Back to Journals » The Application of Clinical Genetics » Volume 16
Prader-Willi and Angelman Syndromes: Mechanisms and Management
Authors Ma VK, Mao R, Toth JN ![]() , Fulmer ML, Egense AS, Shankar SP
, Fulmer ML, Egense AS, Shankar SP ![]()
Received 14 December 2022
Accepted for publication 11 March 2023
Published 6 April 2023 Volume 2023:16 Pages 41—52
DOI https://doi.org/10.2147/TACG.S372708
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Van K Ma,1,2 Rong Mao,3,4 Jessica N Toth,3 Makenzie L Fulmer,3,4 Alena S Egense,1,2 Suma P Shankar1,2,5
1Department of Pediatrics, University of California Davis, Sacramento, CA, USA; 2MIND Institute, University of California Davis, Sacramento, CA, USA; 3Molecular Genetics and Genomics, ARUP Laboratories, Salt Lake City, UT, USA; 4Department of Pathology, University of Utah, Salt Lake City, UT, USA; 5Department of Ophthalmology, University of California Davis, Sacramento, CA, USA
Correspondence: Suma P Shankar, MIND Institute, University of California Davis, 2825 50th Street, Sacramento, CA, 95817, USA, Tel +1 916 703 0235, Fax + 1 916 703 0203, Email [email protected]
Abstract: Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are genetic imprinting disorders resulting from absent or reduced expression of paternal or maternal genes in chromosome 15q11q13 region, respectively. The most common etiology is deletion of the maternal or paternal 15q11q13 region. Methylation is the first line for molecular diagnostic testing; MS-MLPA is the most sensitive test. The molecular subtype of PWS/AS provides more accurate recurrence risk information for parents and for the individual affected with the condition. Management should include a multidisciplinary team by various medical subspecialists and therapists. Developmental and behavioral management of PWS and AS in infancy and early childhood includes early intervention services and individualized education programs for school-aged children. Here, we compare and discuss the mechanisms, pathophysiology, clinical features, and management of the two imprinting disorders, PWS and AS.
Keywords: imprinting disorders, uniparental disomy, chromosome 15, developmental delay, hyperphagia, obesity
Plain Language Summary
Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are genetic conditions that result from a decrease or lack of expression of inherited material from the father or mother on chromosome 15, respectively. DNA methylation is the mechanism used by cells to control what genes are turned on to make RNA and protein; a number of laboratory tests are available to make a definitive diagnosis of PWS or AS. Management of PWS and AS involve specialists from multiple disciplines. Children would benefit from appropriate medical management, early intervention services, and specialized academic instruction and related services as appropriate.
Introduction
Imprinting disorders result from molecular changes that alter the regulation, dosage or sequence of genes in the imprinted regions of chromosomes. In mammals, a subset of genes have parent-of-origin-dependent expression, meaning there is monoallelic expression from one of the two parental autosomes. Also known as differential epigenetic marking, genomic imprinting works to silence genes through DNA methylation without altering the primary nucleotide sequence. Preferential expression of genes on either the maternal or paternal allele results primarily from methylation of cytosine molecules at CpG dinucleotides, key regulatory elements of genes. Methylation profiles are established in the female and male germ lines during gametogenesis, and the offspring of these individuals inherit the methylation pattern. The imprint is subsequently erased in the gametes of these offspring and a new genomic imprint is established, a process known as intergenerational epigenetic inheritance.1,2
More than 100 imprinted genes have been identified, many of which are arranged in clusters called imprinted domains. Alteration of the expression and function of imprinted genes can lead to imprinting defects, manifesting as congenital disorders that may affect human development and have lifelong impacts on health. Since imprinting disorders are a consequence of parental inheritance and not of the sex of the offspring, genomic imprinting affects both female and male offspring. Imprinting disorders have been linked to several autosomes, including chromosomes 6, 7, 8, 11, 12, 14, 15, and 20, with the most observed imprinting disorders mapping to 11p15 and 15q11q13 (latter shown in Figure 1). Abnormal imprinting and loss of heterozygosity have also been shown to contribute to acquired malignancies such as myeloid leukemia.3 The main types of molecular genetic changes underlying imprinting disorders are pathogenic sequence variants in imprinted genes, copy number variants, inheritance of both homologous chromosomes or segments of a chromosome from a single parent (uniparental disomy), and epigenetic changes that affect the regulation of imprinted loci (epimutations). Two of the most common genetic imprinting disorders are Prader-Willi syndrome (PWS) and Angelman syndrome (AS), a result of the absence or reduced expression of paternal or maternal genes in chromosome 15q, respectively.
