Back to Journals » International Journal of Nanomedicine » Volume 19
Preclinical Activity of Two Paclitaxel Nanoparticle Formulations After Intraperitoneal Administration in Ovarian Cancer Murine Xenografts
Authors Demuytere J ![]() , Carlier C, Van de Sande L, Hoorens A
, Carlier C, Van de Sande L, Hoorens A ![]() , De Clercq K, Giordano S, Morosi L, Matteo C, Zucchetti M, Davoli E, Van Dorpe J
, De Clercq K, Giordano S, Morosi L, Matteo C, Zucchetti M, Davoli E, Van Dorpe J ![]() , Vervaet C, Ceelen W
, Vervaet C, Ceelen W
Received 12 July 2023
Accepted for publication 30 November 2023
Published 17 January 2024 Volume 2024:19 Pages 429—440
DOI https://doi.org/10.2147/IJN.S424045
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. RDK Misra
Jesse Demuytere,1,* Charlotte Carlier,1,* Leen Van de Sande,1 Anne Hoorens,2 Kaat De Clercq,3 Silvia Giordano,4 Lavinia Morosi,4 Cristina Matteo,4 Massimo Zucchetti,4 Enrico Davoli,4 Jo Van Dorpe,2 Chris Vervaet,3 Wim Ceelen1
1Department of GI Surgery, Ghent University Hospital, and Cancer Research Institute Ghent (CRIG), Ghent, Belgium; 2Department of Pathology, Ghent University Hospital, Ghent, Belgium; 3Laboratory of Pharmaceutical Technology, Ghent University, Ghent, Belgium; 4Department of Environmental Health Sciences, Istituto di Ricerche Farmacologiche Mario Negri - IRCCS, Milano, Italy
*These authors contributed equally to this work
Correspondence: Wim Ceelen, Department of GI Surgery, Ghent University Hospital, Route 1275, Corneel Heymanslaan 10, Ghent, B-9000, Belgium, Tel +32 9 332 6251, Email [email protected]
Background: Epithelial ovarian cancer is associated with high mortality due to diagnosis at later stages associated with peritoneal involvement. Several trials have evaluated the effect of intraperitoneal treatment. In this preclinical study, we report the efficacy, pharmacokinetics and pharmacodynamics of intraperitoneal treatment with two approved nanomolecular formulations of paclitaxel (nab-PTX and mic-PTX) in a murine ovarian cancer xenograft model.
Methods: IC50 was determined in vitro on three ovarian cancer cell lines (OVCAR-3, SK-OV-3 and SK-OV-3-Luc IP1). EOC xenografts were achieved using a modified subperitoneal implantation technique. Drug treatment was initiated 2 weeks after engraftment, and tumor volume and survival were assessed. Pharmacokinetics and drug distribution effects were assessed using UHPLC-MS/MS and MALDI imaging mass spectrometry, respectively. Pharmacodynamic effects were analyzed using immunohistochemistry and transmission electron microscopy using standard protocols.
Results: We demonstrated sub-micromolar IC50 concentrations for both formulations on three EOC cancer cell lines in vitro. Furthermore, IP administration of nab-PTX or mic-PTX lead to more than 2-fold longer survival compared to a control treatment of IP saline administration (30 days in controls, 66 days in nab-PTX treated animals, and 76 days in mic-PTX animals, respectively). We observed higher tissue uptake of drug following nab-PTX administration when compared to mic-PTX, with highest uptake after 4 hours post-treatment, and confirmed this lower uptake of mic-PTX using HPLC on digested tumor samples. Furthermore, apoptosis was not increased in tumor implants up to 24h post-treatment.
Conclusion: Intraperitoneal administration of both nab-PTX and mic-PTX results in a significant anticancer efficacy and survival benefit in a mouse OC xenograft model.
Keywords: ovarian, peritoneal, paclitaxel, nanomedicine
Introduction
Epithelial ovarian cancer (EOC) is the 8th most common cause of cancer death worldwide, accounting for an estimate of over 150,000 deaths per year. This particularly high mortality is mainly attributed to diagnosis at an advanced (eg FIGO III or IV) stage, with disease commonly involving the peritoneum.1,2
Current standard treatment for advanced EOC consists of both complete cytoreductive surgery and systemic paclitaxel and carboplatin-based chemotherapy.3 Several randomized clinical trials have aimed to evaluate the effect of intraperitoneal (IP) administration of chemotherapy, particularly the platinum component, with positive results.4,5 However, the future role of IP therapy remains to be established.6
The peritoneal cavity is ideally situated to allow for locoregional therapies, due to the ease by which it can be accessed. Additionally, the peritoneal-plasma barrier confers a favorable pharmacokinetic profile, including slower clearance, and higher possible local doses of chemotherapeutic drugs.7 Other potential advantages of IP therapy over systemic therapy include the possibility of an increased capacity to reach small lesions with less-developed vasculature, and lower systemic effects.8,9 Paclitaxel (PTX), a member of the taxane group, is particularly suited for IP administration on a theoretical basis, due to a described peritoneal/plasma ratio of 1000, and significant hepatic first-pass metabolism.10
PTX is FDA approved for the systemic treatment of several solid cancers, including ovarian, breast and lung cancer.11 Due to its high hydrophobicity, it is solubilized in a mixture (1:1) of Cremophor®EL (CrEl) and dehydrated ethanol, known as solvent-based PTX (sb-PTX).11 Paclitaxel administration leads to the assembly of tubulins into dysfunctional microtubules, causing chromosomal missegregation at clinically observed concentrations, leading to mitotic arrest and apoptosis in rapidly proliferating tumor cells.11–14
A major disadvantage of IV sb-PTX therapy is the wide range of side-effects and pharmacokinetic interactions associated with the use of CrEl as a vehicle, as dose-independent effects leading to toxicity have been widely reported.15,16 In response to this, nanoparticle-bound PTX formulations were developed to provide a more favorable side-effect profile.7,9
Nanoparticle-based drug delivery mechanisms are of particular interest in IP therapy, as they confer the possibility of prolonged peritoneal residence time, might allow dose intensification by lowering systemic absorption thereby decreasing systemic toxicity, and allow for tailored approaches to locoregional tumor treatments.9 However, only a few nanocarriers have been approved by the FDA for intravenous (IV) use in cancer treatment, including liposomal doxorubicin (Doxil®) and albumin-bound PTX (Abraxane®). None of these agents have been approved specifically for IP administration.17
Two nanoscale PTX formulations are currently approved for clinical use: nanoparticle albumin-bound PTX (nab-PTX or Abraxane®) is an albumin-bound 130 nm particular PTX formulation which is FDA approved for the IV treatment of advanced non‐small cell lung cancer metastatic breast cancer, and metastatic pancreatic cancer, while micellar PTX (mic-PTX, Genexol®-PM), a biodegradable 20–50 nm polymeric micellar PTX formulation, is approved in South Korea for the treatment of breast cancer and small cell lung cancer, and is currently the subject of several Phase I and II trials in solid tumors.17,18 Several preclinical studies have compared these nanomolecular formulations to their parent drug, sb-PTX, for intraperitoneal use, and demonstrated increased anti-tumor effects, pharmacological advantages, and better tissue penetration.19,20 Given these findings, there is a clear rationale to further investigate different nanomolecular drug formulations of PTX for use in IP therapy.
