Back to Journals » Drug Design, Development and Therapy » Volume 8
Preclinical profile of cabazitaxel
Authors Vrignaud P, Semiond D, Benning V, Beys E, Bouchard H, Gupta S
Received 27 March 2014
Accepted for publication 9 May 2014
Published 13 October 2014 Volume 2014:8 Pages 1851—1867
DOI https://doi.org/10.2147/DDDT.S64940
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Patricia Vrignaud,1 Dorothée Semiond,2 Veronique Benning,2 Eric Beys,2 Hervé Bouchard,3 Sunil Gupta4
1Sanofi Oncology, Vitry-sur-Seine, France; 2Sanofi DSAR, Alfortville, France; 3Sanofi LGCR, Vitry-sur-Seine, France; 4Sanofi Oncology, Cambridge, MA, USA
Abstract: First-generation taxanes have changed the treatment paradigm for a wide variety of cancers, but innate or acquired resistance frequently limits their use. Cabazitaxel is a novel second-generation taxane developed to overcome such resistance. In vitro, cabazitaxel showed similar antiproliferative activity to docetaxel in taxane-sensitive cell lines and markedly greater activity in cell lines resistant to taxanes. In vivo, cabazitaxel demonstrated excellent antitumor activity in a broad spectrum of docetaxel-sensitive tumor xenografts, including a castration-resistant prostate tumor xenograft, HID28, where cabazitaxel exhibited greater efficacy than docetaxel. Importantly, cabazitaxel was also active against tumors with innate or acquired resistance to docetaxel, suggesting therapeutic potential for patients progressing following taxane treatment and those with docetaxel-refractory tumors. In patients with tumors of the central nervous system (CNS), and in patients with pediatric tumors, therapeutic success with first-generation taxanes has been limited. Cabazitaxel demonstrated greater antitumor activity than docetaxel in xenograft models of CNS disease and pediatric tumors, suggesting potential clinical utility in these special patient populations. Based on therapeutic synergism observed in an in vivo tumor model, cabazitaxel is also being investigated clinically in combination with cisplatin. Nonclinical evaluation of the safety of cabazitaxel in a range of animal species showed largely reversible changes in the bone marrow, lymphoid system, gastrointestinal tract, and male reproductive system. Preclinical safety signals of cabazitaxel were consistent with the previously reported safety profiles of paclitaxel and docetaxel. Clinical observations with cabazitaxel were consistent with preclinical results, and cabazitaxel is indicated, in combination with prednisone, for the treatment of patients with hormone-refractory metastatic prostate cancer previously treated with docetaxel. In conclusion, the demonstrated activity of cabazitaxel in tumors with innate or acquired resistance to docetaxel, CNS tumors, and pediatric tumors made this agent a candidate for further clinical evaluation in a broader range of patient populations compared with first-generation taxanes.
Keywords: XRP6258, CNS tumors, mCRPC, pediatric tumor, taxane resistance, xenograft
Introduction
Since the initial approval of paclitaxel (Taxol®; Bristol-Myers Squibb, New York City, NY, USA) in 1992,1,2 the first-generation taxanes paclitaxel and docetaxel (Taxotere®; Sanofi, Paris, France) have altered the treatment paradigm for a wide variety of solid tumors, including breast, lung, prostate, gastric, and ovarian cancers.3,4 Despite demonstrating significant antitumor activity as monotherapy or in combination regimens, clinical use of first-generation taxanes is frequently limited by innate or acquired resistance.5–7 In prostate cancer, the majority of patients will eventually acquire resistance to docetaxel therapy.8
Cabazitaxel (Jevtana®, Sanofi) is a novel second-generation semisynthetic taxane that was identified through a preclinical screen of 450 molecules derived from 10-deacetylbaccatin-III, with the aim of identifying a compound with activity in both taxane-sensitive and taxane-resistant tumors.9 In the pivotal Phase III TROPIC study (NCT00417079), cabazitaxel combined with prednisone significantly extended overall survival compared with mitoxantrone plus prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC) previously treated with a docetaxel-containing regimen.10 This led to cabazitaxel’s approval in 2010, in combination with prednisone, for the treatment of patients with hormone-refractory metastatic prostate cancer who have previously received docetaxel-based therapy.11,12
This review article presents an overview of the preclinical properties of cabazitaxel, including its development, mechanism of action, antitumor activity in a range of in vitro and in vivo tumor models, pharmacokinetics (PK), and metabolic and toxicity profiles, as well as a summary of its clinical development.
Taxanes’ mechanism of action and resistance mechanisms
Mechanism of action
Taxanes are microtubule inhibitors that induce cellular apoptosis through the stabilization of microtubules.7 Microtubules are major components of the cytoskeleton, with critical roles in a variety of cellular processes including maintenance of cell shape, intracellular transport, cell signaling, and cell division.7,13–15 It is this pivotal role in mitosis that makes microtubules a key cellular target for anticancer therapeutics.7
Microtubules are highly dynamic polymers of tubulin, continually undergoing assembly and disassembly within the cell. Taxanes inhibit microtubule function by binding to tubulin molecules, promoting their polymerization, and stabilizing microtubules. Suppression of microtubule dynamics leads to a block in mitosis and, ultimately, tumor cell death.7,13,14
Resistance mechanisms
Innate or acquired resistance to first-generation taxanes is frequently observed in different tumor types, resulting in treatment failure. Multiple potential mechanisms of taxane resistance have been identified in preclinical studies, and it is likely that several of these contribute to a resistant phenotype.6,7,16–19
Two mechanisms in particular have frequently been associated with the development of resistance to taxanes; however, it is worth noting that these are yet to be validated in patient samples, and their clinical relevance is not fully understood.6,7 In preclinical studies, resistance commonly results from overexpression of members of the ATP-binding cassette family of transporters, of which P-glycoprotein, encoded by the multidrug resistance gene (ATP-binding cassette, sub-family B [MDR/TAP], member 1; ABCB1), is the best known.20 Docetaxel and paclitaxel are substrates of P-glycoprotein, which acts as a drug efflux pump, decreasing intracellular drug levels and limiting cytotoxicity.6,7,21,22 Resistance may also arise from spontaneously acquired mutations in tubulin, the cellular target of taxanes, resulting in changes to the tubulin binding site or altered microtubule dynamics.6,7,23
Clinical data suggest that additional mechanisms may contribute to taxane resistance in patients, including the altered expression of specific tubulin isotypes,17 and expression or binding of microtubule-regulatory proteins,18 loss of functional p53,16 dysfunctional regulation of apoptotic and intracellular signaling (eg, HER2 overexpression),19 and decreased tumor cell permeability.24
A number of potential predictive markers for taxane resistance have been identified, including the mitotic spindle checkpoint proteins Aurora A, BUBR1, MAD2, and synuclein-γ, and cell cycle proteins such as BRCA1;18 however, conflicting results have been reported clinically.24
The development of alternative therapies able to overcome taxane resistance has been the focus of considerable attention.