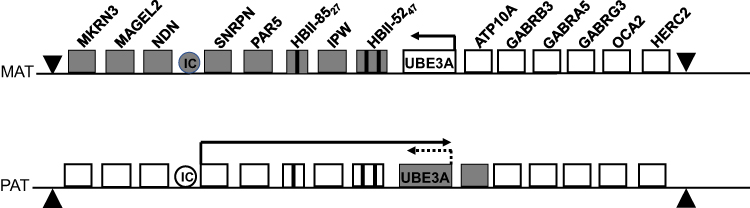 |
Figure 1 Diagram of maternal (MAT; top) and paternal (PAT; bottom) regions of human chromosome 15q11-q13. Clear boxes represent actively expressed genes; grey boxes represent those whose expression has been silenced through genomic imprinting (maternal allele) or through expression of the antisense transcript (paternal UBE3A). |
De Novo Interstitial Deletions of the Chromosomal Region 15q11q13
PWS and AS critical region is located on the proximal arm of chromosome 15 (15q11q13) and is approximately 5–6 Mb in length.4,5 Genes within this region are differentially imprinted depending on parental origin. Approximately 70% of all PWS/AS cases are caused by a 15q11q13 deletion on either the paternal or maternal chromosome, respectively.6 Interstitial deletions occur in the PWS/AS critical region due to low copy (250–400 kb) tandem repeats, which flank common break points and are subject to non-homologous recombination during meiosis.7,8
PWS arises from the loss of function or expression of paternally derived genes. Several genes which are preferentially or exclusively expressed from the paternal chromosome have been described: SNURF-SNRPN, MKRN3, NDN, MAGEL2, NPAP1, PWRN1, SNORD116, IPW, and SNORD115. On the maternal chromosome, these genes have methylated CpG islands in their promoter which leads to silencing of the maternal allele.9–11 As a result, a large deletion of 15q11q13 on the paternal chromosome leads to no functional expression of these genes and results in PWS.
AS arises from the loss of function of the maternally derived ubiquitin ligase E3A, UBE3A, which is preferentially expressed by the maternal chromosome in the neuronal cells. UBE3A is not differentially regulated and is instead regulated indirectly through a paternally derived antisense transcript which prevents expression of the paternal UBE3A.12,13 Thus, when the maternal UBE3A is lost due to a large deletion of 15q11q13, paternal gene expression remains blocked and results in loss of UBE3A expression, leading to AS.
Uniparental Disomy of Chromosome 15
PWS and AS can also be due to uniparental disomy (UPD) of chromosome 15. PWS results when there are two maternal copies of chromosome 15 present, meaning that there is no paternal expression of genes within the critical region. PWS due to UPD accounts for about 25% of all cases and is most often the result of a pre-zygotic event.14
AS due to UPD occurs when there are two paternal copies of chromosome 15, and as a result, there is no expression from the maternally derived UBE3A. UPD accounts for approximately 7% of all cases of AS and most often occurs post-zygotically which can also lead to mosaicism.14,15
Imprinting Defect
Imprinting defects within chromosome 15q11q13 account for approximately 5% of PWS and 3–5% of AS. Small deletions or point mutations in the imprinting center (IC) have been reported to alter the gene expression and result in either PWS or AS.9,16,17 Testing for point mutations within the IC is only available on a research basis.
UBE3A Pathogenic Variants
Intragenic pathogenic variants in the maternally inherited UBE3A located within the chromosome 15q11q13 region account for 10–25% of AS patients. UBE3A, encodes a HECT (homologous to the E6-AP carboxyl terminus) domain E3 ubiquitin ligase that catalyzes the addition of ubiquitin to lysine residues on substrate proteins, leading to the degradation of the ubiquitinated substrate protein.18 The disruption of UBE3A activity leads to inappropriately high levels of these target proteins and consequent neuronal dysfunction. UBE3A is subject to genomic imprinting, with preferential maternal-specific expression in brain and, more specifically, in neurons but not in glia. Despite the critical role that UBE3A plays in human cognitive function and evidence of clinical neurological and behavioral phenotype through mouse model studies, relatively little is known about UBE3A’s role in human nervous system development or how the mutated UBE3A expression leads to the cognitive and language impairment underlying AS.13,19
Diagnostic Work-Up
A testing algorithm has been developed with a focus on, “infant with hypotonia and/or developmental delays”, or “clinical features concerning for PWS or AS” (Figure 2).