In this preclinical study, we report in vitro and in vivo antitumor efficacy, intratumoral PTX penetration, and pharmacodynamic effects of nab-PTX and mic-PTX intraperitoneally, in an EOC xenograft model.
Materials and Methods
Cell Culture
The human EOC cell lines OVCAR-3 (HTB-161) and SK-OV-3 (HTB-77) were purchased from ATCC (Wesel, Germany) and cultured at 37°C in a 5% CO2-containing humidified atmosphere. The SK-OV-3-Luc IP1 cell line, a more aggressive, luciferase positive OC cell line compared to the SK-OV-3 cell line, was created through in vivo selection and cultured at 37°C in a 10% CO2-containing humidified atmosphere. Short tandem repeat profiling was conducted as previously described.21 The SK-OV-3 cell line and SK-OV-3 Luc IP1 cell lines were cultured both in Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies, Merelbeke, Belgium), while the OVCAR-3 cell line was cultured in RPMI 1640 Medium supplemented with Gibco GlutaMAXTM (Life Technologies, Merelbeke, Belgium). All mediums were supplemented with 2% penicillin/streptomycin (Life Technologies) and 10% FCS (Sigma-Aldrich, Overijse, Belgium).
Cell Viability Assay
The CellTiter-Glo® 2.0 luminescent assay was performed to measure the cell viability based on the quantification of adenosine triphosphate (ATP). Cells were seeded as monolayers in opaque 96-well plates (Thermo Scientific, Merelbeke, Belgium) at a density of 4.0×103 cells/well. Subsequently, after 24h of incubation, we exposed the cells to 10 µL of nab-PTX or mic-PTX at five 10-fold dilutions ranging from 0.001 to 10 µM for 72h. Untreated cells were used as control. After 72h of incubation, 100µL of CellTiter Glo® 2.0 reagent (Promega, Leiden, The Netherlands) was administered to each well. Luminescence was measured after 10 min using a Paradigm Detection platform and analyzed with the Soft Max Pro 6.1 software (BIO-RAD Laboratories, Hemel Hempstead, United Kingdom). Three CellTiter-Glo® 2.0 independent assays, with three replicates, were performed.
Subperitoneal Ovarian Xenografts
Ethical approval for this study was obtained from the Ethics Committee on Animal Research and Testing, Ghent University (approval number ECD17/15). The present study followed international, national and institutional guidelines for humane animal treatment and complied with EU directive 2010/63/EU.
Female athymic, nude Foxn1nu mice (ENVIGO, Horst, the Netherlands) of 6 weeks old and an average weight of 20 g were conditioned one week before the start of each study. Peritoneal ovarian xenografts were achieved as previously described.22 All mice were bilaterally engrafted in the subperitoneal (SP) space with 5.0×105 SK-OV-3 Luc IP1 cells, dissolved in 50µL BD matrigel (BD Life Sciences, De Pinte, Belgium).23
To assess the in vivo antitumor efficacy of nab-PTX and mic-PTX, drug treatment was initiated 2 weeks after bilateral SP injection with designation of the first treatment as day 0. Six mice per group were treated IP at days 0, 4, 8, 12 and 16 with either nab-PTX (45 mg PTX/Kg), mic-PTX (45 mg PTX/Kg), or saline. The physical condition of mice was followed up during and after treatment. Animals were euthanized when a bodyweight loss of 20% at any point in time or 15% maintained for 72h was observed. Upon euthanasia, tumor nodules were excised and measured. Furthermore, the simplified peritoneal cancer index (sPCI), in which the abdomen is divided into 7 anatomical regions, and scored according to the maximum diameter of tumor nodules per region up to a maximum of 21, was assessed.24,25 Tumor volumes (mm³) were estimated according to the simplified formula for a spheroid 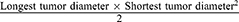 .26 Survival time was recorded starting from bilateral SP SK-OV-3 Luc IP1 injection (day −14).
.26 Survival time was recorded starting from bilateral SP SK-OV-3 Luc IP1 injection (day −14).
To determine PTX pharmacokinetics, drug distribution, and pharmacodynamic effects, another group of 24 mice (12/group) was treated once IP 2 weeks after bilateral SP injection with either nab-PTX or mic-PTX (both 45mg PTX/kg). Three mice of each treatment were sacrificed at 1, 4, 8 and 24 hours after IP injection and both tumor nodules were excised.