Cabazitaxel development
Paclitaxel and docetaxel are semisynthetic derivatives of 10-deacetylbaccatin-III,25,26 a natural paclitaxel precursor molecule that can be extracted easily and sustainably from the needles of the European yew tree (Figure 1).26 In light of the clinical limitations that result from taxane resistance, a large-scale preclinical screening process was undertaken that aimed to identify a taxane derivative with equivalent efficacy to docetaxel in docetaxel-sensitive tumors, but greater activity than docetaxel in tumors that are docetaxel-resistant.9
 | Figure 1 Chemical structure of 10-deacetylbaccatin III, docetaxel, and cabazitaxel. |
In total, 450 candidate molecules were designed and generated for preclinical assessment, based on preclinical comparative structure–activity relationships of paclitaxel and docetaxel. Structural modifications initially focused on the side chain, as this was considered critical for potency, with subsequent modifications to other functional groups within the baccatin moiety.9 The antitumor potential of the taxane derivatives was assessed over three stages: in vitro activity against microtubules; in vitro activity in resistant cell lines; and in vivo activity in a tumor model.24 The in vivo assessments included evaluation in a B16/TXT melanoma resistance model, which was developed through repeat exposure to docetaxel in mice bearing the docetaxel-sensitive B16 tumor, to allow evaluation of the taxane derivatives in a clinically relevant setting. This model mimics the clinical development of docetaxel resistance, where tumors initially respond to treatment, but develop resistance progressively over time.24
Initial attempts to modify the C-3′N–Boc and C-3′ phenyl groups within the side chain of docetaxel resulted in derivatives demonstrating either reduced in vitro potency or failure to improve activity in docetaxel-resistant cell lines (Figure S1).9,27 Alterations to the C-2/C-4 and oxetane ring regions of the baccatin moiety were also evaluated, but failed to increase activity in the in vitro and/or in vivo resistance models.9
Cabazitaxel is a dimethyl derivative of docetaxel, bearing methoxy groups in place of hydroxyl groups at positions C-7 and C-10 (Figure 1).9 In both docetaxel-sensitive and docetaxel-resistant cell lines, these structural alterations resulted in the greatest increase in in vitro potency, without significantly increasing toxicity at the maximum tolerated dose, in contrast to other C-7/C-10 modifications (Figure S1).9 These modifications confer two advantages on cabazitaxel over docetaxel. Firstly, cabazitaxel, which is a P-glycoprotein substrate, has a higher lipophilicity than docetaxel (logP 3.9 versus 3.2),9,28 resulting from the conversion of two secondary alcohols to more lipophilic ethers. This may result in increased cell penetration through passive influx, consequently leading to better activity in resistant cell lines where permeability of the plasma membrane may be altered.24,28–30 This hypothesis was recently confirmed in an experiment in which drug uptake into MCF7 breast adenocarcinoma cells, which do not overexpress P-glycoprotein, was faster for cabazitaxel than for docetaxel.31 Secondly, cabazitaxel has an improved ability to cross the blood–brain barrier (BBB) compared with docetaxel, offering potential benefit in patients with tumors of the central nervous system (CNS).9,29,32,33
Accordingly, during the screening of taxane derivatives and subsequent preclinical evaluation, cabazitaxel has demonstrated equivalent efficacy to docetaxel for stabilizing microtubules in vitro, greater potency than docetaxel in cell lines resistant to taxanes and other chemotherapeutics, activity superior to docetaxel in in vivo CNS disease models, broad-spectrum antitumor activity against a range of murine and human tumors, and in vivo activity in tumor models that are not sensitive, or are poorly sensitive, to docetaxel.9,24,33
In vitro activity
Microtubule stabilization
Cabazitaxel has shown equivalent potency to docetaxel for stabilization of microtubules in vitro. Both cabazitaxel and docetaxel induced a similar reduction in lag time for tubulin assembly (lag time to 50% aggregation 0–0.1 μmol/L) and stabilization of microtubules against cold-induced depolymerization (concentration producing 50% cell killing 0.1–0.25 μmol/L), indicating that cabazitaxel has a cytotoxic mechanism of action similar to that of docetaxel.24
Antiproliferative activity
In cell lines sensitive to chemotherapy, cabazitaxel had similar antiproliferative activity to docetaxel, achieving comparable 50% inhibitory concentration (IC50) values across a range of murine and human cell types (0.004–0.041 μmol/L for cabazitaxel versus 0.008–0.079 μmol/L for docetaxel) (Table 1).24
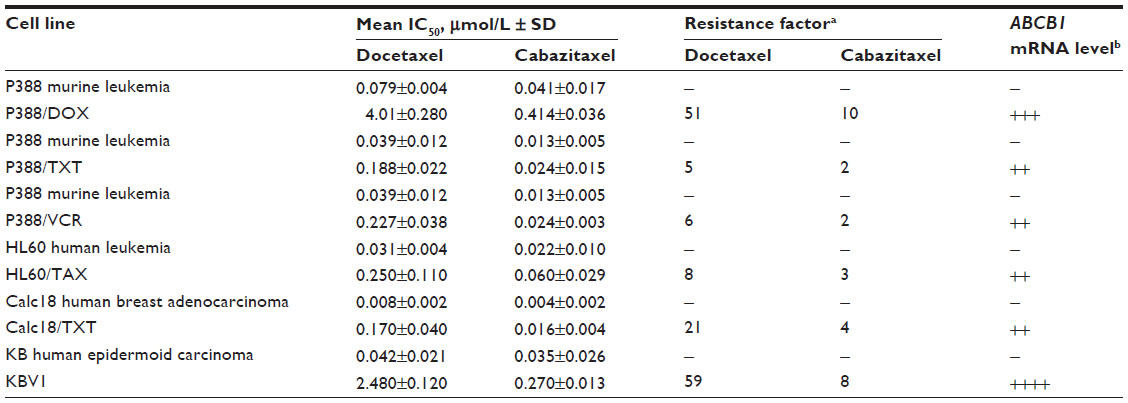 | Table 1 In vitro antiproliferative effects of cabazitaxel and docetaxel against sensitive and P-glycoprotein-expressing resistant cell lines |
In a panel of cell lines bearing acquired resistance to taxanes or to the chemotherapeutic agents doxorubicin, vincristine, or vinblastine, cabazitaxel showed markedly greater antiproliferative activity than docetaxel (IC50 ranged from 0.016–0.414 μmol/L for cabazitaxel versus 0.17–4.01 μmol/L for docetaxel).24 Resistance factors, an indication of the difference in drug concentrations needed to inhibit resistant versus sensitive/parental cell lines, ranged from 2–10 for cabazitaxel and 5–59 for docetaxel in these P-glycoprotein-expressing cell lines (Table 1).24
In murine and human cell lines with resistance mechanisms other than P-glycoprotein overexpression, no cross-resistance to cabazitaxel was observed.24
In vivo activity
Plasma pharmacokinetics
The PK profile of cabazitaxel was evaluated in healthy and tumor-bearing mice, and healthy rats and dogs (Table 2) (Sanofi, data on file, 2010).
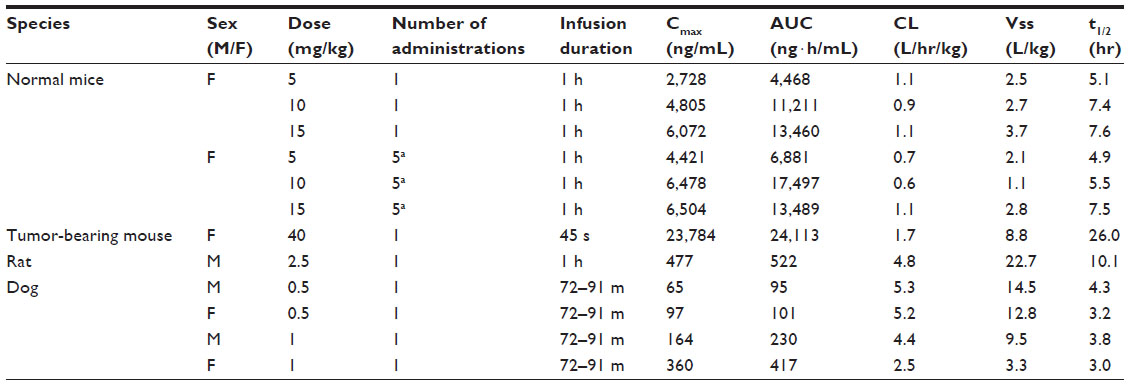 | Table 2 Pharmacokinetic parameters of cabazitaxel in normal and tumor-bearing mice, rats, and dogs |
Absorption
Exposure to cabazitaxel increased with dose after single or repeated intravenous (IV) administration in all species. The increase in exposure was approximately dose-proportional in mice and more than dose-proportional in rats and dogs. No plasma accumulation was observed in mice, rats, or dogs after administration every 5 days, weekly, or every 3 weeks. No sex effect was observed in rats and dogs (Sanofi, data on file, 2010).
Distribution
Plasma protein binding of cabazitaxel was very high in mice (99.3%) and high in rats (95.5%), rabbits (91.4%), dogs (97.1%), and humans (91.9%) (Sanofi, data on file, 2010). Following a single IV administration, cabazitaxel exhibited a very large volume of distribution at steady state in both healthy (2.5–3.7 L/kg) and tumor-bearing mice (8.8 L/kg), in rats (22.7 L/kg), and in dogs (3.3–14.5 L/kg) (Sanofi, data on file, 2010).