 |
Figure 2 (A) Algorithm for genetic testing in an infant with hypotonia and/or developmental delays, and suspected clinical diagnosis of PWS/AS. (B) Recommended follow-up parental testing, based on molecular etiology of proband, to establish recurrence risk [Targeted testing to assess for maternal UBE3A variant; karyotype or FISH to assess for translocation/cytogenetic abnormality in mother or father]. |
mPCR+MLPA
Large deletions in the imprinted PWS/AS critical region on 15q11q13 are the most common cause for PWS and AS.6 Analysis of methylation status of the SNRPN locus using MS-PCR establishes either hypermethylation (PWS) or hypomethylation (AS); however, it cannot distinguish between a deletion or UPD. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) provides more information by simultaneously detecting variation in both copy number and methylation status.20 An MS-MLPA kit (ME028-D1) that is commercially available through MRC Holland (https://www.mrcholland.com/technology/mlpa) contains probes that detect copy number as well as methylation status. Methylation status is determined by eight methylation-sensitive probes that contain an HhaI recognition site. HhaI cleavage sites, also known as CpG islands, are commonly methylated. When methylated, the HhaI enzyme is unable to digest the DNA. Thus, a PWS patient would have no digestion in the PWS/AS critical region due to hypermethylation, while an AS patient would have complete digestion due to hypomethylation. MS-MLPA can also detect microdeletions in the PWS/AS critical region as well as mosaicism. However, MS-MLPA cannot distinguish between a methylation defect due to UPD or an imprinting defect.
Microarray
DNA microarrays, also called chromosomal microarrays (CMA), are widely used to detect genomic imbalances and are a first-tier test for individuals who present clinically with phenotypic features of autism, developmental delay, or congenital anomalies.21 Two CMA techniques exist: comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP). The CGH array is used to detect copy number gains and losses along chromosomes by comparing fluorescent intensities of labeled DNA across the genome with those of a control DNA sample. The SNP array differs in that it includes a set of probes derived from the genome that show differences of a single base pair at common areas of genomic variation (SNPs) and the absolute fluorescent probe intensities are compared against a reference set. Most clinical CMAs contain copy number probes in addition to SNP probes and are therefore considered to be a hybrid array. In addition to detecting genomic imbalances, the genotype plots generated from the SNP probes offer additional information about balanced mechanisms including UPD (isodisomy), long continuous stretches of homozygosity (LCSH), and mosaicism.22
With deletions being the most common etiology for PWS and AS, both CMA techniques could be used for genetic confirmation and to establish breakpoints. Additionally, use of genomic microarray may shed light on other genomic imbalances that may be causative for a patient’s clinical presentation in scenarios where a specific diagnosis cannot be made. Fluorescence in situ hybridization (FISH) may also be used to visualize a deletion within 15q11q13 in metaphase chromosomes in instances where CMA or other methodologies are not available, or in exceedingly rare cases of PWS or AS resulting from a structural abnormality. Of the two CMA techniques, only the SNP array can be used to detect cases of UPD or LSCH that could be a result of UPD. It is important to note that the CMA SNP platform is only able to discern UPD or LSCH in cases of isodisomy, where the two chromosome homologs or segments are identical copies of one parental homolog. In cases of heterodisomy, where there is uniparental inheritance of both parental homologs, there is a complete lack of LCSH and would not be detectable. For that reason, methylation-specific molecular assays provide more sensitivity in establishing genetic confirmation for PWS and AS patients.21,23
UBE3A Pathogenic Variants Detection
UBE3A (NM_130839.5) is comprised of 13 coding exons and codes for 873 amino acids. Alternative splicing of this gene results in three transcript variants encoding three isoforms with different N-termini. There are over 500 pathogenic or likely pathogenic variants of UBE3A documented in ClinVar.24
Single-Gene Testing
Sequencing analysis of UBE3A can detect small intragenic deletions/insertions, missense, nonsense, and splice site variants which account for 90% of disease-causing pathogenic variants. This assay is limited to detecting single-exon, multi-exon, or whole-gene deletions/duplications. If no variant is detected by sequencing, the next step is to perform gene-targeted deletion/duplication analysis for UBE3A; one example is using MLPA where the overall sensitivity is greater than 99%.20
Multigene (Next Generation Sequencing) Panels
UBE3A has been included in multigene panels that include AS as a differential diagnosis to improve sensitivity. AS has a phenotype which overlaps with other diseases and is most commonly included in epilepsy, autism, and intellectual deficiency multi-gene panels.
Management
PWS and AS may present at different ages from birth to later childhood. PWS individuals may present as a neonate with hypotonia and feeding difficulties, hyperphagia around 2 years of age, or later on with developmental and behavior concerns, or hormonal disturbances due to hypothyroidism or obesity.25 AS individuals usually present with global development delays including severe speech and language delays, gait abnormalities, feeding difficulties, and seizures in early childhood.26 Management of both these conditions involves multiple medical specialists and therapists with ideal care given through multidisciplinary teams. Table 1 and Table 2 summarize the recommendations for management of PWS and AS.