Tumor Samples and Histology
Excised tumor nodules were either snap-frozen in liquid nitrogen, stored at −80°C and shipped in dry ice for imaging of PTX penetration, or fixed in 4% PFA and embedded in paraffin for routine histology. In addition, tumor nodules were marked with a yellow pigment (Davidson marking system, Bradley products, Bloomington MN, USA) at the non-peritoneal side after excision to distinguish the portion of tissue directly in contact with the peritoneal fluid. For routine histology, tumor sections of 5 µm were cut with a microtome (Microm HM355S, Thermo Scientific, Rockford, IL, USA). Haematoxylin and eosin and cleaved caspase-3 stainings were performed according to the standard protocols (ready-to-use DAKO Envision+ system-HPR kit (K04011)) for analysis of tissue structure and apoptosis, respectively.
After light microscopic analysis (Olympus BX43F, Olympus, Tokyo, Japan), slides were digitized using a whole slide imaging scanner (Pannoramic 250, 3DHistech, Hungary) and were analyzed using 3DHistech’s Caseviewer 2.3 software. To quantify active caspase 3 staining, whole slide images of three subsequent sections per mouse were analyzed in Qupath, an open-source whole slide imaging software analysis suite,27 utilizing the positive cell detection function, after comparison to manual counting. Settings are detailed in Supplementary Table 1.
For transmission electron microscopy (TEM), tumor samples were fixed in a mixture of 4% PFA (VWR, Leuven, Belgium), 5% glutaraldehyde (VWR) and 0.1M sodium cacodylate buffer (Merck, VWR) and embedded in epon-812 (Aurion, Wageningen, Nederland). Semithin sections of 1 µm were cut, stained with toluidine blue and analyzed with a light microscope (Olympus BX53, Tokyo, Japan). Three to five regions of interest were analyzed per sample to quantify mitosis. Ultrathin sections of 60 nm were cut of the tumor sections of interest and contrasted with uranyl acetate (FLUKA, Sigma-Aldrich) and lead citrate (Merck), followed by imaging with a Zeiss TEM900 Transmission electron microscope (TEM, Carl Zeiss, Oberkochen, Germany) at 50kV.
Pharmacokinetic Analysis of Nab-PTX and Mic-PTX in Plasma and Tissue
Plasma
Aliquots (10 μL each) of whole blood samples were pipetted onto PKI Bioanalysis Cards (PerkinElmer Health Sciences, USA) and left to dry for at least 3 h at room temperature prior to analysis. The entire dry blood spot (DBS) was punched out and collected into a 1.5-mL Eppendorf tube. Extraction was performed by adding 200 μL of IS working solution. The sample tubes were closed and thermostatted at 37 °C for 20 min whilst continuously being shaken by a Biosan TS-100 Thermo shaker (Biosan, Riga, Latvia) at 500 rpm. Then, 100 μL of the extract was added to 100 μL water and vortex-mixed for 30s. After transferring the resulting solution to an autosampler vial, a volume of 10 μL was injected into the UHPLC-MS/MS system (instrumentation and set-up can be found in the Supplementary Methods).
Tissue
The total concentration of PTX in samples was determined by high-performance liquid chromatography (HPLC) coupled to a triple quadrupole mass spectrometer (API4000 system, AB SCIEX, MA, USA) coupled with a Series 200 autosampler and micropump (Perkin Elmer, MA, USA). The extraction procedure for PTX in tumor nodules was a modification of the method described by Ansaloni et al28 and is discussed in Supplementary Methods.
Mass Spectrometry Imaging of Paclitaxel
The spatial distribution of PTX in tumor tissues was determined on three frozen tissue sections of 10µm per sample by mass spectrometry imaging (MSI), according to the method that was recently published by Morosi et al.29 Instrumentation is described in Supplementary Methods.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism™ 8 (GraphPad software, Ca, USA). IC50 was calculated using nonlinear regression. For analysis of in vitro cytotoxicity, two-way ANOVA was used, while other in vivo data were analyzed using the Kruskal-Wallis and the Mann–Whitney U-test. For the analysis of mitosis, unpaired samples t-test was performed. Animal survival was estimated using the Kaplan–Meier method, and survival differences between groups were evaluated using the log-rank (Mantel-Cox) test. The threshold for significance was set at p <0.05.
Results
In vitro Cytotoxicity of Nab-PTX and Mic-PTX in Ovarian Cancer Cells
Both PTX formulations showed antitumoral activity after 72h of incubation for all cell lines in a cell viability assay, with sub-micromolar IC50 concentrations for all groups. IC50 differed significantly between treatment type and cell lines, with a significant interaction between these factors (p=0.002) (Figure 1). The most susceptible cell lines were SK-OV-3 and SK-OV-3 Luc IP1 for Nab-PTX and Mic-PTX, respectively (Table 1).
 |
Table 1 Mean IC50 (nM) (Mean, [95% CI]) of Nab-PTX and Mic-PTX in Different Ovarian Cancer Cell Lines |
 |
Figure 1 Bar chart demonstrating mean sub micromolar IC50 concentrations in several OC cell lines. Whiskers indicate standard deviation. Asterisks indicate P values: **P ≤ 0.01; ****P ≤ 0.0001. |
Both PTX Formulations Reduce Peritoneal Tumor Burden and Increase Median Survival After Repeated IP Administration in vivo
When reaching the humane endpoint, mice were euthanized and tumor burden and survival were assessed. Intraperitoneal administration of mic-PTX in a murine EOC model significantly reduced the peritoneal tumor burden, including the number of affected peritoneal regions as expressed by the sPCI (p = 0.034). Additionally, mean tumor volume was significantly decreased compared to the control group (1491mm3 vs 454mm3, p = 0.042). Similarly, IP nab-PTX reduced sPCI (p = 0.08) and tumor volume (1491mm3 vs 581mm3, p = 0.09) (Figure 2A). Compared to controls, animals treated with both IP PTX formulations showed a significantly longer median survival (30 days in controls, 66 days in nab-PTX treated animals, and 76 days in mic-PTX animals, respectively) (p = 0.0007) (Figure 2B).
