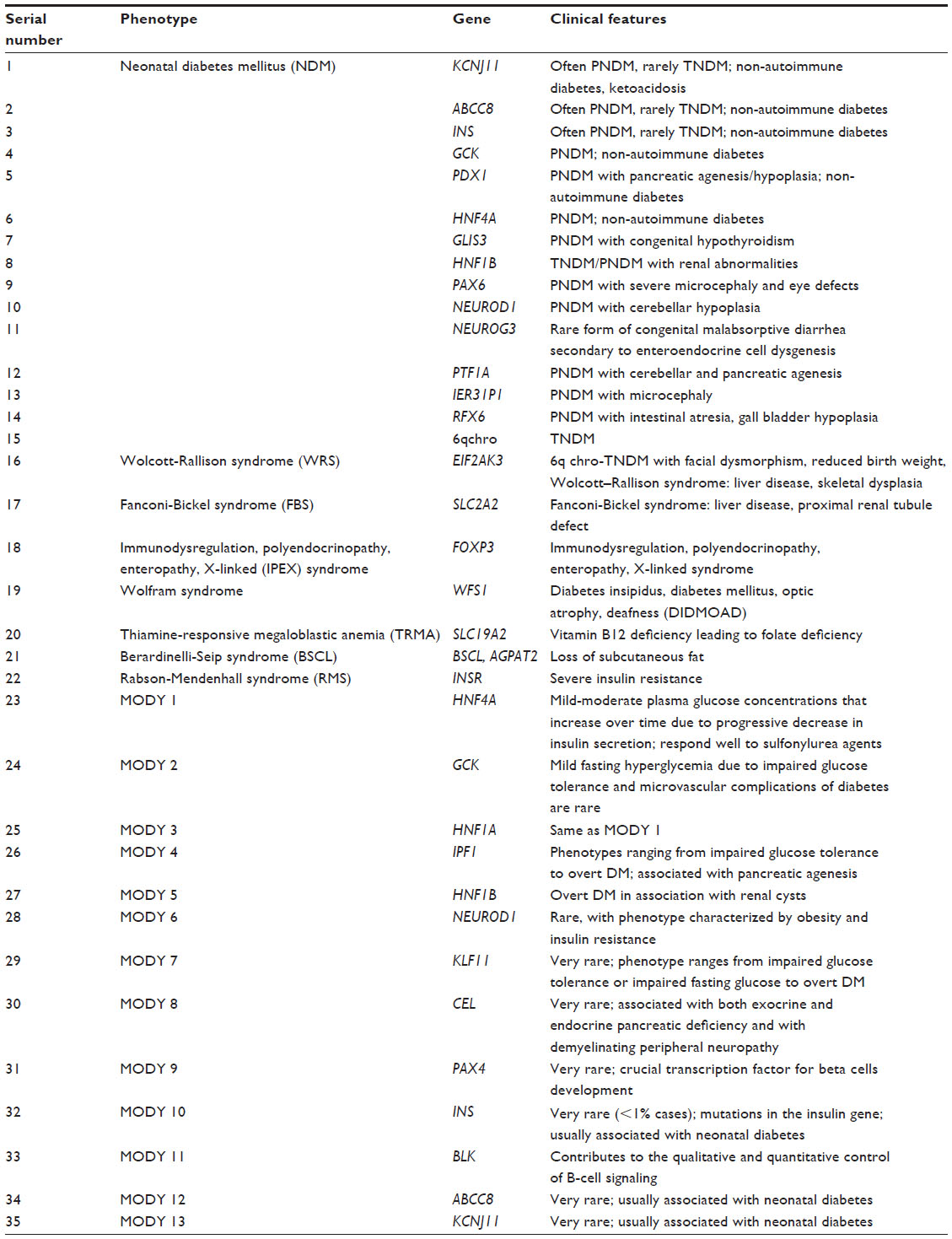 | Table 2 Genetics of monogenic diabetes |
In MODY, the clinical pattern is characterized by young age at diagnosis – usually below 25 years – with a marked family history of diabetes in multi-generations due to autosomal dominant inheritance, and negativity for pancreatic autoantibodies such as glutamic acid decarboxylase 65 and islet antigen 2.17 NDM is a rare form of monogenic diabetes with onset below 6 months of age,18 and glutamic acid decarboxylase antibody-negative NDM is of two types: permanent NDM,19 requiring lifelong treatment; and transient NDM, where the diabetes may spontaneously remit (before 1 year of age) but will often relapse, usually during adolescence or early adulthood.20
MODY and NDM are caused by mutations in a single gene. Predictive testing for these mutations is very informative since the risk of disease between carriers and noncarriers of mutations is substantially different, with the former being high and the latter approximating the population average. With a large difference in disease risk between carriers and noncarriers, genetic testing can be useful for predicting disease, and targeting preventive or therapeutic interventions to the relatively small group of individuals at increased risk.
A number of subtypes of MODY exist based on the gene involved in the disease subtype. A more prevalent subtype is the HNF1A-MODY. Based on genetic characterization of the HNF1A-MODY patient, the antidiabetic treatment can be tailored. These patients show much better response to sulfonylureas than to metformin, as sulfonylureas act on the beta cells and increase the insulin secretion.21 In the case of GCK-MODY, diet control is mostly sufficient to manage the affected subjects.