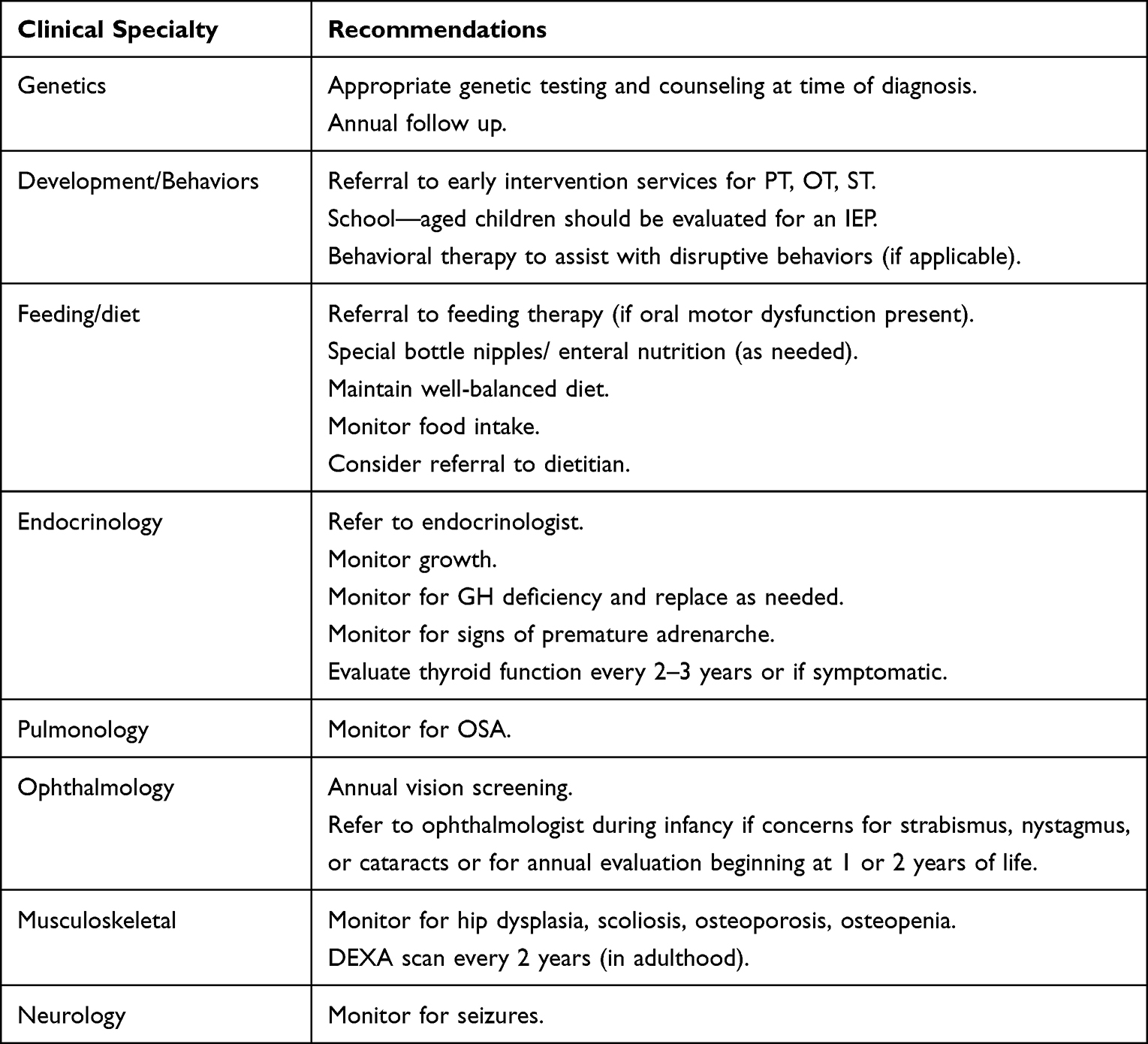 |
Table 1 Health Recommendations for PWS. |
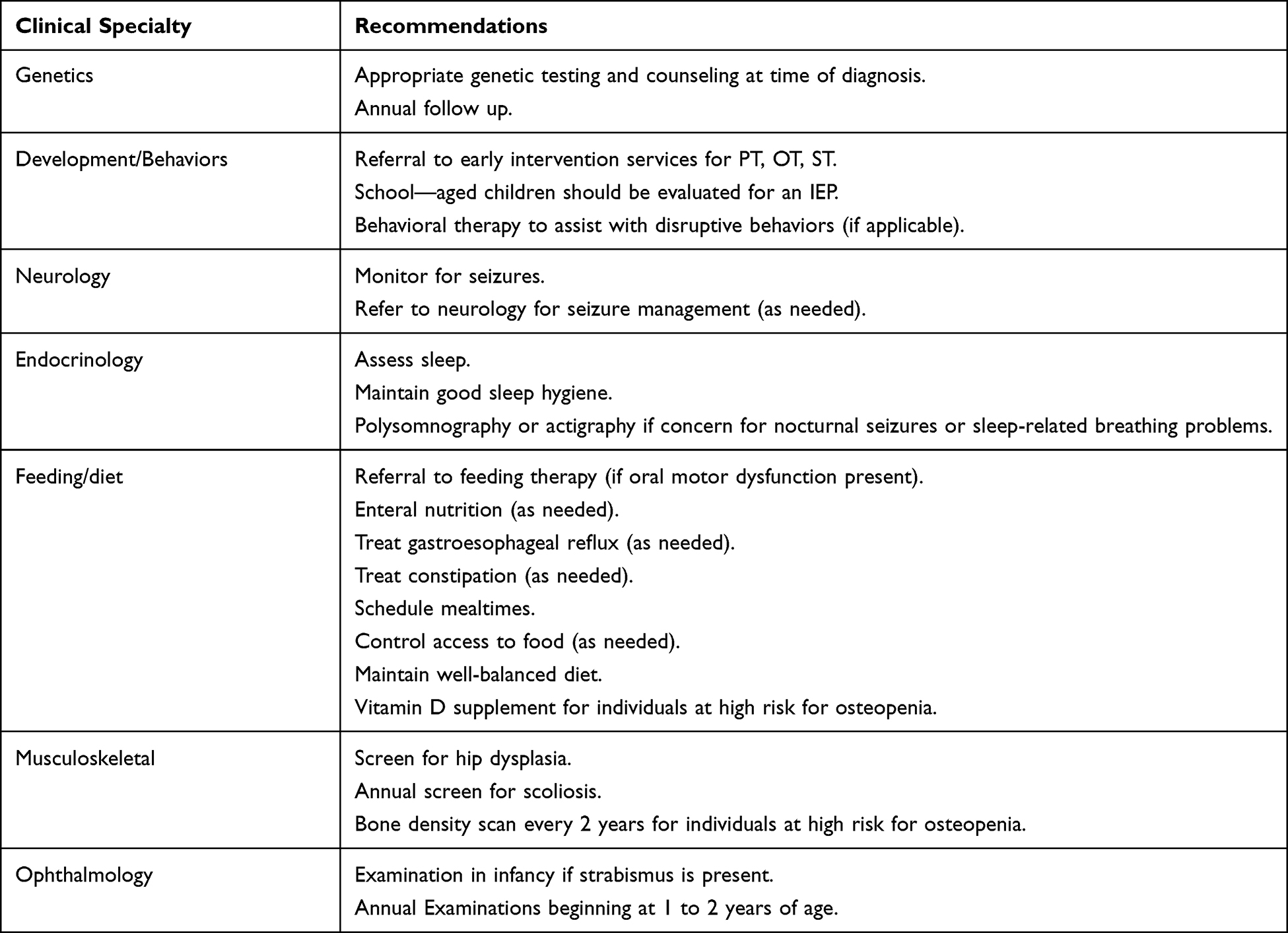 |
Table 2 Health Recommendations for AS. |
Genetic Counseling
Genetic counseling is an important component of PWS/AS management. Genetic counselors (GCs) provide psychosocial support and education to a family through the entire testing and diagnostic process. Specifically, topics may include counseling about the diagnosis, the complexities of PWS/AS genetics and genetic testing for PWS/AS. GCs facilitate discussions to help patients/families make the most informed decision about next steps in testing to clarify the genetic subtype of PWS/AS and associated recurrence risk. They provide resources including PWS/AS family support associations/organizations, whose information can also be found online.25,26 Families may interact with GCs in a prenatal setting for a positive family history or abnormal prenatal test for PWS/AS, or most commonly in a general or pediatric genetics setting.
For parents of a child with PWS or AS, recurrence risk for another child with the respective condition is most commonly <1%.27,28 However, rare genetic subtypes have a 50% recurrence risk (such as inheritance of a pathogenic variant in an imprinted gene) and theoretically up to 100% (15/15 Robertsonian translocation).20,29 While much less common, a female with PWS due to chromosome deletion or IC deletion has a 50% chance to have child with AS; males have not been reported to father children.25
Distinguishing the specific molecular class of PWS/AS etiology provides a more defined recurrence risk, allowing for family planning discussions including preconception/prenatal options for parents, at-risk siblings, other at-risk family members, and in rare cases, the patient themselves. It may also provide genotype-phenotype information.30,31 In some cases, after the initial genetic etiology is identified, follow-up testing for individuals with PWS/AS and if necessary, parents, to further elucidate the molecular subtype of proband’s diagnosis may be required. This may include karyotype/FISH in proband and/or parents to evaluate for translocation, UPD studies, IC deletion study in proband and/or parents, or targeted variant analysis of UBE3A variant in a parent (Figure 2).20
Prader-Willi Syndrome
Feeding/Diet
Neonatal
Infancy is characterized by severe hypotonia leading to decreased movement, lethargy, weak cry, and feeding and swallowing difficulties that may result in failure to thrive (FTT). Infants may benefit from special bottle nipples or enteral tube feeds and speech pathologists can assist with feeding and swallowing difficulties.25 Hypotonia typically improves over time.