 |
Figure 2 (A) Violin plots of tumor volume (left) and sPCI score (right) of ovarian cancer xenografts, showing a significant decrease in both for treated groups compared to controls. (B) Kaplan-Meier survival plot of mice treated with nab-PTX, mic-PTX or saline. (N=6/group). |
Both PTX Formulations Had No Effect on Apoptosis Up to 24h After a Single IP Injection
We performed histological analysis and confirmation of tumor presence on HE-stained samples of excised tissue. Manual analysis of cleaved caspase 3 staining was performed in a semi-quantitative fashion, and demonstrated no significant difference in staining intensity up to 24 hours post IP-injection of either mic-PTX or nab-PTX. Subsequently, we quantified cleaved caspase 3-staining using automated positive cell detection using the Qupath software package on whole-slide images. We calculated the positive cell percentage, expressed as the percentage of positive cells compared to the total number of detected cells. Both mic-PTX and nab-PTX did not significantly increase the percentage of apoptotic cells compared to control tissues up until 24 hours post-treatment (Figure 3). Furthermore, we observed no significant differences between both treatment groups.
 |
Figure 3 Bar chart plotting percentage of Cleaved Caspase 3-positive cells per timepoint (n=3) for Nab-PTX-treated xenografts (left) and Mic-PTX-treated xenografts (right). For both groups, no increase in cleaved caspase-3 signal was observed compared to controls. |
Treatment with Nab-PTX or Mic-PTX Induce Mitotic Catastrophe in vivo
We further assessed the pharmacodynamic effects of nab-PTX and mic-PTX using toluidine blue semithin sections of SK-OV-3 Luc IP1 peritoneal metastases of our xenograft model (Figure 4). Although variance between mice was high, the mean mitotic index increased clearly 24h after nab-PTX administration when compared to controls (respectively 18% and 2%, p<0.001), with little effect after 2 hours. A similar albeit weaker trend was observed after 24h of mic-PTX administration when compared to controls (11% and 2%, p=0.083) (Figure 5).
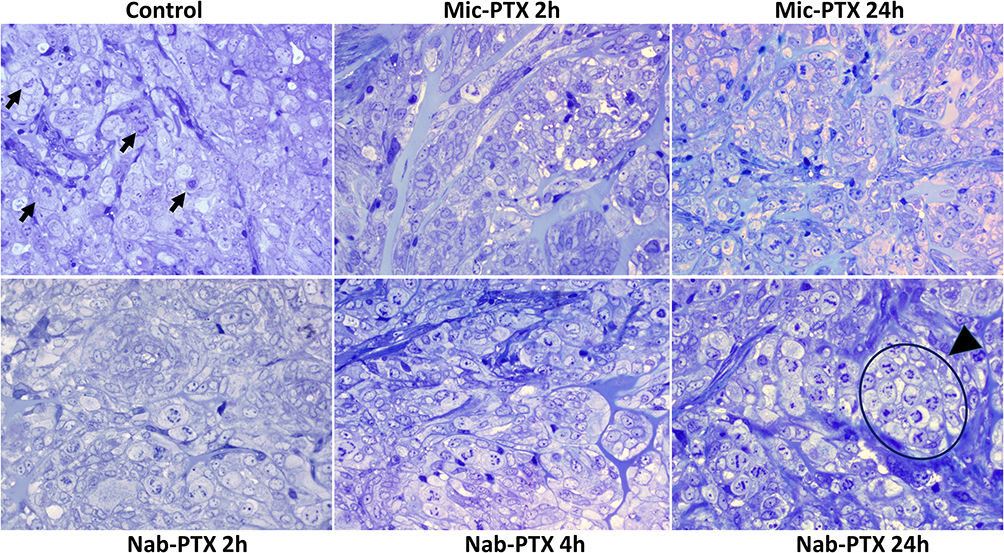 |
Figure 4 Toluidine blue stained semithin sections of glutaraldehyde fixed resin embedded SK-OV-3 Luc IP1 tumor xenografts, exposed to saline, to Nab-PTX for 2h, 4h and 24h and to Mic-PTX for 2 and 24h. Mic-PTX treated xenografts demonstrated a lightly increased mitotic count. Nab-PTX induced mitotic catastrophe in xenografts. The mitotic index increased with increasing exposure to Abraxane (24h > 4h > 2h > control). Some mitotic figures are indicated in the control condition (arrowhead); a large cluster of mitotic cells is indicated in the condition where cells are exposed to Abraxane for 24h (arrowhead). Mitotic cells in cultures exposed to Abraxane displayed abnormal DNA condensation. There was no increase in apoptotic cells. |
 |
Figure 5 Bar charts of mean mitotic percentage per treatment group as determined on semithin sections. We observed large variance between tumors. Nab-PTX treated tumors exhibit significantly increased mitotic counts after 24 hours, with a smaller effect noted for Mic-PTX treated tumors. No effect on mitotic count is observed after 2 hours for both formulations. Whiskers indicate standard deviation. |
Transmission Electron Microscopy
Transmission electron microscopy (TEM) images confirmed these mitotic catastrophes (MCs) in detail (Figure 6). Numerous aberrant mitotic figures displaying abnormal DNA condensation were observed after IP injection of nab-PTX in SP tumor nodules. Higher magnification of aberrant mitotic cells showed an intact cell membrane and preserved mitochondria, indicating that the primary mechanism of cell death by nab-PTX was determined by MC and not by necrosis. The characteristics of DNA condensation are consistent with MC, and there was no apparent increase in cells displaying the characteristic chromatin condensation and margination of apoptotic cells, consistent with our observation of no increase in cleaved caspase-3 stained cells.