GCK-MODY is frequently misdiagnosed and clinical diagnosis depends on the age of the patient: slim children with GCK mutations are often diagnosed to be in the initial stages of type 1 diabetes; pregnant women with GCK mutations are diagnosed as gestational diabetics; and older patients with GCK mutations are diagnosed as having T2D. Therefore, identification of GCK mutations is imperative in the diagnosis and treatment of the condition. The involvement of glucokinase gene mutations in the evolution of gestational diabetes and subsequent development of T2D in adulthood has been shown in various studies.22 Women with glucokinase mutations often present with gestational diabetes, as their asymptomatic hyperglycemia is detected by routine testing in pregnancy. The diagnosis of a glucokinase gene mutation is very important for both mother and child. In the absence of this knowledge, treatment with insulin could lead to macrosomia in the fetus. These subjects need to be treated by diet alone. It is thus possible to tailor the treatment strategy based on genetic screening of GCK mutations.
Yet another very important example of tailoring treatment based on genetic prediction is permanent NDM. Conventional treatment for neonatal diabetes has been to give insulin injections, however this is not warranted in patients with KCNJ11 mutations. Patients with KCNJ11 mutations are characterized by a very good response to sulfonylurea treatment – indeed, even better than with insulin injections. Here the action of sulfonylurea corrects the mechanism underlying this type of diabetes by closing the activated potassium channel of the beta cells; hence exogenous insulin is not necessary for these patients.23,24 In a recent study we identified KCNJ11 and ABCC8 mutations in Indian NDM children with onset of diabetes below 6 months of age. As the KATP mutations are sulfonylurea responsive, children with KCNJ11 (Cys42Arg and Arg201Cys) and ABCC8 (Val86Ala, Asp212Tyr, and Pro254Ser) gene mutations were also successfully shifted from insulin injections to oral sulfonylurea drugs.25
Genetic prediction of polygenic or multifactorial T2D
T2D is a classic example of a common multifactorial disease in which both genetic and nongenetic factors play an important role.26 In Mendelian disorders, rare genetic variants usually referred to as mutations, confer a major portion of disease risk. A precise genotype–phenotype correlation is possible in monogenic disorders and hence genetic testing to assess the probability of disease occurring in individuals and the first-degree relatives of an affected proband is feasible. It has to be noted that for predicting risk for disease in general populations, monogenic mutations causing Mendelian disorders have very limited value because of their infrequency. For analysis of risk in populations, genome-wide associations are used as a tool based on the “common disease – common variant” hypothesis. In polygenic diabetes, one looks for the associated susceptibility alleles with modest effect, rather than for sequence differences with strong causal effects.
The predictive value of testing for a single genetic variant is limited for a multifactorial disease such as T2D. This is because T2D results from an interplay of a number of genetic and nongenetic factors. These risk variants are generally common (>1%), and hence carriers and noncarriers have disease risks that are only slightly higher or lower, respectively, than the population average, and the differences in disease risk are also small. Since multiple genetic variants are involved in T2D, simultaneous testing for these variants can be performed, resulting in genetic profiling. Genetic profiling has the ability to predict disease risk as a function of the combined effects of genetic variants. Not all genetic factors predict disease in the same manner. Therefore, an individual’s disease risk is dependent on both the number of risk genotypes carried and the specific risk carried by each genotype. As can be expected, genotypes more strongly associated with disease contribute more to a person’s disease risk.
Three attributes compare the risk or odds of disease in carriers versus noncarriers of risk genotypes. One is the relative risk, which by definition is the ratio of the disease risk in carriers divided by the disease risk in noncarriers. Risk difference is defined as the absolute difference between the disease risk of carriers and noncarriers. The odds ratio by definition is the ratio of the odds of disease in carriers divided by the odds of disease in noncarriers; it also explains the ratio of odds of the risk genotype in individuals who will develop the disease from those who will not. Risk models have been developed including exclusively the genetic variants, or genetic variants added to clinical risk factors.
More than 1,000 genome-wide association studies (GWAS) have been carried out by analyzing hundreds of thousands of single-nucleotide polymorphisms (SNPs) in very large samples,27,28 which have identified several loci associated with many common diseases (http://www.genome.gov/gwastudies).29 Common variants with minor allele frequency >5% have been looked into. The results of these studies have shown the best associated genes to have an odds ratio of 1.1–1.5.30 Moreover, a large number of loci are needed to significantly influence any single disease. Using these data for prediction of development of polygenic T2D is very tricky. Although unlike clinical markers genetic markers do not change with time, and hence possess the advantage of identifying high-risk individuals several decades before the disease onset, enabling early prevention, their predictive power has so far been rather limited. To date, 75 susceptibility loci associated with T2D and metabolic traits have been identified,31 mostly in European, African, and South Asian populations. In various ethnic populations at least about 20 SNPs have been firmly replicated.32–37 However, data from these studies explain only a small proportion of T2D susceptibility. A high proportion of missing heritability is yet to be unravelled.38 Table 3 summarizes the recent GWAS in T2D published from 2011 to date. Previous studies have investigated the predictive value of the genomic results, either based on GWAS or replication studies. A few have been reviewed in the following paragraphs. A comparison of genetic prediction and traditional clinical markers from the most positively associated TCF7L2 gene and yet another important gene, namely the CDKAL1 gene, in our own population has shown that genetic markers do not add any predictive advantage over conventional clinical factors, as shown in Table 4.