Childhood
Early childhood is characterized by excessive eating and environmental modifications can be helpful to limit excessive food intake. Children should follow closely with a pediatric nutritionist to ensure a well-balanced diet and adequate nutrition. Individuals with PWS should also be monitored during meals and encouraged to chew and eat slowly as they are at risk for aspiration and choking.25,32 Consuming a glass of water prior to food consumption is encouraged.32
Endocrinology
Individuals with PWS should be closely followed by an endocrinologist as they may have associated hypothalamic and pituitary dysfunction. Growth hormone (GH) is most effective when started at an early age, thought to be related to the role of insulin-like growth factor 1 on brain development and maturation.32 GH replacement therapy can help normalize height, increase lean body mass, and decrease fat.33 IGF-1 level should be routinely monitored to titrate GH doses to optimize benefits and limit risks. Hypogonadism is present in both males and females and presents as genital hypoplasia early in life, incomplete pubertal development in adolescence, and infertility.34 Given the prevalence of cryptorchidism in males, human chorionic gonadotropin (HCG) should be considered in conjunction with an evaluation by a pediatric urologist for surgery.35 Replacement sex hormones should be considered at an appropriate age to reflect normal pubertal development and can also help improve bone density.32 Hormone levels should be monitored regularly, and replacement doses titrated as appropriate.
Central hypothyroidism may develop; thyroid function should be evaluated in the first several months of life and screened every 2–3 years or sooner if symptomatic, with a low threshold to initiate treatment.35 The relationship between central adrenal insufficiency and PWS is unclear, and the incidence varies significantly. As a result, there is less consensus on appropriate screening schedules. However, some evidence suggests that individuals with PWS may not be able to mount an adequate stress response and providers should consider screening for adrenal function and/or consider stress dose steroids during periods of significant illness.32
Individuals with PWS have impaired thirst and temperature regulation.33 Special care should be taken during extreme temperatures, and participation in sports and outdoor activities, including additional fluid intake and rest when needed. Antibiotics should not be delayed if there is clinical suspicion for infectious processes as body temperatures do not increase.32
Obesity and Obesity-Related Medical Complications
Obesity and obesity-related medical problems such as type II diabetes mellitus are a significant risk for individuals with PWS. Children and adolescents with PWS who have a body mass index greater than 95th percentile should have routine blood glucose and hemoglobin A1C and/or oral glucose tolerance tests.
Sleep
Sleep apnea (central and obstructive) and excessive daytime sleepiness is common in PWS. Obesity-related obstructive sleep apnea (OSA) should be managed to prevent associated complications; behavioral specialists may assist with compliance of oxygen or continuous positive airway pressure therapy. Those with excessive daytime sleepiness despite management of OSA should receive multiple sleep latency testing.32 Dysregulation of REM sleep and abnormal theta waves has been identified in PWS, supporting the role of genomic imprinting in sleep regulatory mechanisms.36 In addition, while there have been reports of sudden death in individuals with PWS while taking GH, the evidence is inconsistent. As a precaution, individuals with PWS should receive a polysomnography before and 8–10 weeks after starting growth hormone.32
Orthopedics
Approximately 10–20% of individuals with PWS will have hip dysplasia, noted at birth or later in infancy.25,33 Regardless of age, individuals should be monitored for scoliosis and radiographs ordered as clinically indicated. Symptoms vary in severity, typically occurring in infancy or more commonly in early adolescence. Individuals with PWS are at increased risk for fractures as osteoporosis and osteopenia are common.25,33 Adults should be monitored for osteoporosis with a dual-energy x-ray absorptiometry (DEXA) every 2 years.25
Development/Behaviors
Motor and language development are typically delayed in PWS and most persons with PWS have mild intellectual disability.25 Children with PWS should receive early intervention services including physical therapy (PT), occupational therapy (OT), and speech therapy (ST); PT and OT can address feeding challenges and speech and articulation difficulties, and PT can assist with hypotonia and core strength. Children may benefit from following up with a neurodevelopmental specialist to help address developmental concerns as children may also have learning disabilities and poor academic performance. School-aged children should be evaluated for an individualized education plan (IEP) to receive specialized instruction and services. Behavioral therapy can assist with social functioning, anxiety, and compulsive behaviors.