The PK of cabazitaxel was also evaluated in mice bearing advanced-stage (>400 mm3) mammary MA16/C adenocarcinomas.24 Cabazitaxel was administered at the highest nontoxic dose (HNTD) of 40 mg/kg. Drug uptake into the tumor was both rapid and sustained, with maximum drug concentrations in tumor tissue reached within 15 minutes, and a 40-fold greater concentration of cabazitaxel within the tumor versus plasma after 48 hours (Figure 2).24
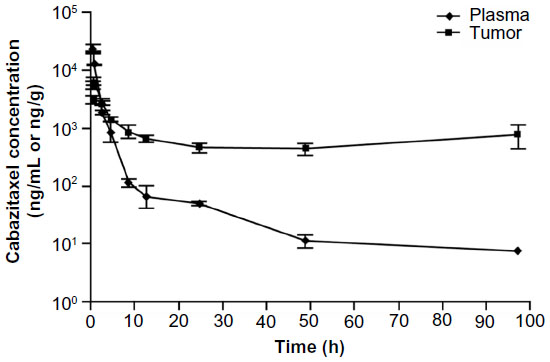 | Figure 2 Pharmacokinetics of 40 mg/kg cabazitaxel (highest nontoxic dose) in plasma and tumor tissues in mice. |
In this model, exposure to cabazitaxel was 1.6-times greater in the tumor compared with plasma during the 48 hours following treatment administration, and 2.9-times greater over the entire experimental period (168 hours). Concentrations of cabazitaxel above the cellular antiproliferative IC50 were sustained for 24 hours in the plasma and for 96 hours in tumor tissue.24
Brain distribution of cabazitaxel was assessed in mice, rats, and dogs. Cabazitaxel penetrated rapidly in the brain, with similar relative exposure between brain and blood across the different species.33
Metabolism
Cabazitaxel metabolism has been compared across multiple species. In vivo, cabazitaxel was the major circulating compound in all species including humans (≥65% of the total radioactivity area under the curve). Cabazitaxel undergoes extensive biotransformation, with less than 2.5% of the administered dose excreted unchanged in studies of mice, rats, dogs, and humans. The two main metabolic pathways, accounting for 40%, 54%, and 45% of the dose excreted in humans, dogs, and mice, respectively, corresponded to two O-demethylations at the C-7 and C-10 positions, one leading to 7-O-demethyl-cabazitaxel (pathway B) and the other to 10-O-demethyl-cabazitaxel (pathway A) (Figure 3; Table 3). In male and female rats, the main pathway corresponded to t-butyl-hydroxylation on the lateral chain (pathway C), accounting for 49%–55% of the dose excreted. This pathway was also abundant in mice, representing 41% of the dose, and was found in humans (21% of the dose). Another pathway consisting of the cleavage of the parent drug (pathway D) was very minor in all species, including humans (<0.3% of the dose). In contrast, unchanged cabazitaxel was the major circulating compound in plasma in all species, including humans, accounting for ≥65% of the total radioactivity area under the curve.34
 | Figure 3 Proposed schematic of the principal metabolic pathways of cabazitaxel. |
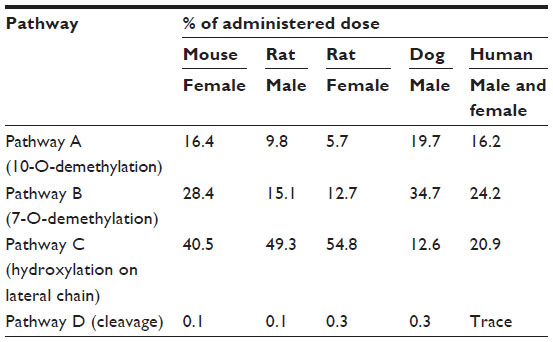 | Table 3 Relative contribution of different metabolic pathways to cabazitaxel metabolism across species |
Excretion
The PK of cabazitaxel following a single IV infusion in normal mice, tumor-bearing mice, and in dogs is generally characterized by a biphasic elimination (Sanofi, data on file, 2010). Plasma clearance was high in rats (4.8 L/h/kg) and dogs (2.5–5.3 L/h/kg) and moderate in normal (0.9–1.1 L/h/kg) and tumor-bearing mice (1.7 L/h/kg), compared with respective hepatic blood flow. The terminal half-life was moderately long in dogs (3.0 to 4.3 hours), long in normal mice (5.1–7.6 hours) and rats (10 hours), and extremely long in tumor-bearing mice (26 hours). It should be noted that a higher dose was given to tumor-bearing mice, which was reflected in quantifiable levels occurring at later sampling times than in normal mice (Sanofi, data on file, 2010).
Cabazitaxel excretion was nearly complete (91%–95% of the administered dose) in mice, rats, and dogs, and radioactivity was largely excreted in the feces in all three species (87%–91% of the administered dose), with minimal excretion via the urinary route (1%–4%). In bile-duct-cannulated male rats, radioactivity was mainly excreted via the biliary route, accounting for 65% of the administered dose within 48 hours. Similarly, in humans the majority of the radioactivity was recovered in feces (around 76% of the administered dose), with 4% of the dose found in urine.34
In summary, the disposition of cabazitaxel was assessed in a range of animal species, and the distribution, metabolic pathways, and elimination processes documented in animals are consistent with those observed in humans (Sanofi, data on file, 2010). Thus, the animals studied are likely to represent good models for the disposition of cabazitaxel in humans. The finding that unchanged cabazitaxel is the main circulating compound in plasma indicates that analysis of the parent drug is appropriate for PK and pharmacodynamics studies of cabazitaxel.
Antitumor activity
Prostate cancer models
The efficacy of taxanes in prostate cancer, in addition to their impact on cell division, may in part relate to inhibitory effects on androgen receptor signaling.35–37 In mice bearing a docetaxel-sensitive cell line-derived castrate-resistant prostate cancer xenograft (DU145), cabazitaxel was found to be highly active, inducing 100% complete tumor regressions (complete response [CR]) and 83% long-term tumor-free survival (TFS).24
Cabazitaxel was compared with docetaxel and abiraterone acetate (a specific inhibitor of CYP17) in a patient-derived prostate tumor xenograft with induced resistance to castration (HID28). At equivalent dose levels (20 mg/kg), cabazitaxel demonstrated greater antitumor efficacy than docetaxel.38 Cabazitaxel inhibited tumor growth with a percentage tumor volume change (calculated as the percentage ratio between the mean tumor volume of a treated and a control group) of 1.4% at Day 35 compared with 16.7% for docetaxel (Figure 4).38 Complete and partial remissions, respectively, were achieved in six out of ten and four out of ten mice receiving cabazitaxel, and in one out of ten and two out of ten mice treated with docetaxel.38 No antitumor activity was demonstrated with abiraterone acetate (50 mg/kg orally administered daily for 21 days).38 These data provide support for the clinical development of cabazitaxel for the first-line treatment of mCRPC.
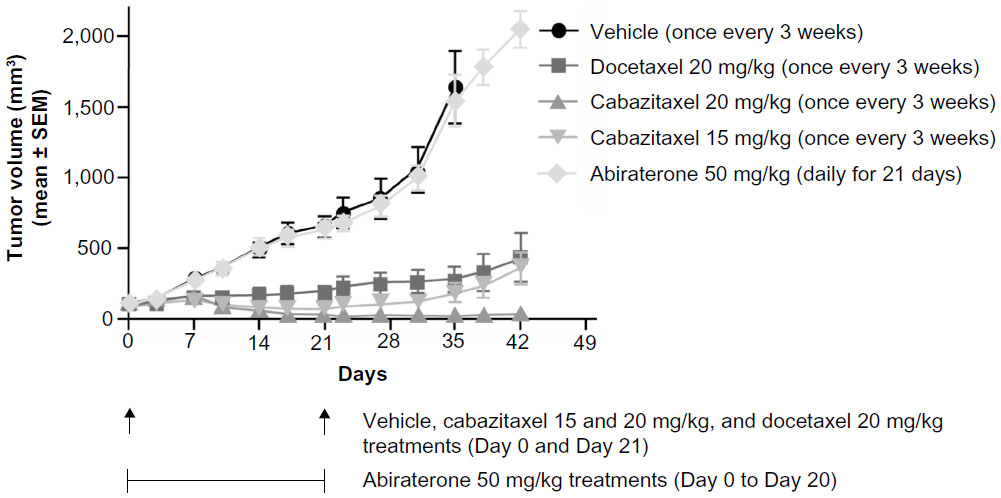 | Figure 4 Antitumor activity of cabazitaxel, docetaxel, and abiraterone in a docetaxel-sensitive hormone-refractory prostate cancer xenograft model. |
Docetaxel-sensitive models
Cabazitaxel demonstrated broad-spectrum activity in murine and human xenograft models (Table 4). Excellent antitumor activity was observed in murine B16 melanoma, colon adenocarcinoma C51, and mammary adenocarcinoma MA16/C and MA17/A tumors.24 In a range of advanced disease models, including human colon HCT 116 and HT-29, lung A549 and NCI-H460, pancreas MIA PaCa-2, head and neck SR475, and kidney Caki-1 xenografts, administration of cabazitaxel achieved CR. In seven of eleven such models, CR levels were 100%. In seven models, CR resulted in TFS at study completion, with 80 to 100% TFS in five of them.24
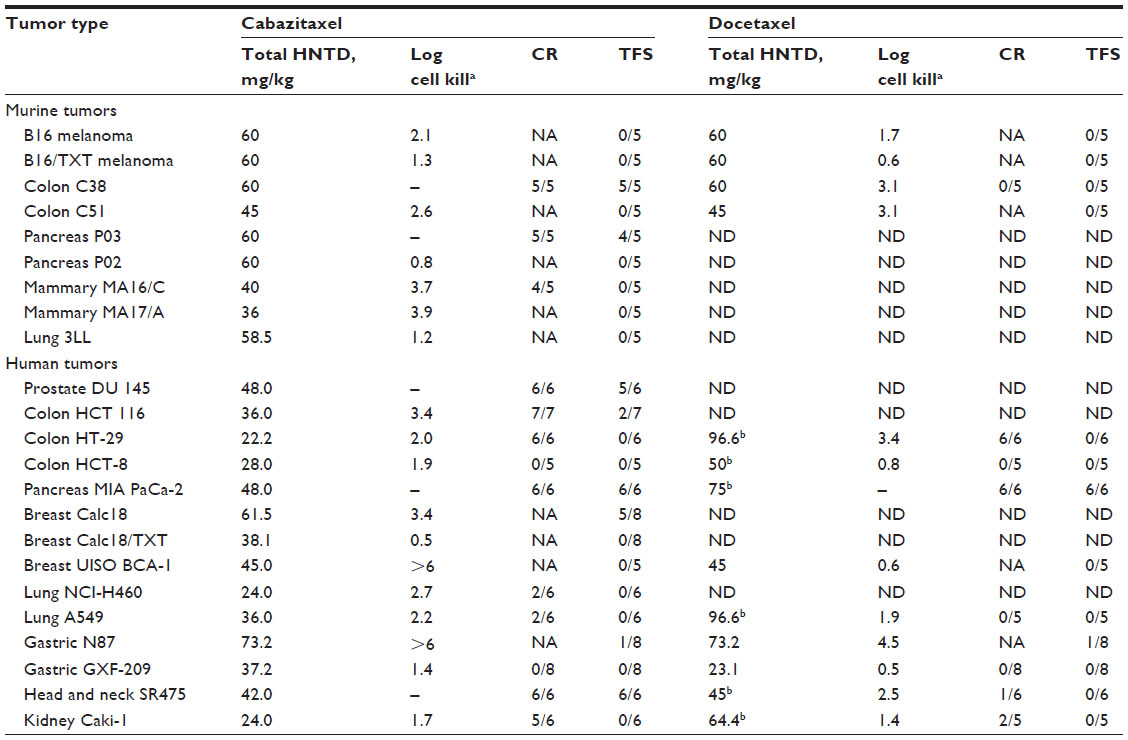 | Table 4 Dose–response antitumor activity of cabazitaxel and docetaxel in mice bearing murine and human docetaxel-sensitive and -resistant tumors |
In most cases, the antitumor activity of cabazitaxel was similar to that of docetaxel, as assessed by log cell kill values derived from measurements of tumor growth (log cell kill = tumor growth delay/3.32× tumor doubling time).24 Despite this, docetaxel induced CR in just four of seven evaluable xenograft models (human colon HCT-29, pancreas MIA PaCa-2, head and neck SR475, and kidney Caki-1), and TFS at study completion was only observed in mice bearing human pancreas MIA PaCa-2 xenografts.