 |
Figure 6 Ultrathin sections of glutaraldehyde fixed resin embedded SK-OV-3 Luc IP1 tumor xenografts exposed to saline (A)(scale bar 5000 nm), or to Nab-PTX for 24h ((B) scale bar 2000 nm; (C) scale bar 500 nm). Arrows indicate the cell membrane (blue), the nuclear membrane (red) and the mitochondria (black). In xenografts exposed to Nab-PTX numerous mitotic figures were observed, often arranged in clusters. Mitotic cells in cultures exposed to Nab-PTX displayed abnormal DNA condensation. Higher magnification (rightmost figure) of such a mitotic cell shows that this cell has an intact cell membrane and preserved mitochondria, indicating mitotic catastrophe rather than apoptosis or necrosis. |
Pharmacokinetics in Plasma and Tissue After Single IP Administration
The systemic absorption dynamics after single IP administration were similar between nab-PTX and mic-PTX, with no significant differences in Tmax, Cmax and area under the curve. Peak plasma concentrations were approximately 20–30 mg/mL. In resected tissue, mic-PTX cleared from the tissue faster, with similar Tmax and Cmax values. However, total drug concentrations were higher after nab-PTX compared to mic-PTX (AUC0–48h of 247 versus 153 µg/g*h, respectively) (Figure 7 and Table 2).
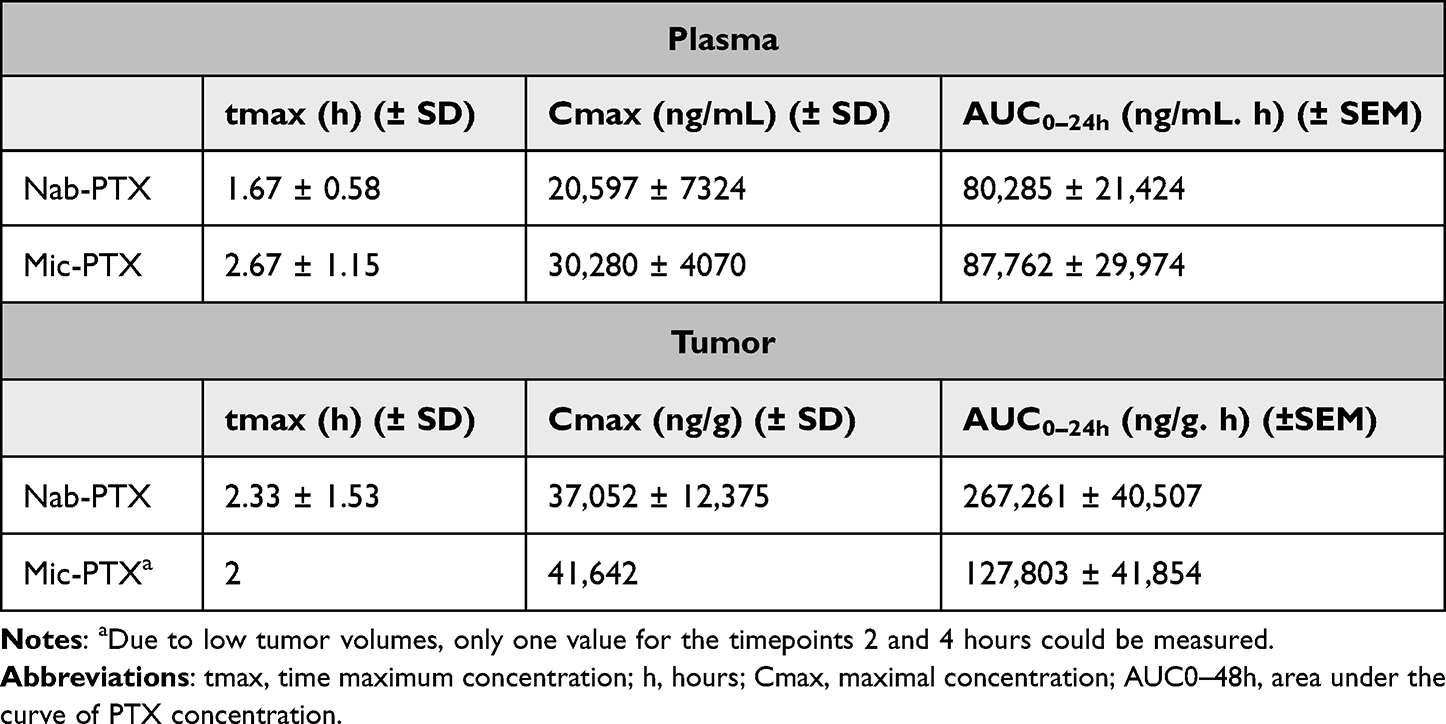 |
Table 2 Mean PK Parameters of Paclitaxel in Plasma and Tumor Tissue |
 |
Figure 7 Line plots of PTX pharmacokinetics in plasma (left) and tumor samples (right), sampled at 1,2,4,8 and 24 hours. Similar pharmacokinetic behavior between both formulations can be noted, with a more rapid clearance of mic-PTX from tumor compared to nab-PTX, and higher peak nab-PTX values. Whiskers indicate standard error of mean. |
Nab-PTX Demonstrates Higher Tissue Uptake as Quantified by PTX Imaging Mass Spectrometry
Paclitaxel penetration in tumor tissues was assessed by matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) 1, 4, 8 and 24 hours after treatment with nab-PTX (45 mg PTX/kg) or mic-PTX (45 mg PTX/kg). Semiquantitative assessment using pseudocolor maps of PTX tissue uptake showed, after 4 hours, a more pronounced tissue uptake after IP nab-PTX delivery compared to mic-PTX, concurrent with the pharmacokinetic data measured in tissue (Figure 8).