Clinical Trials
Since individuals with PWS are thought to have a deficit of oxytocin producing neurons in the paraventricular nucleus of the hypothalamus, intranasal OT is currently being studied as treatment for PWS symptoms. Results thus far have been inconclusive. One study reported improvements in socialization, anxiety, and repetitive behaviors, and decrease in appetite.37 Another study reported improvements in eating and social behaviors (anger, sadness, and conflicts) in children 11 years of age and younger.38 OT has also been shown to improve feeding and social skills in infants.39 A Phase III study of carbetocin, an analog of oxytocin, has been completed but results were insufficient for FDA approval (NCT03649477). Glucagon-like peptide-1 receptor agonists, traditionally used to treat diabetes mellitus, have also been shown to have benefits in glycemic, appetite, and weight control in PWS.40 Results from a phase III study of diazoxide choline showed improvements in participants with severe baseline hyperphagia and significant improvements in body composition.41 A phase II/III study of cannabidiol oral solution (NCT05098509) and unacylated ghrelin analog (AZP-531) is underway (NCT03790865).
Angelman Syndrome
Development/Behaviors
Individuals with AS have global developmental delay and intellectual disability; the severity of cognitive delay is multifactorial. Referrals should be made to early interventional services for PT, OT, and ST, and these services should continue throughout life. ST should focus on nonverbal forms of communication such as signing, and augmentative and alternative communication devices.26 School-aged children should have an IEP that focuses on maximizing educational potential and improving communication and motor skills. Appropriate classroom and school placement should be determined based on shared decision-making between caregivers and teachers. A functional behavioral assessment should be completed for children with behavioral challenges to identify and understand challenging behaviors to create a behavior intervention plan. Techniques, such as applied behavioral analysis, are recommended to reduce disruptive and/or self-injurious behaviors and improve adaptive skills.
Neurological
Epilepsy occurs in up to 90% of individuals with AS and is often refractory to anti-epileptic drugs (AED).42 While there have been no published guidelines on the use of various AED, broad-spectrum AED are generally recommended. Consensus recommendation is to start with clobazam or levetiracetam as the first-line treatment.42 Valproate should be avoided as it can cause an increase in tremors and regression of motor skills that resolves after tapering off valproate.43 Care should be taken to not overtreat with AED in the setting of abnormal movements or when electroencephalogram (EEG) abnormalities persist despite no clinical seizures.26 While mice models have found that cannabidiol decreases seizures and EEG abnormalities and anecdotal caregiver reports are promising, there has been no clinical research supporting its use.44 Ketogenic diet (KD) and low glycemic index therapy (LGIT) have also been shown to assist in cases of refractory epilepsy. KD is recommended for infants and children with feeding tubes and LGIT is first line for other children; LGIT should be switched to KD if it is not completely effective.42
Sleep
Individuals with AS should be routinely screened for sleep difficulties. Sleep challenges, such as increased sleep latency, and nighttime and early morning awakenings, are present in up to 80% of individuals with AS.45 A detailed sleep diary and bedtime routine should be reviewed if sleep is a concern. Polysomnography is indicated if there is concern for sleep-related breathing problems, nocturnal seizures, or unusual behaviors during sleep. For individuals who are not able to tolerate polysomnography, actigraphy is a non-invasive alternative means for measuring sleep parameters and motor activity.
Feeding/Diet
Involvement of a feeding therapist early on is recommended since AS may present with oral motor dysfunction. Sucking and swallowing difficulties can lead to FTT in infancy. Tongue protrusion and dyspraxia of swallowing and breathing during feeding may also contribute to FTT and aspiration. Enteral nutrition should be considered if conservative management is not successful or for those with a history of aspiration pneumonia.42 More than a quarter of children with AS exhibit hyperphagia, ranging from no intrinsic limitation on food consumption to actively searching for food and nonfood items.46 Suggested strategies include scheduled mealtimes and eating a well-balanced diet. Food should not be used as a reward.
Orthopedics
Children should be routinely screened for hip dysplasia and radiographs in frog leg position should be completed as necessary. Orthotics may be used for individuals with subluxed or pronated ankles; corrective osteotomy is controversial as recovery is often poor.42 PT is highly recommended to assist with mobility and improving range of motion. The incidence of scoliosis in children with AS is up to 20% and up to 50% in adults.26 Early use of thoracolumbosacral orthosis is recommended in individuals who are non-ambulatory.26 While surgical intervention for scoliosis has been shown to improve quality of life, it should be approached with caution as there is a high rate of surgical complications.47 A bone density scan is recommended every two years to assess bone health for those at higher risk including non-ambulatory individuals, and those taking AED, on KD, with a history of fractures without clear trauma, and with late onset of puberty.42 Vitamin D supplementation, daily exercise, and regular sunlight exposure are advised to help with bone density.