Models resistant or poorly sensitive to docetaxel
Cabazitaxel was evaluated in the B16/TXT acquired-resistance model, which was developed by repeatedly exposing mice bearing the docetaxel-sensitive murine B16 melanoma to docetaxel at the HNTD (60 mg/kg per passage), with a total of 27 passages over 17 months required to obtain a fully docetaxel-resistant tumor. Cabazitaxel demonstrated antitumor activity against B16/TXT, which does not overexpress P-glycoprotein, but was not active against the P-glycoprotein-overexpressing tumor Calc18/TXT in which docetaxel resistance was induced in vitro.24
Interestingly, cabazitaxel showed activity against a number of murine and human xenograft models bearing innate resistance to docetaxel, including the human breast tumor UISO BCA-1. In this early-stage model, cabazitaxel achieved a log cell kill value of greater than 6, compared with 0.6 with docetaxel. Moreover, docetaxel did not delay tumor growth at its HNTD (15 mg/kg per injection; log cell kill 0.6, P>0.5), whereas cabazitaxel was highly active both at its HNTD (15 mg/kg per injection; log cell kill >6, P=0.0016) and at the dose level below (9.3 mg/kg per injection; log cell kill 4.4, P=0.0016).24
Taken together, these findings made cabazitaxel a candidate for further clinical evaluation in patients who have relapsed following taxane treatment, as well as in those with innately docetaxel-refractory tumors.24
Special populations
CNS tumors
The activity of first-generation taxanes in patients with tumors of the CNS is limited,39–43 due to poor penetration of these drugs across the BBB under normal physiologic conditions.44,45 This is a result of both the physical barrier formed by brain endothelial cell tight junctions at the BBB44 and the expression of a range of efflux transporters, including P-glycoprotein and other members of the multidrug resistance protein family, that limit uptake of transporter substrates into the brain to protect brain tissue from toxic insult.44–46
Preclinical data suggest that uptake of cabazitaxel into the brain is greater than that observed with first-generation taxanes, potentially resulting from saturation of an efflux process at the BBB.33,47 Cabazitaxel uptake across the BBB was threefold higher in P-glycoprotein-deficient mice compared with control mice, suggesting that efflux from the brain may be mediated by P-glycoprotein, amongst other transporters.47 Further evidence of saturation of an efflux process at the BBB was provided by the observation that brain exposure increased nonproportionally at doses of cabazitaxel greater than 10 μg/mL.47
PK analyses of the CNS have shown that cabazitaxel is able to rapidly penetrate the brains of mice, rats, and dogs following IV infusion. In mice bearing advanced MA16/C tumors grafted subcutaneously, the maximum drug concentration reached within the brain was considerably lower than in subcutaneous tumor tissue or plasma. However, due to the more sustained presence of cabazitaxel in the brain, overall exposure was 2.3-fold and 3.7-fold greater than in tumor tissue and plasma, respectively, at 0–48 hours.33 The relationship between blood and brain exposure was highly consistent across the three animal species, suggesting that a similar relationship may also be seen in humans.33
Studies using in vivo CNS disease models demonstrated greater activity for cabazitaxel than docetaxel. Cabazitaxel had enhanced antitumor activity compared with docetaxel in two human intracranial glioblastoma xenograft models when compared at their respective HNTDs, as shown by greater increases in life span.33 Superior activity with cabazitaxel was observed both at early stages of tumor growth, before the BBB is disrupted, and during advanced stages, consistent with an inherently enhanced ability to penetrate the brain.33
In situ brain perfusion using wild-type mice also demonstrated a two- to threefold greater brain penetration with cabazitaxel than with paclitaxel or docetaxel (unpublished data).47
Based on these results, the potential of cabazitaxel to be an effective agent against CNS tumors should be evaluated further in clinical trials.
Pediatric patients
First-generation taxanes, despite showing activity in preclinical pediatric sarcoma models,48 have achieved limited success in clinical trials in pediatric tumors,49–52 and their efficacy has never been fully established. The potential utility of cabazitaxel in pediatric patients has been investigated using cell-line derived and patient-derived pediatric sarcoma xenograft models. In five of six such models, cabazitaxel induced significantly greater tumor growth inhibition and tumor regression compared with equivalent doses of docetaxel.33 A Phase I dose-escalation study evaluating cabazitaxel in pediatric patients with refractory solid tumors, including tumors of the CNS, is currently ongoing (NCT01751308).53
Combination chemotherapy
Docetaxel or paclitaxel in combination with cisplatin have shown improved efficacy compared with cisplatin-based therapy alone, or with alternative cisplatin- or taxane-based combination regimens. Improved outcomes with such approaches have been documented in multiple tumor types.54–57 Both docetaxel and paclitaxel are indicated, in combination with cisplatin, for the treatment of a variety of solid tumors.4,58
To evaluate preclinically whether cabazitaxel in combination with cisplatin may offer enhanced antitumor activity over monotherapy, this combination was assessed in a murine C51 colon adenocarcinoma xenograft model, which was selected based on its platinum sensitivity.33,59 The combination of cabazitaxel and cisplatin showed greater antitumor activity than either agent alone, although the combination toxicity index of this preclinical combination is low, requiring a reduction in dose of 50%–65% versus the single agent dose to avoid additional toxicity.33 This indicates that reduced dosages could be administered in future if required. Therapeutic synergism was observed regardless of the sequence of administration, with the two agents given simultaneously or sequentially, of either cabazitaxel followed by cisplatin or cisplatin followed by cabazitaxel.33
The preclinical assessment of cabazitaxel with other potentially synergistic chemotherapeutics is also warranted.
Cabazitaxel in combination with cisplatin has been investigated in a Phase I clinical trial in patients with advanced solid tumors (NCT00925743). Stable disease was observed in a high proportion of patients, and the combination had a manageable safety profile consistent with that of a platinum/taxane combination.60 A Phase I/II study that aimed to determine the maximum tolerated dose, safety, and PK of cabazitaxel in combination with gemcitabine, however, was terminated following extensive dose-limiting toxicities (NCT01001221).61
Safety
The safety of cabazitaxel was investigated in a series of nonclinical studies that included general toxicology assessments with a wide variety of administration schedules in mice, rats, and dogs, as well as genotoxicity evaluation using in vitro and in vivo tests, in vivo fertility and embryofetal toxicity studies in rats and/or rabbits, and a comprehensive range of safety pharmacology assays (Sanofi, data on file, 2010).