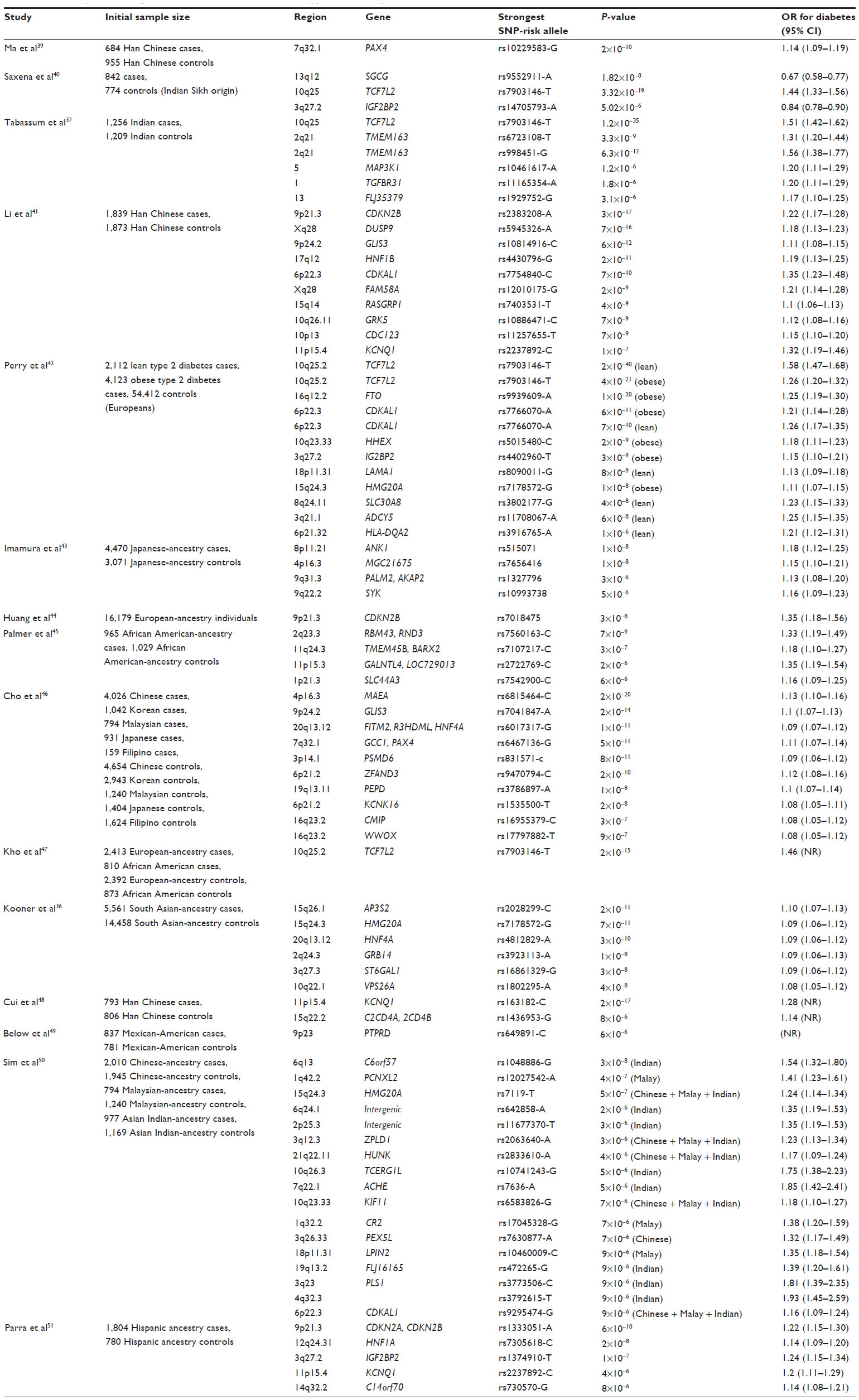 | Table 3 Summary of recent genome-wide association studies in type 2 diabetes published from 2011 till date |
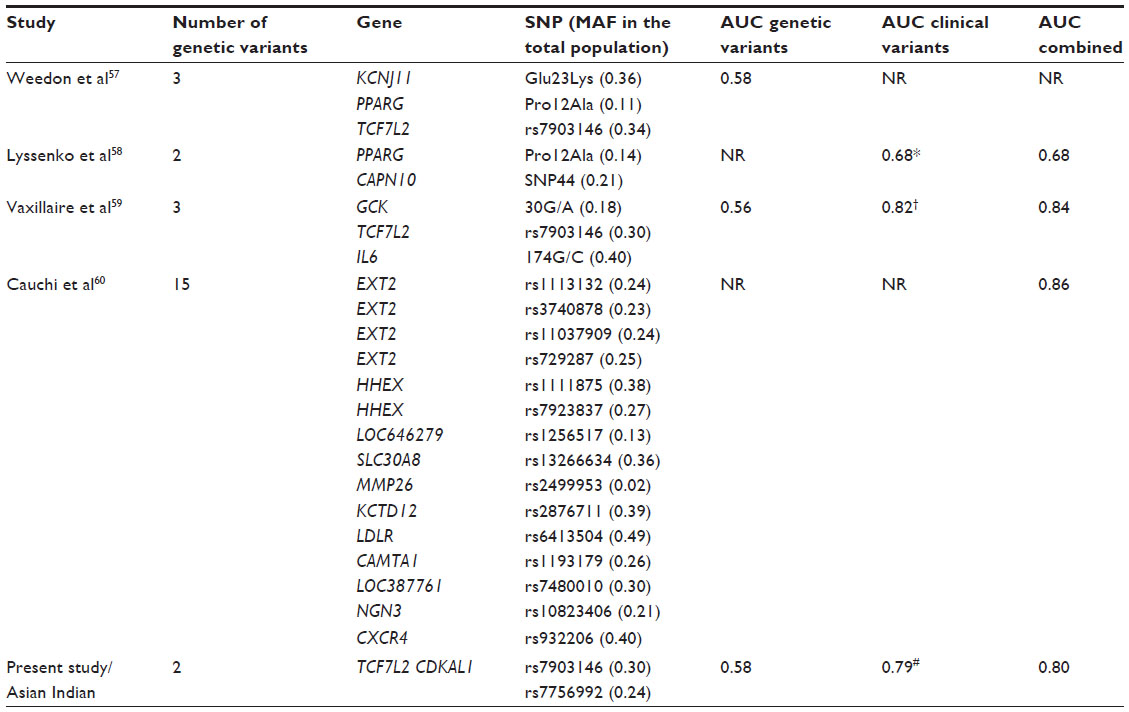 | Table 4 Overview of diagnostic accuracies obtained from earlier empirical studies on genetic risk variants and type 2 diabetes |
Mohan et al52 compared the effectiveness and costs of screening for undiagnosed T2D using oral glucose tolerance testing (OGTT) alone, or following a positive result from the IDRS, or following a positive result from genotyping of the TCF7L2 polymorphisms in Asian Indians. In subjects without known diabetes (n=961) recruited from the Chennai Urban Rural Epidemiology Study (CURES),53 OGTT, IDRS, and genotyping of rs12255372(G/T) and rs7903146(C/T) of TCF7L2 polymorphisms was done. IDRS includes four parameters: age, abdominal obesity, family history of T2D, and physical activity. Seventy-two subjects were identified with newly diagnosed diabetes (NDD) by OGTT, using World Health Organization criteria.54 IDRS screening (cut off ≥60) yielded 413 positive subjects, which included 54 (75%) of the NDD subjects identified by OGTT. Genotyping yielded 493 positive subjects, which only included 36 (50%) of the 72 NDD subjects identified by OGTT, showing less discriminatory power. Screening with both SNPs missed 27 (37.5%) NDD subjects identified by IDRS. In contrast, IDRS missed only nine (12.5%) of the NDD subjects identified by genotyping. The conclusion of the study was that a simple IDRS is more effective and far less expensive for screening of undiagnosed T2D compared with genotyping TCF7L2 SNPs, the strongest genetic marker for T2D so far. In this study only one gene was considered. The scenario could turn out to be somewhat different if additional powerful SNPs were included. However, at the present time it appears unlikely that even a combination of genes can beat a set of clinical markers in predicting prevalent or future diabetes.
The evaluation of T2D risk in individuals carrying many risk variants is critical for a potential clinical use of a genetic test in the general population. In our population, the two variants that showed strongest association with T2D are the rs7903146 SNP of the TCF7L2 gene and the rs7756992 SNP of the CDKAL1 gene. Hence we have considered these two SNPs for analysis. We calculated a weighted genetic score based on these two SNPs using the following formula:55
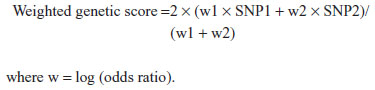
An ROC curve analysis was then performed to explore the discriminatory power of the weighted genetic score (WGS) in predicting the presence of diabetes (Figure 1).