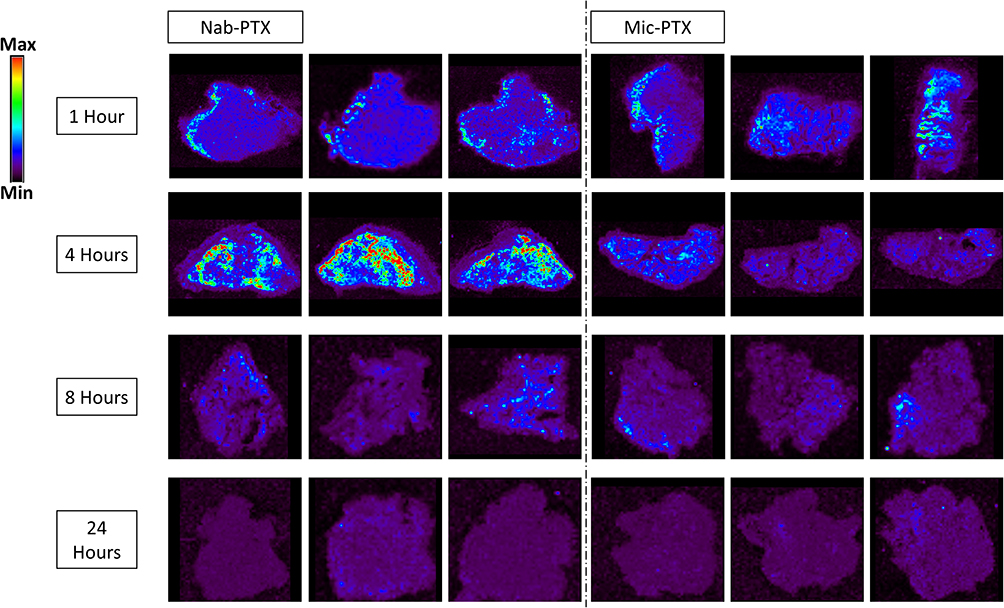 |
Figure 8 Mass spectrometry imaging visualization of paclitaxel distribution (m/z 284.2) in tumor tissues from mice treated with nab-PTX or mic-PTX. We can observe more intense and widespread PTX signal intensity in the nab-PTX treated xenografts, particularly around the 4h timepoint. |
Discussion
We investigated the preclinical efficacy and pharmacokinetics of two PTX nanoformulations, namely nab-PTX (or Abraxane®) and mic-PTX (or Genexol®-PM) in the context of peritoneal metastases due to ovarian cancer. We demonstrated sub-micromolar IC50 concentrations for both formulations on three EOC cancer cell lines in vitro. Furthermore, in a murine model of EOC PM, IP administration with either nab-PTX or mic-PTX leads to more than 2-fold longer survival compared to a control treatment of IP saline administration. This in vivo efficacy was further supported by similar decreases in tumor volume and sPCI score when reaching the humane endpoint, indicating enhanced local control of disease. Pharmacokinetic analysis was carried out, indicating similar systemic uptake of both nab-PTX and mic-PTX, with no significant differences in uptake profile. We subsequently analyzed the tissue distribution and pharmacodynamics of PTX following a single IP administration of both drugs using MALDI imaging mass spectrometry. Here, we demonstrated higher tissue uptake of drug following nab-PTX administration when compared to mic-PTX, with highest uptake after 4 hours post-treatment. We confirmed this lower uptake in tissue of mic-PTX using high-performance liquid chromatography (HPLC) on digested tumor samples, which confirmed a lower total amount of drug uptake for mic-PTX.
Our in vitro findings were largely in line with those found in literature, with similar IC50 concentrations reported for all three cell lines used.30,31 However, we observed differing sensitivity of our SK-OV-3 Luc IP1 cell line to both formulations when compared to SK-OV-3, potentially due to changes acquired during in vivo selection. Furthermore, the mechanisms of uptake of nab-PTX and mic-PTX differ. Albumin-dependent effects influence tumoral uptake of nab-PTX,13 while mic-PTX consists of PLA-b-PEG polymer micelles, of which the mechanism of uptake is less well studied. As such acquired changes might impact the in vitro mechanism of uptake of both formulations differently.32 Additionally, Xiao et al reported median OS time 39 and 81 days for control (PBS) and nab-PTX treated mice, respectively, after repeated IP treatment of orthotopic OC xenografts with peritoneal metastases, supporting our observed in vivo efficacy of IP treatment.33 No comprehensive analyses comparing pharmacokinetics and tissue distribution of IP administration for both nab-PTX and mic-PTX have been published. A limitation of our study is the smaller sample size, potentially limiting generalizability of our pharmacokinetic and pharmacodynamic findings.
Nab-PTX has been shown to achieve good tissue penetration in a rabbit HIPEC model.19 Interestingly, tissue uptake of nab-PTX was higher compared to mic-PTX, with the highest concentration reached after 4 hours post-treatment. This effect might be partly explained by a delayed (>24h) release of PTX from the micellar formulation into the peritoneal cavity, due to the high affinity of PTX for the hydrophobic core of the polymeric micelles.34 In contrast, nab-PTX dissociates more easily after IP administration owing to its reversible non-covalent binding.35 Additionally, interactions with the tumor microenvironment (TME) are increasingly important in understanding tumoral accumulation of nab-PTX, due to the presence of albumin-binding proteins such as secreted protein acidic and rich in cysteine (SPARC) in the TME. These proteins bind albumin in the vicinity of the tumor and might as such lead to increased retention of nab-PTX in tumor.13 In a murine model of Ewing sarcoma, knockdown of SPARC leads to faster clearance, and lower total dose of nab-PTX.36 This mechanism might be an important factor in explaining the faster clearance of mic-PTX, when compared to nab-PTX. Also, Desai et al reported up to 4 times higher endothelial transcytosis when comparing nab-PTX to solvent based-PTX. They showed nab-PTX specific effects influencing tumor penetration, and a possible adverse effect of micellar sequestration.26 However, the influence of these effects on transmesothelial transport and their mechanisms is currently unknown.