General toxicology evaluation comprised single-dose and 5-day studies, single-cycle (weekly administration) and 4-week (daily administration) studies, and 5-, 10-, and 13-cycle studies entailing one administration every 3 weeks (Sanofi, data on file, 2010). The main toxicity parameters of cabazitaxel in general toxicology studies are summarized in Table 5 (Sanofi, data on file, 2010).
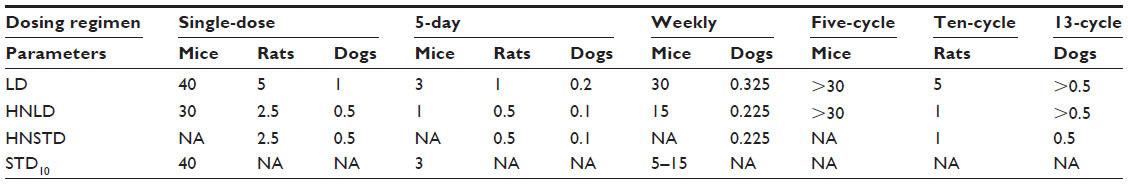 | Table 5 Cabazitaxel preclinical toxicology data summary: main toxicity parameters (mg/kg/day) in mice, rats, and dogs |
As expected for an antimitotic agent, cabazitaxel predominantly affected tissues with a high cell turnover in rats and dogs. The no observable effect levels for the main target organs are presented in Table 6. Microscopic alterations were observed in the bone marrow (resulting in decreased circulating white blood cell count), lymphoid system, gastrointestinal tract, and male reproductive system. In multiple cycle assessments in rats, alopecia correlating with cell degeneration was also seen. Most of these changes were reversible and were considered compatible with 3-weekly administration (Sanofi, data on file, 2010). The findings are consistent with cabazitaxel’s clinical adverse event profile, which is characterized primarily by hematologic disorders such as neutropenia and gastrointestinal disorders such as diarrhea, nausea, and vomiting.11 Organs with lower epithelial tissue turnover were affected in a few instances. In particular, an increased incidence of mitotic figures or single cell necrosis was observed in the liver, adrenal gland, uterus, and eyes (Sanofi, data on file, 2010). The effects observed with cabazitaxel on tissues with high and low cell turnover resembled those reported for other taxane anticancer drugs (Sanofi, data on file, 2010).
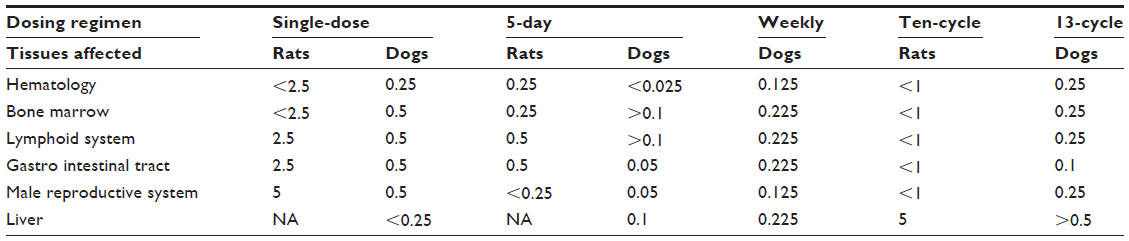 | Table 6 Cabazitaxel preclinical toxicology data summary: NOEL (mg/kg/day) for main target organs in rat and dog toxicity studies |
Adverse reactions with possible clinical relevance included arteriolar/periarteriolar necrosis in the liver, bile duct hyperplasia, and hepatocellular necrosis (Sanofi, data on file, 2010). In clinical studies with cabazitaxel, transient increases in bilirubin and transaminase levels as well as liver injuries have been occasionally reported (Sanofi, data on file, 2010). In the light of these observations, a potential impact on the liver is being investigated further in an ongoing Phase I trial of cabazitaxel in patients with advanced solid tumors and varying degrees of hepatic impairment (NCT01140607). (Sanofi, data on file, 2013).
Similar to what has been reported with other taxanes,25,62,63 peripheral neurotoxicity was observed in rodent models following treatment with cabazitaxel (Table 7). Sciatic nerve degeneration occurred in mice following treatment with a variety of dosing regimens ranging from single-dose to five-cycle administration, and in rats in the single-dose and ten-cycle studies. In mice, these changes were not reversible at 10 or 20 weeks after single administration (Sanofi, data on file, 2010). In the clinical setting, cases of peripheral neuropathy, peripheral sensory neuropathy (eg, paraesthesias, dysesthesias), and peripheral motor neuropathy have been observed in patients receiving cabazitaxel.11 Cross-study comparisons suggest, however, that the incidence and severity of peripheral neuropathy may be lower with cabazitaxel compared with other taxanes.10,64–69
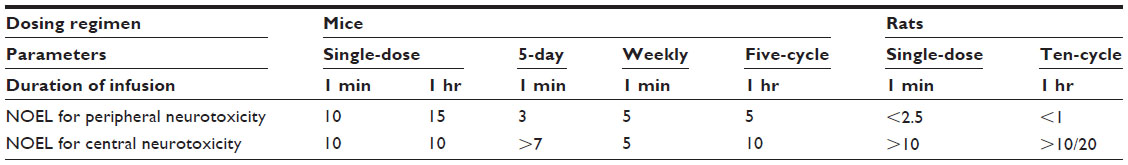 | Table 7 Cabazitaxel preclinical toxicology data summary: NOEL (mg/kg/day) for central and peripheral neurotoxicity in mice and rats |
Central neurotoxicity characterized by degenerative changes to the brain and cervical spinal cord has also been reported in mice following cabazitaxel treatment (Table 7), but was not observed in rats or dogs (Sanofi, data on file, 2010). In radiolabeling studies, a good correlation between the localization of brain lesions (vacuoles) and radioactivity levels was observed, with both concentrated around the ventricles (Figure 5), suggesting that these effects were related to cabazitaxel treatment (Sanofi, data on file, 2010). Similar degenerative changes were observed in the brains of mice treated with IV paclitaxel.70
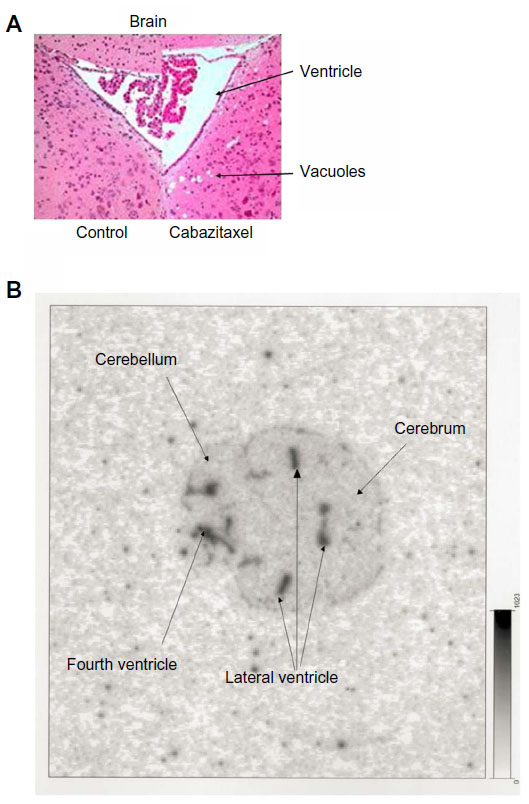 | Figure 5 Mouse brain histopathology and autoradiography. |
Partially reversible eye disorders, characterized by mild subcapsular lens fiber swelling or degeneration, were observed in rats following administration of the highest dose level of 20/10 mg/kg in the ten-cycle toxicity study, but not at lower doses. Such adverse events have not been noted in clinical trials, but may have some relevance to clinical use and are under active surveillance (Sanofi, data on file, 2010).11
In investigations of genotoxicity, cabazitaxel did not induce mutations in the bacterial reverse mutation (Ames) test. Other genotoxicity observations included an increased number of polyploid cells in the in vitro chromosome aberration test, and an increase in micronuclei in the in vivo rat bone marrow micronucleus test. These were consistent with the pharmacologic activity of a microtubule depolymerization inhibitor, and have been observed previously with paclitaxel (Sanofi, data on file, 2010).71
Cabazitaxel did not affect the mating performance or fertility of male or female rats, but did result in degeneration of the male reproductive system after repeated dosing. Administration during early gestation in pregnant rats led to fetal death and low fetal weight associated with a delay in skeletal ossification. Drug exposures in these studies were lower than those in humans receiving clinically relevant doses of cabazitaxel. Thus, cabazitaxel is not recommended for use in pregnant women or those of childbearing age who are not using contraception (Sanofi, data on file, 2010).11
In a series of safety pharmacology assays, no physiologically relevant effects were observed in the CNS, respiratory, gastrointestinal, renal, and cardiovascular systems after IV administration of cabazitaxel (Sanofi, data on file, 2010). Overall, preclinical safety signals of cabazitaxel were consistent with the previously reported safety profiles of paclitaxel and docetaxel.25,62,63 In terms of dose levels, the highest non-lethal doses observed in the single-dose toxicity studies conducted in mice, rats, and dogs with cabazitaxel and docetaxel were approximately 3- to 4-times lower with cabazitaxel than with docetaxel (Table 8) (Sanofi, data on file, 2013). This aligns perfectly with the recommended clinical dose of cabazitaxel (25 mg/m2), which is approximately 3- to 4-times lower than that of docetaxel (75–100 mg/m2).