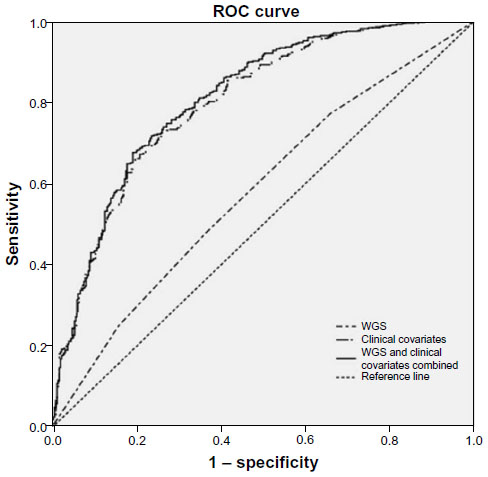 | Figure 1 ROC curve analysis for the weighted genetic score in predicting the presence of diabetes. |
The area under the ROC curve (AUC) is known as the measure of the discriminatory power of a test. A perfect test would have an AUC of 1; a test with no discriminatory power would have an AUC of 0.5.56 The value for the AUC for the two SNP-based WGS was 0.57, whereas the value for the clinical covariates (age, sex, body mass index [BMI], waist circumference) was 0.79. Adding the WGS to the clinical covariates led to a limited improvement in the AUC to 0.80. However, the limitation of this study is the small sample size used for analyses. Table 4 gives an overview of diagnostic accuracies obtained from earlier empirical studies, including ours, on genetic variants and T2D.
It is likely that although a single susceptible SNP is not of value in prediction of diseases such as T2D that are polygenic in nature, with a number of variants contributing in small measures, each SNP is necessary but not sufficient by itself in contributing to the risk of the disease. Although independently the variants may not be useful, combined information from these multiple variants is likely to be beneficial in identifying subjects at high risk or low risk of developing complex diseases.61
The first study to look at the combined predictive value of three common genetic variants (Lys23 of KCNJ11, Pro12 of PPARG, and the T allele at rs7903146 of TCF7L2) that have individually reached genome-wide significance in meta-analysis was that of Weedon et al.57 The study looked at 2,409 T2D cases and 3,668 population-based controls in the white UK population. Subjects with all six risk alleles had an odds ratio of 5.71 (95% CI [confidence interval], 1.15 to 28.3) when compared with those with no risk alleles. The 8.1% of participants that were double-homozygous for the risk alleles at TCF7L2 and Pro12Ala had an odds ratio of 3.16 (95% CI, 2.22 to 4.50), compared with 4.3% with no TCF7L2 risk alleles and either no or one Pro12Ala risk alleles. To evaluate the discriminatory power of the three-SNP combined genetic risk, an ROC curve was plotted and the AUC was 0.58. One reason why the genetic risk did not reach sufficient risk could be because of the number of genetic variants used in the study. The fact that only three genetic variants were included is justifiable because at that point of time only those three genetic variants were reproducibly associated. With the emergence of GWAS, now there are a number of genetic risk variants associated with diabetes, and eventually a number of studies have included more than 15 SNPs to carry out genetic prediction.