Recent research demonstrates that intraperitoneal administration of nanoparticles can be associated with non-specific uptake of these particles in intra-abdominal organs such as liver and spleen and may be a cause of morbidity.37,38 When comparing intraperitoneal administration cabazitaxel and a polyacrylate-based cabazitaxel-loaded nanoparticle in mice, Hyldbakk et al observed significantly higher cabazitaxel exposure for the nanoparticle group in both tumor and intra-abdominal organs. However, when the nanoparticle formulation was administered intravenously, comparable exposure of intra-abdominal organs was observed to the IP administration, with limited exposure of the peritoneum.39 As such, an intraperitoneal administration offers a clear benefit in terms of peritoneal exposure, although non-specific effects should be taken into account. In our cohort, systemic uptake was low for both formulations, and treatment was well tolerated, demonstrating suitability of these agents for intraperitoneal administration.
Paclitaxel is thought to mainly kill cancer cells via inducing mitotic arrest, by stabilizing microtubules polymers. However, recent research indicates a potential alternative mechanism of action, by causing chromosome missegregation.12 We did not observe increased apoptosis, as evidenced by measuring cleaved caspase 3 protein expression up to 24 hours post-treatment, excluding apoptosis as a main mechanism of cell death. Transmission electron microscopy further confirmed the absence of traditional markers of apoptosis or necrosis in nab-PTX and mic-PTX treated tumours. However, we did observe numerous aberrant mitotic figures in several samples displaying abnormal DNA condensation, consistent with mitotic catastrophe.12 However, variability in mitotic counts and mitotic catastrophe was high between mice, while cleaved caspase activity was universally low. Paclitaxel is a potent inducer of mitotic catastrophe, wherein aberrant mitosis leads to eventual cell death.40 While the exact molecular mechanisms of mitotic catastrophe remain to be explored, we show that nab-PTX and mic-PTX exert in vivo anti-tumor effects in an apoptosis-independent manner, potentially relying on mitotic catastrophe.
Conclusion
To conclude, we show that IP administration of both nab-PTX and mic-PTX results in a significant anticancer efficacy and survival benefit in a mouse OC xenograft model. Furthermore, an increased amount of mitotic catastrophes and higher PTX signal intensities are shown in tumor tissue after IP treatment with nab-PTX using TEM and MALDI-MSI, respectively. Further research will aim to elucidate the pharmacodynamic effects of mic-PTX and to verify the difference in drug uptake after IP injection of both PTX formulations.
Acknowledgments
Wim Ceelen is a senior clinical researcher from the Fund for Scientific Research – Flanders (FWO). This work was funded in part by a grant from the Flemish League against Cancer (Kom op Tegen Kanker). This paper is based on the thesis of Dr Charlotte Carlier. It has been published on the institutional website of Ghent University: https://biblio.ugent.be/publication/8575482.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68(4):284–296. doi:10.3322/caac.21456
2. Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med. 2017;14(1):9–32. doi:10.20892/j.issn.2095-3941.2016.0084
3. Ledermann JA, Raja FA, Fotopoulou C, Gonzalez-Martin A, Colombo N, Sessa C. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):24–32. doi:10.1093/annonc/mdt333
4. Hess LM, Benham-Hutchins M, Herzog TJ, et al. A meta-analysis of the efficacy of intraperitoneal cisplatin for the front-line treatment of ovarian cancer. Int J Gynecol Cancer. 2007;17(3):561–570. doi:10.1111/j.1525-1438.2006.00846.x
5. Jaaback K, Johnson N, Lawrie TA. Intraperitoneal chemotherapy for the initial management of primary epithelial ovarian cancer. Cochrane Database Syst Rev. 2016;1:CD005340. doi:10.1002/14651858.CD005340.pub4
6. Monk BJ, Chan JK. Is intraperitoneal chemotherapy still an acceptable option in primary adjuvant chemotherapy for advanced ovarian cancer? Ann Oncol. 2017;28:viii40–viii45. doi:10.1093/annonc/mdx451
7. Van de Sande L, Cosyns S, Willaert W, Ceelen W. Albumin-based cancer therapeutics for intraperitoneal drug delivery: a review. Drug Deliv. 2020;27(1):40–53. doi:10.1080/10717544.2019.1704945
8. Francis P, Rowinsky E, Schneider J, Hakes T, Hoskins W, Markman M. Phase I feasibility and pharmacologic study of weekly intraperitoneal paclitaxel: a gynecologic oncology group pilot study. J Clin Oncol. 1995;13(12):2961–2967. doi:10.1200/JCO.1995.13.12.2961
9. Dakwar GR, Shariati M, Willaert W, Ceelen W, De Smedt SC, Remaut K. Nanomedicine-based intraperitoneal therapy for the treatment of peritoneal carcinomatosis — mission possible? Adv Drug Deliv Rev. 2017;108:13–24. doi:10.1016/j.addr.2016.07.001
10. Markman M. Intraperitoneal antineoplastic drug delivery: rationale and results. Lancet Oncol. 2003;4(5):277–283. doi:10.1016/s1470-2045(03)01074-x
11. Stage TB, Bergmann TK, Kroetz DL. Clinical pharmacokinetics of paclitaxel monotherapy: an updated literature review. Clin Pharmacokinet. 2018;57(1):7–19. doi:10.1007/s40262-017-0563-z
12. Weaver BA, Bement W. How taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. doi:10.1091/mbc.E14-04-0916
13. Yardley DA. nab-paclitaxel mechanisms of action and delivery. J Control Release. 2013;170(3):365–372. doi:10.1016/j.jconrel.2013.05.041