 | Table 8 Comparison of HNLDs (mg/kg) in single-dose toxicity studies in mice, rats, and dogs with cabazitaxel and docetaxel |
Clinical development
The safety and efficacy of cabazitaxel have been evaluated in a number of clinical studies,10,72–76 including the pivotal randomized Phase III TROPIC study that led to the approval of cabazitaxel for the treatment of patients with mCRPC who had previously received docetaxel therapy.10–12
Several Phase I/II clinical studies were conducted in patients with advanced solid tumors who had previously received chemotherapy and in patients with metastatic breast cancer previously treated with taxane therapy.10,72–75,77 Results indicated that cabazitaxel had an acceptable safety profile and possible antitumor activity in patients with prior exposure to taxanes. These clinical observations were consistent with preclinical studies that showed the marked antiproliferative activity of cabazitaxel in a wide range of docetaxel-resistant cell lines and tumor xenografts.24
In two Phase I dose-finding studies, cabazitaxel was administered to patients with advanced solid tumors as a 1-hour infusion every 3 weeks.72,74 In both of these studies, the dose-limiting toxicities were hematologic toxicity (primarily neutropenia) and diarrhea, and similar cabazitaxel dosing was recommended for subsequent clinical investigations (20 mg/m2 and 25 mg/m2).
The clinical PK profile of cabazitaxel is similar to that of docetaxel, displaying linear PK over tested dose ranges and a triphasic elimination profile.9,62,72,74 A population PK model for cabazitaxel was developed using pooled data from 170 patients who participated in five Phase I–III clinical trials,78 which revealed a PK profile comprising rapid initial and intermediate phases (population half-lives of 4.4 minutes and 1.6 hours, respectively) followed by a long terminal phase (population half-life = 95.1 hours).9,10,72–75 Compared with docetaxel, cabazitaxel demonstrated a longer terminal half-life, higher plasma clearance (population PK estimate 48.5 L/h; 26.4 L/h/m2 for a median body surface area [BSA] of 1.84 m2), and greater mean volume of distribution at steady state (population PK estimate 4,870 L; 2,640 L/m2 for a median BSA of 1.84 m2).9,78
Population PK modeling has shown that a three-compartment structural kinetic model with first-order elimination from the central compartment best fits the concentration–time profile of both cabazitaxel78 and docetaxel.79 However, PK modeling (using estimates of population intercompartmental constants) showed the presence of a deeper peripheral compartment for cabazitaxel than for docetaxel. This deeper compartment is in slow equilibrium with the central compartment and was the main contributor to the very large steady-state volume of distribution and very long elimination half-life of cabazitaxel. These data suggest that cabazitaxel is eliminated more slowly than docetaxel. Mass balance studies in humans suggested that excretion is slightly slower for cabazitaxel (76% of the administered dose of 14C was recovered in feces, with only approximately 4% recovered in the urine over 2 weeks)34 than for docetaxel (80% of the administered dose of 14C was excreted in the feces, with approximately 5% recovered in the urine over 7 days).80 However, the reported slower elimination/excretion profile observed for cabazitaxel has no appreciable impact on observed adverse events, as there is no evidence of delayed or cumulative toxicity with cabazitaxel administration.
The population data showed that BSA was the only evaluated factor that impacted cabazitaxel PK, with no need for dose alteration in special populations (based on age, sex, race, and mild-to-moderate renal impairment).9,78 Furthermore, concurrent administration of cabazitaxel with prednisone or prednisolone (unpublished data), or with capecitabine, did not impact its PK profile.9,76
Phase III TROPIC study
Based on the promising results reported from early-phase clinical studies, including antitumor activity in docetaxel-refractory mCRPC, cabazitaxel was compared with mitoxantrone in a randomized open-label Phase III trial (TROPIC; NCT00417079) in 755 patients with mCRPC that had progressed on previous docetaxel-based therapy.10,74,75 Patients were randomized to cabazitaxel IV 25 mg/m2 (n=378) or mitoxantrone 12 mg/m2 (n=377) administered every 3 weeks, both in combination with oral prednisone (or prednisolone) 10 mg daily.10
Patients receiving cabazitaxel plus prednisone/prednisolone had a significantly longer median overall survival compared with mitoxantrone (15.1 months versus 12.7 months), corresponding to a 30% reduction in the risk of death (hazard ratio 0.70, 95% confidence interval 0.59–0.83; P<0.0001);10 this was the first demonstration of a survival advantage for any agent in the second-line setting. The cabazitaxel arm also experienced longer median progression-free survival (2.8 months versus 1.4 months; hazard ratio 0.74, 95% confidence interval 0.64–0.86; P<0.0001) and a higher rate of objective tumor response (14.4% versus 4.4%; P=0.0005) and prostate-specific antigen response (39.2% versus 17.8%; P=0.0002).
The most common adverse events associated with cabazitaxel were hematologic, with grade ≥3 neutropenia, leukopenia, and anemia occurring in 82%, 68%, and 11% of patients, respectively. Diarrhea (6%) was the most frequent nonhematologic grade ≥3 adverse event.10 Peripheral neuropathy, an adverse event observed in preclinical studies of taxanes in rodents as well as in clinical studies of other taxanes (Sanofi, data on file, 2010),25,62,63 occurred in 14% of patients receiving cabazitaxel; however, grade 3 peripheral neuropathy was rare, occurring in just three patients (1%).10 This compares favorably with first-generation taxanes, with reported incidence of grade 3/4 sensory neuropathy of 0%–7% for docetaxel and 2%–33% for paclitaxel in Phase III clinical studies in a number of tumor types.64–69 If neuropathy develops, treatment should be delayed until the improvement of symptoms, and dose reduction is recommended for persistent cases of grade ≥2 severity.11 More patients receiving cabazitaxel died within 30 days of the last infusion compared with mitoxantrone (5% versus 2%), and patients in this arm were more likely to have a dose reduction (12% versus 4%) or treatment delay (28% versus 15%).10 The most frequent cause of treatment-related death in the cabazitaxel arm was neutropenia and associated complications,10 and, as such, proactive management of hematologic adverse events is recommended.9,11,12 Prophylactic use of granulocyte colony-stimulating factor (G-CSF) was not permitted in the TROPIC study, but may also be beneficial for the management of neutropenia.9,11,12 Indeed, in global compassionate-use and early-access programs for cabazitaxel, in which G-CSF use was recommended as per American Society of Clinical Oncology (ASCO) guidelines,81 a similar incidence of neutropenic complications in patients with prophylactic G-CSF use and in patients without G-CSF use at baseline suggests that adequate risk mitigation of such hematological adverse events can be achieved with G-CSF in patients at risk of developing neutropenia.82
Based on the positive results from this study, cabazitaxel was approved for use in combination with prednisone by the US Food and Drugs Administration, European Medicines Agency, and other national health authorities for the treatment of patients with metastatic hormone-refractory prostate cancer previously treated with a docetaxel-containing treatment regimen.11,12
Conclusion
Cabazitaxel may have clinical potential in a broader range of patient populations compared with first-generation taxanes, as shown by demonstrated in vivo antitumor activity in tumors with innate or acquired resistance to docetaxel, CNS tumors, and pediatric tumors. In clinical studies to date, cabazitaxel confirmed its antitumor activity and demonstrated a safety profile that can be managed well with appropriate therapy, and this has been confirmed in real-world safety studies. Results from the Phase III TROPIC trial led to the approval of cabazitaxel in patients with mCRPC who have received prior docetaxel-based therapy. As such, cabazitaxel is one of the increasing number of agents available for the treatment of mCRPC. While the optimal sequence of agents is yet to be definitively determined, given the favorable efficacy and tolerability profile of cabazitaxel, it is likely to continue to play a considerable role in the mCRPC treatment paradigm.
Cabazitaxel is also under clinical investigation in a number of studies and indications, including in the Phase III PROSELICA trial, which is evaluating a reduced dose of cabazitaxel (20 mg/m2) compared with the standard dose 25 mg/m2 (NCT01308580), and the Phase III FIRSTANA trial comparing cabazitaxel with docetaxel as first-line therapy for patients with mCRPC (NCT01308567).