One such study is that of Cauchi et al,60 carried out in a French population. About 15 T2D-associated SNPs identified by GWAS were selected for the study and the cumulative genetic risk of carrying risk alleles on T2D prevalence was determined. Subjects with at least 18 risk alleles had approximately nine-fold higher risk of developing T2D compared with the reference group, with an AUC of 0.86. However, this was not calculated for genes and clinical characteristics separately.
Another study by van Hoek et al62 in a Caucasian population investigated 18 polymorphisms from GWAS studies on T2D and found nine SNPs in nine different gene loci to be associated with T2D in their population. Researchers predicted T2D based on genetic polymorphisms alone (AUC =0.6), clinical characteristics (age, sex, and BMI) alone (AUC =0.66), and both together (AUC =0.68). The study demonstrated the lack of improvement in discriminatory accuracy of disease prediction even when gene markers and clinical characteristics were combined. A similar study was published by Lango et al63 on subjects from the Genetics of Diabetes Audit and Research Tayside study in Scotland, selecting a set of 18 SNPs (the majority of them were same as selected by van Hoek et al62), studying their association with diabetes, and assessing the predictive value of genetic testing. Of individuals with >24 risk alleles, 1.2% had an odds ratio of 4.2 (95% CI, 2.11– 8.56) against the 1.8% with 10–12 risk alleles. The AUC for genetic variants alone was 0.60; for age, BMI, and sex it was 0.78; and adding the two groups (genetic risk variants and clinical characteristics) only marginally increased the AUC to 0.80. The discriminatory power to predict T2D thus did not improve after addition of genetic risk variants.
Genetic risk calculation based on the number of risk alleles carried does not take into account the effect size of each risk allele. In an attempt to account for variability in allelic contribution, Lin et al55 constructed an additive genetic risk score in the population-based cross-sectional CoLaus study in Switzerland,64 taking into consideration the most replicated SNPs within 15 T2D susceptibility genes, and weighting each SNP with its reported effect. Adding the weighted genetic score to the clinical covariates led to a limited yet significant improvement in the AUC to 0.87 (P=0.002).
The one advantage of genetic risk calculation is that the genotype does not change over the life course, while risk factors for T2D such as overweight, dyslipidemia, elevated fasting glucose, and even parental history of diabetes may not manifest early in life. Genotype information in young adulthood might therefore have greater predictive value over clinical risk predictors. Based on this hypothesis, Vassy et al65 carried out a study where data from the Coronary Artery Risk Development in Young Adults study was used to examine whether 38 common genetic variants known to be associated with diabetes in cross-sectional adult case-control studies66,67 predicted the onset of T2D and improved diabetes prediction models based on clinical risk factors alone. The authors found that the addition of genetic score did not improve T2D risk prediction over the risk factors already measured in the model. One of the main limitations of the study as cited by the authors was that the SNPs selected were those that showed association with T2D at the genome-wide significant level, and the majority of them were not actually the causal variants and were in intronic regions of the genome. Franks,68 in his commentary on the study by Vassy et al,65 brings out a very important point on why this study could not prove that genetic risk models perform better at younger ages. If the risk alleles for specific loci truly vary by age, genetic risk algorithms derived in adulthood will be inappropriate for use in younger populations, and algorithms that are specific to this younger age group, where effect alleles are coded appropriately, will be required. It is to be noted that the risk alleles for eleven of the 38 SNPs studied by Vassy et al contrast those reported in the published literature.
The genetic risk prediction models have so far not been very successful. There could be a number of reasons for this. Possibly the genes identified so far are not strong enough for prediction. It is possible that newer and stronger genes for T2D might be identified in future. Inclusion of these genes might improve the predictive value. Moreover, most of the SNPs used in prediction models may not be the causal variants; they might be in linkage disequilibrium with the causal variant. It is possible that identification of the causal SNPs might further improve prediction.62 The genetic risk prediction models for T2D examined to date have focused on common gene variants, and it would be wrong to conclude at this stage whether rare variants will or will not be clinically useful for prediction.68
Sanghera et al69 examined the role of nine most significant SNPs reported in GWAS – PPARG2 (rs1801282), IGF2BP2 (rs4402960), CDK5 (rs7754840), SLC30A8 (rs13266634), CDKN2A (rs10811661), HHEX (rs1111875), TCF7L2 (rs10885409), KCNJ11 (rs5219), and FTO (rs9939609) – in an Asian Sikh community from North India. They found that four of the nine SNPs from PPARG2, IGF2BP2, TCF7L2, and FTO showed significant association with T2D. However, in this study the authors did not explore the possibility of genetic prediction using these four SNPs. Most of the studies that have investigated the predictive value of multiple genetic variants in T2D are in Caucasian populations.55,57–65,6874,75 It remains to be seen whether studies in other populations yield different results.