14. Risinger AL, Riffle SM, Lopus M, Jordan MA, Wilson L, Mooberry SL. The taccalonolides and paclitaxel cause distinct effects on microtubule dynamics and aster formation. Mol Cancer. 2014;13(1):41. doi:10.1186/1476-4598-13-41
15. Gelderblom H, Verweij J, Nooter K, Sparreboom A, Cremophor EL. The drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. doi:10.1016/s0959-8049(01)00171-x
16. Stinchcombe TE. Nanoparticle albumin-bound paclitaxel: a novel Cremphor-EL®-free formulation of paclitaxel. Nanomedicine. 2007;2(4):415–423. doi:10.2217/17435889.2.4.415
17. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med. 2019;4(3):e10143. doi:10.1002/btm2.10143
18. Werner ME, Cummings ND, Sethi M, et al. Preclinical evaluation of Genexol-PM, a nanoparticle formulation of paclitaxel, as a novel radiosensitizer for the treatment of non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2013;86(3):463–468. doi:10.1016/j.ijrobp.2013.02.009
19. Coccolini F, Acocella F, Morosi L, et al. High penetration of paclitaxel in abdominal wall of rabbits after hyperthermic intraperitoneal administration of nab-paclitaxel compared to standard paclitaxel formulation. Pharm Res. 2017;34(6):1180–1186. doi:10.1007/s11095-017-2132-4
20. Kinoshita J, Fushida S, Tsukada T, et al. Comparative study of the antitumor activity of Nab-paclitaxel and intraperitoneal solvent-based paclitaxel regarding peritoneal metastasis in gastric cancer. Oncol Rep. 2014;32(1):89–96. doi:10.3892/or.2014.3210
21. De Vlieghere E, Carlier C, Ceelen W, Bracke M, De Wever O. Data on in vivo selection of SK-OV-3 Luc ovarian cancer cells and intraperitoneal tumor formation with low inoculation numbers. Data Brief. 2016;6:542–549. doi:10.1016/j.dib.2015.12.037
22. Carlier C, Strese S, Viktorsson K. Preclinical activity of melflufen (J1) in ovarian cancer. Oncotarget. 2016;7(37):59322–59335. doi:10.18632/oncotarget.11163
23. De Vlieghere E, Gremonprez F, Verset L. Tumor-environment biomimetics delay peritoneal metastasis formation by deceiving and redirecting disseminated cancer cells. Biomaterials. 2015;54:148–157. doi:10.1016/j.biomaterials.2015.03.012
24. Ceelen WP, Van Nieuwenhove Y, Van Belle S, Denys H, Pattyn P. Cytoreduction and hyperthermic intraperitoneal chemoperfusion in women with heavily pretreated recurrent ovarian cancer. Ann Surg Oncol. 2012;19(7):2352–2359. doi:10.1245/s10434-009-0878-6
25. Swellengrebel HAM, Zoetmulder FAN, Smeenk RM, Antonini N, Verwaal VJ. Quantitative intra-operative assessment of peritoneal carcinomatosis – a comparison of three prognostic tools. Eur J Surg Oncol. 2009;35(10):1078–1084. doi:10.1016/j.ejso.2009.02.010
26. Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–1324. doi:10.1158/1078-0432.CCR-05-1634
27. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. doi:10.1038/s41598-017-17204-5
28. Ansaloni L, Coccolini F, Morosi L. Pharmacokinetics of concomitant cisplatin and paclitaxel administered by hyperthermic intraperitoneal chemotherapy to patients with peritoneal carcinomatosis from epithelial ovarian cancer. Br J Cancer. 2015;112(2):306–312. doi:10.1038/bjc.2014.602
29. Morosi L, Spinelli P, Zucchetti M, et al. Determination of paclitaxel distribution in solid tumors by nano-particle assisted laser desorption ionization mass spectrometry imaging. PLoS One. 2013;8(8):e72532. doi:10.1371/journal.pone.0072532
30. Louage B, Nuhn L, Risseeuw MDP. Well-defined polymer–paclitaxel prodrugs by a grafting-from-drug approach. Angew Chem Int Ed Engl. 2016;55(39):11791–11796. doi:10.1002/anie.201605892
31. Kim SC, Kim DW, Shim YH. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72(1–3):191–202. doi:10.1016/S0168-3659(01)00275-9
32. Ma P, Mumper RJ. Paclitaxel nano-delivery systems: a comprehensive review. J Nanomed Nanotechnol. 2013;4(2):1000164. doi:10.4172/2157-7439.1000164
33. Xiao K, Luo J, Fowler WL. A self-assembling nanoparticle for paclitaxel delivery in ovarian cancer. Biomaterials. 2009;30(30):6006–6016. doi:10.1016/j.biomaterials.2009.07.015
34. Lavasanifar A, Samuel J, Kwon GS. Poly(ethylene oxide)-block-poly(L-amino acid) micelles for drug delivery. Adv Drug Deliv Rev. 2002;54(2):169–190. doi:10.1016/S0169-409X(02)00015-7
35. Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int J Nanomed. 2009;4(1):99–105. doi:10.2147/ijn.s3061
36. Pascual-Pasto G, Castillo-Ecija H, Unceta N, et al. SPARC-mediated long-term retention of nab-paclitaxel in pediatric sarcomas. J Control Release. 2022;342:81–92. doi:10.1016/j.jconrel.2021.12.035
37. Baldwin P, Ohman AW, Tangutoori S, Dinulescu DM, Sridhar S. Intraperitoneal delivery of NanoOlaparib for disseminated late-stage cancer treatment. IJN. 2018;13:8063–8074. doi:10.2147/IJN.S186881
38. Burns JM, Shafer E, Vankayala R, Kundra V, Anvari B. Near infrared fluorescence imaging of intraperitoneal ovarian tumors in mice using erythrocyte-derived optical nanoparticles and spatially-modulated illumination. Cancers. 2021;13(11):2544. doi:10.3390/cancers13112544
39. Hyldbakk A, Fleten KG, Snipstad S, et al. Intraperitoneal administration of cabazitaxel-loaded nanoparticles in peritoneal metastasis models. Nanomedicine. 2023;48:102656. doi:10.1016/j.nano.2023.102656
40. Zhao S, Tang Y, Wang R, Najafi M. Mechanisms of cancer cell death induction by paclitaxel: an updated review. Apoptosis. 2022;27(9):647–667. doi:10.1007/s10495-022-01750-z
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.