Acknowledgments
The authors gratefully acknowledge Drs M-C Bissery, L Calvet, C Combeau, A Commerçon, F Lavelle, P Lejeune, JF Riou, M-L Risse, SS Sidhu, P Detilleux, G Sanderink, and D Weitz for their contribution to the preclinical development and analysis of cabazitaxel, and Drs L Bourre, D Nicolle, M-E Legrier, V Yvonnet, J Charpentier, M-F Poupon, C Geffriaud-Ricouard, S Oudard, and JG Judde for their contribution to the HID28 prostate cancer xenograft study. The authors received editorial support from Ben Caldwell of MediTech Media, funded by Sanofi.
Disclosure
All authors are employees of Sanofi and hold Sanofi stock. The authors report no other conflicts of interest in this work.
References
No authors listed. FDA approves treatment IND protocol for taxol. Clin Pharm. 1992;11:912. | |
Label and Approval History – Taxol [webpage on the Internet]. Silver Spring: US Food and Drug Administration; 2011. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=020262&DrugName=TAXOL&ActiveIngred=PACLITAXEL&SponsorApplicant=HQ%20SPCLT%20PHARMA&ProductMktStatus=1&goto=Search.Label_ApprovalHistory. Accessed July 29, 2014. | |
Sanofi U.S.LLC. TAXOTERE® (Docetaxel) Injection Concentrate, Intravenous Infusion (IV) Prescribing Information 2013; USA. Bridgewater: Sanofi U.S.LLC; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/020449s071lbl.pdf. Accessed July 29, 2014. | |
Mead Johnson Oncology Products. TAXOL® (Paclitaxel) Injection. Princeton: Bristol-Meyers Squibb Company; 2011. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf. Accessed July 29, 2014. | |
Chien AJ, Moasser MM. Cellular mechanisms of resistance to anthracyclines and taxanes in cancer: intrinsic and acquired. Semin Oncol. 2008;35:S1–S14. | |
Fojo AT, Menefee M. Microtubule targeting agents: basic mechanisms of multidrug resistance (MDR). Semin Oncol. 2005;32:S3–S8. | |
Perez EA. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009;8:2086–2095. | |
O’Neill AJ, Prencipe M, Dowling C, et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol Cancer. 2011;10:126. | |
Bouchard H, Semiond D, Risse ML, Vrignaud P. Novel taxanes: Cabazitaxel case study. In: Fischer J, Ganellin CR, Rotella DP, editors. Analogue-Based Drug Discovery III, first edition.Weinheim, Germany. Wiley-VCH Verlag GmbH & Co. KGaA, 2013;13:319–341. | |
de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–1154. | |
Sanofi. JEVTANA® (Cabazitaxel) Injection, Summary of Product Characteristics. Bridgewater: Sanofi; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002018/WC500104764.pdf. Accessed July 29, 2014. | |
Sanofi U.S.LLC. JEVTANA® (Cabazitaxel) Injection, Prescribing Information, FDA. Bridgewater: Sanofi U.S.LLC; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/201023s003lbl.pdf. Accessed July 29, 2014. | |
Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anticancer Agents. 2002;2:1–17. | |
Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. | |
Nogales E. Structural insight into microtubule function. Annu Rev Biophys Biomol Struct. 2001;30:397–420. | |
Giannakakou P, Poy G, Zhan Z, et al. Paclitaxel selects for mutant or pseudo-null p53 in drug resistance associated with tubulin mutations in human cancer. Oncogene. 2000;19:3078–3085. | |
Kavallaris M, Kuo DY, Burkhart CA, et al. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest. 1997;100:1282–1293. | |
McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008;1785:96–132. | |
Modi S, DiGiovanna MP, Lu Z, et al. Phosphorylated/activated HER2 as a marker of clinical resistance to single agent taxane chemotherapy for metastatic breast cancer. Cancer Invest. 2005;23:483–487. | |
ABCB1 ATP-binding cassette, sub-family B (MDR/TAP), member 1 [webpage on the Internet]. Bethesda: NCBI; 2014. Available from: http://www.ncbi.nlm.nih.gov/gene/5243. | |
Nightingale G, Ryu J. Cabazitaxel (jevtana): a novel agent for metastatic castration-resistant prostate cancer. P T. 2012;37:440–448. | |
Horwitz SB, Cohen D, Rao S, Ringel I, Shen HJ, Yang CP. Taxol: mechanisms of action and resistance. J Natl Cancer Inst Monogr. 1993;(15):55–61. | |
Yin S, Bhattacharya R, Cabral F. Human mutations that confer paclitaxel resistance. Mol Cancer Ther. 2010;9:327–335. | |
Vrignaud P, Sémiond D, Lejeune P, et al. Preclinical antitumor activity of cabazitaxel, a semi-synthetic taxane active in taxane-resistant tumors. Clin Cancer Res. 2013;19:2973–2983. | |
Lavelle F, Bissery MC, Combeau C, Riou JF, Vrignaud P, André S. Preclinical evaluation of docetaxel (Taxotere). Semin Oncol. 1995;22:3–16. | |
Denis JN, Greene AE, Guenard D, Gueritte-Voegelein F, Mangatal L, Potier P. Highly efficient, practical approach to natural taxol. J Am Chem Soc. 1988;110:5917–5919. | |
Holton RA, Somoza C, Kim H-B, et al. First total synthesis of Taxol. 1. Functionalization of the B ring. J Am Chem Soc. 1994;116:1597–1598. | |
Sanofi U.S.LLC. Taxotere® Material Safety Data Sheet. Bridgewater: Sanofi U.S.LLC; 2010. Available from: http://bdipharma.com/MSDS/Sanofi-Aventis/Taxotere%208-10.pdf. Accessed July 29, 2014. | |
Roche VF. Cancer and chemotherapy. In: Lemke TL, Williams DA, Roche VF, Zito SW, editors. Foye’s Principles of Medicinal Chemistry, seventh edition. Baltimore, MD: Lippincott Williams, Wilkins, 2013;37:1199–1266. | |
Montaudon D, Vrignaud P, Londos-Gagliardi D, Robert J. Fluorescence anisotropy of cell membranes of doxorubicin-sensitive and -resistant rodent tumoral cells. Cancer Res. 1986;46:5602–5605. | |
Azarenko O, Smiyun G, Mah J, Wilson L, Jordan MA. Antiproliferative mechanism of action of the novel taxane cabazitaxel as compared with the parent compound docetaxel in MCF7 breast cancer cells. Mol Cancer Ther. 2014;13:2092–2103. | |
Paller CJ, Antonarakis ES. Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des Devel Ther. 2011;5:117–124. | |
Semiond D, Sidhu SS, Bissery M-C, Vrignaud P. Can taxanes provide benefit in patients with CNS tumors and in pediatric patients with tumors? An update on the preclinical development of cabazitaxel. Cancer Chemother Pharmacol. 2013;72:515–528. | |
Ridoux L, Semiond D, Vincent C, et al. A Phase I open-label study investigating the disposition of [14C]-cabazitaxel in patients with advanced solid tumors. Anticancer Drugs. Submitted March 2014. | |
Darshan MS, Loftus MS, Thadani-Mulero M, et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011;71:6019–6029. | |
Gan L, Chen S, Wang Y, et al. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009;69:8386–8394. | |
Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010;70:7992–8002. | |
Bourre L, Nicolle D, Legrier M-E, et al. Evaluation of the response to cabazitaxel of a docetaxel-responsive hormone-refractory prostate tumor xenograft model (HID28). In: American Society of Clinical Oncology Annual Meeting; June 1–5, 2012; Chicago, Illinois. Abstract e15161. | |
Chamberlain MC, Kormanik P. Salvage chemotherapy with paclitaxel for recurrent primary brain tumors. J Clin Oncol. 1995;13:2066–2071. | |
Chamberlain MC, Kormanik P. Salvage chemotherapy with taxol for recurrent anaplastic astrocytomas. J Neurooncol. 1999;43:71–78. | |
Forsyth P, Cairncross G, Stewart D, Goodyear M, Wainman N, Eisenhauer E. Phase II trial of docetaxel in patients with recurrent malignant glioma: a study of the National Cancer Institute of Canada Clinical Trials Group. Invest New Drugs. 1996;14:203–206. | |
Hurwitz CA, Strauss LC, Kepner J, et al. Paclitaxel for the treatment of progressive or recurrent childhood brain tumors: a pediatric oncology phase II study. J Pediatr Hematol Oncol. 2001;23:277–281. | |
Prados MD, Schold SC, Spence AM, et al. Phase II study of paclitaxel in patients with recurrent malignant glioma. J Clin Oncol. 1996;14:2316–2321. | |
Deeken JF, Loscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13:1663–1674. | |