Genetic risk scores alone, consisting of between two and 40 SNPs, have C-statistics for T2D prediction ranging from 0.54 to 0.68. In the Framingham Offspring Study, cumulative T2D incidence over 28 years of follow-up increased significantly with genotype risk score, and each 1-point increase in the score increased the odds of T2D over 8 years by 12%. The group with the highest genotype scores had an odds ratio for T2D of 2.6 compared with those with the lowest scores. However, the clinical T2D prediction models that consist of basic demographic, clinical, and laboratory predictors have C-statistics ranging from 0.66 in the Rotterdam Study70 to 0.90 in the Framingham Offspring Study,46,48 values superior to what genotype scores alone have yet achieved. Moreover, the addition of genotype risk scores to clinical prediction models only modestly improves the C-statistic. For example, the C-statistic improves from 0.903 to 0.906 with the addition of a 40-SNP score to the clinical model in the Framingham Offspring Study and from 0.74 to 0.75 in the larger Malmö Preventive Project.75
Thus genetic testing will likely not have a role in clinical T2D prediction unless its addition to prediction models correctly reclassifies individuals as having lower or higher risk than previously thought based on patient phenotype, and unless the prevention strategies targeted for the individuals change as a result of the marginal information value afforded by the genetic test. Hence, at this time we do not support commercial exploration of genetic testing for T2D.
Family history and heritability of T2D
If genetic information does not improve T2D prediction compared with clinical prediction models, the next reasonable question is how genetic information compares with family history of T2D alone, itself a strong risk predictor of T2D. In the pre-genomic era, twin and family studies played an important role in separating the putative environmental and genetic components of T2D. Twin studies have estimated the genetic proportion of variance in T2D to be between 25% and 40%,71,72 which suggests that both genetic and nongenetic factors contribute substantially to an individual’s T2D risk. Having one parent with T2D doubles an individual’s risk, and having two affected parents can increase an individual’s risk up to sixfold.73 Family history does correlate with T2D genetic risk: data from the PPP-Botnia and Framingham Offspring Studies show that T2D genetic risk scores increase slightly with the degree of T2D family history.74
However, the known T2D genetic variants do not account for the strong relationship between family history and T2D risk. In the Malmö Preventive Project, a self-reported first-degree family history of diabetes carried an odds ratio for incident T2D of 1.62 after adjustment for clinical predictors. When added to this multivariate model, the genetic risk score was an independent predictor of T2D (odds ratio 1.12 per 1-point increase in score) but the effect of family history was unchanged (odds ratio 1.65).74 However, the evidence above demonstrates that, compared with currently identified genetic variants, family history remains a more powerful T2D predictor as it likely captures the genetic and environmental determinants of T2D risk, just like the clinical risk scores do.
Conclusion
Unfortunately, the application of GWAS data for predicting T2D in the clinical setting has been disappointing thus far.75–77 Genetic risk scores developed based on the strong associations from GWAS have not had much clinical utility in predicting incident events when genetic information was added to models based on classical, nongenetic factors. It has to be borne in mind that there are yet unidentified genetic markers with greater effect size than the ones known so far, which might have a greater impact on the risk of T2D and hence possess greater predictive value. The complex gene–gene interactions that might play a role in common diseases62 have to be considered when creating prediction models with genetic factors. Similarly, the genetic and nongenetic factor interaction should also be taken into account. In fact, preliminary evidence points to age78 and BMI79–83 as potential modifiers of genetic effect on the risk of T2D. In conclusion, at the present time traditional clinical markers outperform genetic information and are not costly. It is possible, however, with further discoveries and improvements in technology that this could change, and the dream of personalized genomics may yet become a reality one day.
Disclosure
The authors note no conflict of interest.
References
International Diabetes Federation. IDF Diabetes Atlas. 5th ed. Brussels, Belgium: International Diabetes Federation; 2011. | |
Lindström J, Neumann A, Sheppard KE, et al. Take action to prevent diabetes – the IMAGE toolkit for the prevention of type 2 diabetes in Europe. Horm Metab Res. 2010;42(1):S37–S55. | |
Ackermann RT, Marrero DG. Adapting the Diabetes Prevention Program lifestyle intervention for delivery in the community: the YMCA model. Diabetes Educ. 2007;33(1):69, 74–75, 77–78. | |
Kilkkinen A, Heistaro S, Laatikainen T, et al. Prevention of type 2 diabetes in a primary health care setting. Interim results from the Greater Green Triangle (GGT) Diabetes Prevention Project. Diabetes Res Clin Pract. 2007;76(3):460–462. | |
Saaristo T, Peltonen M, Keinänen-Kiukaanniemi S, et al. National type 2 diabetes prevention programme in Finland: FIN-D2D. Int J Circumpolar Health. 2007;66(2):101–112. | |
Schwarz PE, Schwarz J, Schuppenies A, Bornstein SR, Schulze J. Development of a diabetes prevention management program for clinical practice. Public Health Rep. 2007;122(2):258–263. | |
Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D’Agostino RB Sr. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Arch Intern Med. 2007;67(10):1068–1074. | |
Schmidt MI, Duncan BB, Bang H, et al. Identifying individuals at high risk for diabetes: the Atherosclerosis Risk in Communities study. Diabetes Care. 2005;28(8):2013–2018. | |
Bergmann A, Li J, Wang L, Schulze J, Bornstein SR, Schwarz PE. A simplified Finnish diabetes risk score to predict type 2 diabetes risk and disease evolution in a German population. Horm Metab Res. 2007;39(9):677–682. | |
Schulze MB, Hoffmann K, Boeing H, et al. An accurate risk score based on anthropometric, dietary, and lifestyle factors to predict the development of type 2 diabetes. Diabetes Care. 2007;30(3):510–515. | |
Schwarz PE, Li J, Reimann M, et al. The Finnish Diabetes Risk Score is associated with insulin resistance and progression towards type 2 diabetes. J Clin Endocrinol Metab. 2009;94(3):920–926. | |
Mohan V, Deepa R, Deepa M, Somannavar S, Datta M. A simplified Indian Diabetes Risk Score for screening for undiagnosed diabetic subjects. J Assoc Physicians India. 2005;53:759–763. | |
Willems SM, Mihaescu R, Sijbrands EJ, van Dujin CM, Janssens AC. A methodological perspective on genetic risk prediction studies in type 2 diabetes: recommendations for future research. Curr Diab Rep. 2011;11(6):511–518. | |
Wolfs MGM, Hofker MH, Wijmenga C, van Haeften TW. Type 2 diabetes mellitus: new genetic insights will lead to new therapeutics. Curr Genomics. 2009;10(2):110–118. | |
Vaxillaire M, Froguel P. Monogenic diabetes in the young, pharmacogenetics and relevance to multifactorial forms of type 2 diabetes. Endocrine Reviews. 2008;29(3):254–264. | |
Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200–213. | |
Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 2011;34(8):1878–1884. | |
Aguilar-Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev. 2008;29(3):265–291. | |
Ellard S, Flanagan SE, Girard CA, et al. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet. 2007;81(2):375–382. | |
Temple IK, Gardner RJ, Mackay DJ, Barber JC, Robinson DO, Shield JP. Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes. 2000;49(8):1359–1366. | |
Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51(Suppl 3):S368–S376. | |
Freathy RM, Hayes MG, Urbanek M, et al. Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study: common genetic variants in GCK and TCF7L2 are associated with fasting and postchallenge glucose levels in pregnancy and with the new consensus definition of gestational diabetes mellitus from the International Association of Diabetes and Pregnancy Study Groups. Diabetes. 2010;59(10):2682–2689. | |
Babenko AP, Polak M, Cavé H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355(5):456–466. | |
Pearson ER, Flechtner I, Njølstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations.N Engl J Med. 2006;355(5):467–477. | |
Jahnavi S, Poovazhagi V, Mohan V, et al. Clinical and molecular characterization of neonatal diabetes and monogenic syndromic diabetes in Asian Indian children. Clin Genet. 2013;83(5):439–445. | |
McCarthy M, Menzel S. The genetics of type 2 diabetes. J Clin Pharmacol. 2001;51(3):195–199. | |
Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445(7130):881–885. | |
Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40(5):638–645. | |
Genome.gov. [webpage on the Internet]. Available from http://www.genome.gov/gwastudies. Accessed April 2, 2014. | |
Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331–1336. | |
Sanghera DK, Blackett PR. Type 2 diabetes genetics: beyond GWAS. J Diabetes Metab. 2012;3(198). | |
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes.Nat Genet. 2007;39(6):770–775. | |
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proença C, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. 2009;41(1):89–94. | |
Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316(5829):1336–1341. | |
Chidambaram M, Radha V, Mohan V. Replication of recently described type 2 gene variants in a South Indian population. Metabolism. 2010;59(12):1760 –1766. | |
Kooner JS, Saleheen D, Sim X, et al. Genome-wide association study in people of South Asian ancestry identifies six novel susceptibility loci for type 2 diabetes. Nat Genet. 2011;43(10):984–989. | |
Tabassum R, Chauhan G, Dwivedi P, et al. Genome-wide association study for type 2 diabetes in Indians identifies a new susceptibility locus at 2q21. Diabetes. 2013;62(3):977–986. | |
Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. | |
Ma RC, Hu C, Tam CH, et al. Genome-wide association study in a Chinese population identifies a susceptibility locus for type 2 diabetes at 7q32 near PAX4. Diabetologia. 2013;56(6):1291–1305. | |
Saxena R, Saleheen D, Been LF, et al. Genome-wide association study identifies a novel locus contributing to type2diabetes susceptibility in Sikhs of Punjabi origin from India. Diabetes. 2013;62(5):1746–1755. | |
Li H, Gan W, Lu L, et al. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes. 2013;62(1):291–298. | |
Perry JR, Voight BF, Yengo L, et al. Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLoS Genet. 2012;8(5):e1002741. | |
Imamura M, Maeda S, Yamauchi T, et al. A single-nucleotide polymorphism in ANK1 is associated with susceptibility to type 2 diabetes in Japanese populations. Hum Mol Genet. 2012;21(13):3042–3049. | |
Huang J, Ellinghaus D, Franke A, Howie B, Li Y. 1000 genomes-based imputation identifies novel and refined associations for the Wellcome Trust Case Control Consortium phase 1 data. Eur J Hum Genet. 2012;20(7):801–805. | |
Palmer ND, McDonough CW, Hicks PJ, et al. A genome-wide association search for type 2 diabetes genes in African Americans. PLoS One. 2012;7(1):e29202. | |
Cho YS, Chen CH, Hu C, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2011;44(1):67–72. | |
Kho AN, Hayes MG, Rasmussen-Torvik L, et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. J Am Med Inform Assoc. 2012;19(2):212–218. | |
Cui B, Zhu X, Xu M, et al. A genome-wide association study confirms previously reported loci for type 2 diabetes in Han Chinese. PLoS One. 2011;6(7):e22353. | |