Fellner S, Bauer B, Miller DS, et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J Clin Invest. 2002;110:1309–1318. | |
Kemper EM, Verheij M, Boogerd W, Beijnen JH, van Tellingen O. Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. Eur J Cancer. 2004;40:1269–1274. | |
Cisternino S, Bourasset F, Archimbaud Y, Sémiond D, Sanderink G, Scherrmann JM. Nonlinear accumulation in the brain of the new taxoid TXD258 following saturation of P-glycoprotein at the blood-brain barrier in mice and rats. Br J Pharmacol. 2003;138:1367–1375. | |
Izbicka E, Campos D, Marty J, Carrizales G, Mangold G, Tolcher A. Molecular determinants of differential sensitivity to docetaxel and paclitaxel in human pediatric cancer models. Anticancer Res. 2006;26:1983–1988. | |
Blaney SM, Seibel NL, O’Brien M, et al. Phase I trial of docetaxel administered as a 1-hour infusion in children with refractory solid tumors: a collaborative pediatric branch, National Cancer Institute and Children’s Cancer Group trial. J Clin Oncol. 1997;15:1538–1543. | |
Hurwitz CA, Relling MV, Weitman SD, et al. Phase I trial of paclitaxel in children with refractory solid tumors: a Pediatric Oncology Group Study. J Clin Oncol. 1993;11:2324–2329. | |
Mora J, Cruz CO, Parareda A, de Torres C. Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol. 2009;31:723–729. | |
Zwerdling T, Krailo M, Monteleone P, et al. Phase II investigation of docetaxel in pediatric patients with recurrent solid tumors: a report from the Children’s Oncology Group. Cancer. 2006;106:1821–1828. | |
Clinicaltrials.gov [webpage on the Internet]. Cabazitaxel in pediatric and central nervous system tumors. Bethesda: National Institutes of Health; 2014. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01751308. Accessed July 29, 2014. | |
Lorch JH, Goloubeva O, Haddad RI, et al. Induction chemotherapy with cisplatin and fluorouracil alone or in combination with docetaxel in locally advanced squamous-cell cancer of the head and neck: long-term results of the TAX 324 randomised phase 3 trial. Lancet Oncol. 2011;12:153–159. | |
Van Cutsem E, Moiseyenko VM, Tjulandin S, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol. 2006;24:4991–4997. | |
Piccart MJ, Bertelsen K, James K, et al. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: three-year results. J Natl Cancer Inst. 2000;92:699–708. | |
Rosell R, Gatzemeier U, Betticher DC, et al. Phase III randomised trial comparing paclitaxel/carboplatin with paclitaxel/cisplatin in patients with advanced non-small-cell lung cancer: a cooperative multinational trial. Ann Oncol. 2002;13:1539–1549. | |
Sanofi U.S.LLC. TAXOTERE® (Docetaxel) Injection Concentrate, Intravenous Infusion (IV) Prescribing Information. Bridgewater: Sanofi U.S.LLC; 2010. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020449s059lbl.pdf. Accessed May 2014. | |
Vrignaud P. Therapeutic synergism of cabazitaxel in combination with cisplatin in tumor-bearing mice. In: American Association for Cancer Research Annual Meeting; April 2–6, 2011; Orlando, Florida. Abstract 2522. | |
Lockhart AC, Sundaram S, Sarantopoulos J, et al. Phase I trial of cabazitaxel plus cisplatin in patients with advanced solid tumors. Poster 162 presented at: Genitourinary Cancers Symposium; February 2–4, 2012; San Francisco, California. | |
Rixe O, Puzanov I, LoRusso PM, et al. Dose-escalation Phase I study of cabazitaxel (Cbz) + gemcitabine (Gem) in patients (pts) with metastatic or unresectable advanced solid malignancy. Poster 497P presented at: European Society of Medical Oncology Congress; Sept 28 – Oct 2, 2012; Vienna. | |
Bissery MC, Nohynek G, Sanderink GJ, Lavelle F. Docetaxel (Taxotere): a review of preclinical and clinical experience. Part I: Preclinical experience. Anticancer Drugs. 1995;6:339–8. | |
Rowinsky EK, Cazenave LA, Donehower RC. Taxol: a novel investigational antimicrotubule agent. J Natl Cancer Inst. 1990;82:1247–1259. | |
Kudoh S, Takeda K, Nakagawa K, et al. Phase III study of docetaxel compared with vinorelbine in elderly patients with advanced non-small-cell lung cancer: results of the West Japan Thoracic Oncology Group Trial (WJTOG 9904). J Clin Oncol. 2006;24:3657–3663. | |
Jones SE, Erban J, Overmoyer B, et al. Randomized phase III study of docetaxel compared with paclitaxel in metastatic breast cancer. J Clin Oncol. 2005;23:5542–5551. | |
Fossella FV, DeVore R, Kerr RN, et al. Randomized phase III trial of docetaxel versus vinorelbine or ifosfamide in patients with advanced non-small-cell lung cancer previously treated with platinum-containing chemotherapy regimens. The TAX 320 Non-Small Cell Lung Cancer Study Group. J Clin Oncol. 2000;18:2354–2362. | |
Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. | |
Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. | |
Winer EP, Berry DA, Woolf S, et al. Failure of higher-dose paclitaxel to improve outcome in patients with metastatic breast cancer: cancer and leukemia group B trial 9342. J Clin Oncol. 2004;22:2061–2068. | |
Gokhale PC, Newsome JT, Dritschilo A, et al. Toxicity, histopathologic and pharmacokinetic evaluation of liposome-encapsulated paclitaxel and Taxol in mice and rabbits. Poster 2759 presented at: American Association for Cancer Research Annual Meeting; April 10–14, 1999; Philadelphia, Pennsylvania. | |
Tinwell H, Ashby J. Genetic toxicity and potential carcinogenicity of taxol. Carcinogenesis. 1994;15:1499–1501. | |
Diéras V, Lortholary A, Laurence V, et al. Cabazitaxel in patients with advanced solid tumours: Results of a Phase I and pharmacokinetic study. Eur J Cancer. 2013;49:25–34. | |
Fumoleau P, Trigo JM, Isambert N, Sémiond D, Gupta S, Campone M. Phase I dose-finding study of cabazitaxel administered weekly in patients with advanced solid tumours. BMC Cancer. 2013;13:460. | |
Mita AC, Denis LJ, Rowinsky EK, et al. Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res. 2009;15:723–730. | |
Pivot X, Koralewski P, Hidalgo JL, et al. A multicenter phase II study of XRP6258 administered as a 1-h iv infusion every 3 weeks in taxane-resistant metastatic breast cancer patients. Ann Oncol. 2008;19:1547–1552. | |
Villanueva C, Awada A, Campone M, et al. A multicentre dose-escalating study of cabazitaxel (XRP6258) in combination with capecitabine in patients with metastatic breast cancer progressing after anthracycline and taxane treatment: a phase I/II study. Eur J Cancer. 2011;47:1037–1045. | |
Villanueva C, Awada A, Campone M, et al. A multicentre dose-escalating study of cabazitaxel (XRP6258) in combination with capecitabine in patients with metastatic breast cancer progressing after anthracycline and taxane treatment: a phase I/II study. Eur J Cancer. 2011;47(7):1037–1045. | |
Ferron GM, Dai Y, Semiond D. Population pharmacokinetics of cabazitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:681–692. | |
Bruno R, Vivier N, Vergniol JC, De Phillips SL, Montay G, Sheiner LB. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm. 1996;24:153–172. | |
Clarke SJ, Rivory LP. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinet. 1999;36:99–114. | |
Smith TJ, Khatcheressian J, Lyman GH, et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol. 2006;24:3187–3205. | |
Malik Z, Di Lorenzo G, Ardavanis A, et al. Updated safety results from a cohort compassionate-use programme (CUP) and early access programme (EAP) with cabazitaxel (Cbz) plus prednisone (P; Cbz + P) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) previously treated with docetaxel (D). Eur J Cancer. 2013;49(Supplement 2):S1–S1028:abstract 2902. |
Supplementary material
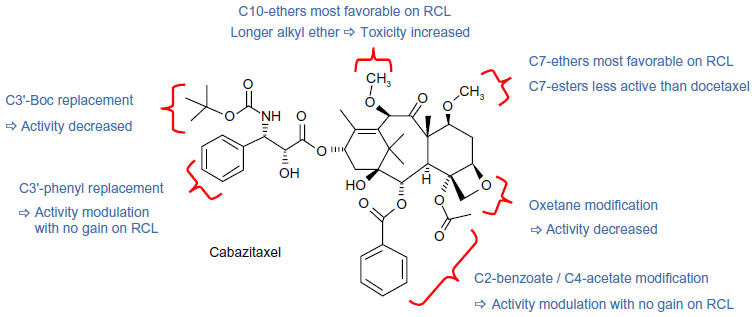 | Figure S1 Structure–activity relationships for cabazitaxel. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.