Below JE, Gamazon ER, Morrison JV, et al. Genome-wide association and meta-analysis in populations from Starr County, Texas, and Mexico City identify type 2 diabetes susceptibility loci and enrichment for expression quantitative trait loci in top signals. Diabetologia. 2011;54(8):2047–2055. | |
Sim X, Ong RT, Suo C, et al. Transferability of type 2 diabetes implicated loci in multi-ethnic cohorts from Southeast Asia. PLoS Genet. 2011;7(4):e1001363. | |
Parra EJ, Below JE, Krithika S, et al. Genome-wide association study of type 2 diabetes in a sample from Mexico City and a meta-analysis of a Mexican-American sample from Starr County, Texas. Diabetologia. 2011;54(8):2038–2046. | |
Mohan V, Goldhaber-Fiebert JD, Radha V, Gokulakrishnan K. Screening with OGTT alone or in combination with the Indian diabetes risk score or genotyping of TCF7L2 to detect undiagnosed type 2 diabetes in Asian Indians. Indian J Med Res. 2011;133:294–299. | |
Deepa M, Pradeepa R, Rema M, Mohan A, Deepa R, Shanthirani S, et al. The Chennai Urban Rural Epidemiology Study (CURES) - study design and methodology (urban component) (CURES-I). J Assoc Physicians India. 2003;51:863–870. | |
Report of the Expert Committee on the Diagnosis and Classification of diabetes mellitus. Diabetes Care. 1997;20:1183–1197. | |
Lin X, Song K, Lim N, et al. Risk prediction of prevalent diabetes in a Swiss population using a weighted genetic score–the CoLaus study. Diabetologia. 2009;52(4):600–608. | |
Hanley JA, McNeil BJ. A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology. 1983;148(3):839–843. | |
Weedon MN, McCarthy MI, Hitman G, et al. Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLoS Med. 2006;3(10):e374. | |
Lyssenko V, Almgren P, Anevski D, et al. Genetic prediction of future type 2 diabetes. PLoS Med. 2005;2(12):e345. | |
Vaxillaire M, Veslot J, Dina C, et al. Impact of common type 2 diabetes risk polymorphisms in the DESIR prospective study. Diabetes. 2008;57(1):244–254. | |
Cauchi S, Meyre D, Durand E, et al. Post genome-wide association studies of novel genes associated with type 2 diabetes show gene-gene interaction and high predictive value. PLoS One. 2008;3(5):e2031. | |
Yang Q, Khoury MJ, Botto L, Friedman JM, Flanders WD. Improving the prediction of complex diseases by testing for multiple disease-susceptibility genes. Am J Hum Genet. 2003;72(3):636–649. | |
van Hoek M, Dehghan A, Witteman JC, et al. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies:a population-based study. Diabetes. 2008;57(11):3122–3128. | |
Lango H, UK Type 2 Diabetes Genetics Consortium, Palmer CN, et al. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes. 2008;57(11):3129–3135. | |
Firmann M, Mayor V, Vidal PM, et al. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc Disord. 2008;8:6. | |
Vassy JL, Dasmahapatra P, Meigs JB, et al. Genotype prediction of adult type 2 diabetes from adolescence in a multiracial population. Pediatrics. 2012;130(5):e1235–e1242. | |
Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40(5):638–645. | |
Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42(7):579–589. | |
Franks PW. Genetic risk scores ascertained in early adulthood and the prediction of type 2 diabetes later in life. Diabetologia. 2012;55(10):2555–2558. | |
Sanghera DK, Ortega L, Han S, et al. Impact of nine common type 2 diabetes risk polymorphisms in Asian Indian Sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTO variants confer a significant risk. BMC Med Genet. 2008;9:59. | |
Vassy JL, Meigs JB. Is Genetic testing useful to predict type 2 diabetes? Best Pract Res Clin Endocrinol Metab. 2012;26(2):189–201. | |
Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance – a population-based twin study. Diabetologia. 1999;42(2):139–145. | |
Kaprio J, Tuomilehto J, Koskenvuo M, et al. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia. 1992;35(11):1060–1067. | |
Meigs JB, Cupples LA, Wilson PW. Parental transmission of type 2 diabetes: the Framingham Offspring Study. Diabetes. 2000;49(12):2201–2207. | |
Vassy JL, Shrader P, Jonsson A, et al. Association between parental history of diabetes and type 2 diabetes genetic risk scores in the PPP-Botnia and Framingham Offspring Studies. Diabetes Res Clin Pract. 2011;93(2):e76–e79. | |
Lyssenko V, Jonsson A, Almgren P, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359(21):2220–2232. | |
de Miguel-Yanes JM, Shrader P, Pencina MJ, et al. Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care. 2011;34(1):121–125. | |
Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359(21):2208–2219. | |
Talmud PJ, Hingorani AD, Cooper JA, et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ. 2010;340:b4838. | |
Prudente S, Morini E, Trischitta V. Insulin signaling regulating genes: effect on T2DM and cardiovascular risk. Nat Rev Endocrinol. 2009;5:682e93. | |
Bacci S, Rizza S, Prudente S, et al. The ENPP1 Q121 variant predicts major cardiovascular events in high-risk individuals: evidence for interaction with obesity in diabetic patients. Diabetes. 2011;60(3):1000–1007. | |
Cornelis MC, Qi L, Zhang C, et al. Joint effects of common genetic variants on the risk for type 2 diabetes in US men and women of European ancestry. Ann Intern Med. 2009;150(8):541–550. | |
Timpson NJ, Lindgren CM, Weedon MN, et al. Adiposity-related heterogeneity in patterns of type 2 diabetes susceptibility observed in genome-wide association data. Diabetes. 2009;58(2):505–510. | |
Prudente S, Dallapiccola B, Pellegrini F, Doria A, Trischitta V. Genetic prediction of common diseases. Still no help for the clinical diabetologist! Nutr Metab Cardiovasc Dis. 2012;22(11):929–936. | |
Hindorff LA, MacArthur J, Morales J, Junkins HA, Hall PN, Klemm AK, and Manolio TA. A Catalog of Published Genome-Wide Association Studies. Available at: http://www.genome.gov/gwastudies. Accessed 24 April, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.