Clinical Trials
Trials of folate, betaine, metofalin, vitamin B12, creatine, and levodopa/carbidopa have not demonstrated significant clinical benefit.48–50 Gaboxadol (OV101), a direct agonist of δ-subunit–containing extrasynaptic γ-aminobutyric acid type A receptors ameliorated some of the neurologic symptoms seen in mice with AS.51 Results from Phase 2 were promising in adolescents and adults but results from the Phase 3 trial were negative (NCT04106557); study and development of the drug has since been halted.51 One open-label trial of minocycline suggested positive effects on language, fine motor skills, and some adaptive behaviors but a randomized placebo-controlled trial showed no significant improvements.52,53 Early results of a crossover trial of exogenous medical food ketone formulation showed improvements in stool consistency and suggest other benefits including fine motor skills (NCT03644693).54 There are also ongoing phase I/II trials of antisense oligonucleotide therapies (NCT04428281, NCT04259281).
Summary
In summary, PWS and AS are imprinting disorders due to absent or reduced expression of the paternal or maternal differentially expressed genes in chromosome 15q11q13 region, respectively. The multiple molecular etiologies of PWS/AS include pathogenic sequence variants in imprinted genes, copy number variants, uniparental disomy, and epimutations, with deletion at 15q11q13 being the most common. For an individual suspected to have PWS/AS, methylation testing is recommended, with MS-MLPA being the most sensitive testing method; CMA is an appropriate first-line test for individuals with developmental delay and/or hypotonia. Evaluation includes molecular testing for the individual and possibly the parents. Genetic counseling is recommended throughout this process. Clinically, individuals with PWS typically present with hypotonia and feeding difficulties in the neonatal period and go on to develop excessive eating behaviors and complications associated with obesity. In contrast, individuals with AS may appear to have normal development starting at birth but go on to demonstrate delays and develop seizures and self-injurious behaviors. Although PWS presents earlier with hyperphagia and obesity, some individuals with AS may present with hyperphagia and obesity as well. Ideally, individuals with PWS/AS should be followed in a multidisciplinary clinic to provide comprehensive medical, developmental, and behavioral management.
Disclosure
Dr. SP Shankar is Albert Rowe endowed chair in Genetics II & receives salary and research support. The authors report no other conflicts of interest in this work.
References
1. Butler MG. Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet. 2009;26(9–10):477–486. doi:10.1007/s10815-009-9353-3
2. Monk D, Mackey DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. 2019;20(4):235–248. doi:10.1038/s41576-018-0092-0
3. O’Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010;115(14):2731–2739. doi:10.1182/blood-2009-10-201848
4. Butler MG, Meaney FJ, Palmer CG, Opitz JM, Reynolds JF. Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet. 1986;23(793–809):793–809. doi:10.1002/ajmg.1320230307
5. Magenis RE, Brown MG, Lacy DA, Budden S, LaFranchi S. Is Angelman syndrome an alternate result of del(15)(q11q13)? Am J Med Genet. 1987;28(4):829–838. doi:10.1002/ajmg.1320280407
6. Procter M, Chou L-S, Tang W, Jama M, Mao R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem. 2006;52(7):1276–1283. doi:10.1373/clinchem.2006.067603
7. Christian SL, Robinson WP, Huang B, et al. Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet. 1995;57(1):40–48.
8. Amos-Landgraf JM, Ji Y, Gottlieb W, et al. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65(2):370–386. doi:10.1086/302510
9. Glenn CC, Saitoh S, Jong MT, et al. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am J Hum Genet. 1996;58(2):335–346.
10. Glenn CC, Driscoll DJ, Yang TP, Nicholls RD. Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Reprod. 1997;3(4):321–332. doi:10.1093/molehr/3.4.321
11. MacDonald HR, Wevrick R. The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet. 1997;6(11):1873–1878. doi:10.1093/hmg/6.11.1873
12. Runte M, Hüttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10(23):2687–2700. doi:10.1093/hmg/10.23.2687
13. Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 2013;9(12):e1004039. doi:10.1371/journal.pgen.1004039
14. Fridman C, Koiffmann CP. Origin of uniparental disomy 15 in patients with Prader-Willi or Angelman syndrome. Am J Med Genet. 2000;94(3):249–253. doi:10.1002/1096-8628(20000918)94:3<249::AID-AJMG12>3.0.CO;2-X
15. Robinson WP, Christian SL, Kuchinka BD, et al. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin Genet. 2000;57(5):349–358. doi:10.1034/j.1399-0004.2000.570505.x
16. Ohta T, Gray TA, Rogan PK, et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am J Hum Genet. 1999;64(2):397–413. doi:10.1086/302233
17. Mabb AM, Judson MC, Zylka MJ, Philpot BD. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011;34(6):293–303. doi:10.1016/j.tins.2011.04.001
18. Hosoki K, Takano K, Sudo A, Tanaka S, Saitoh S. Germline mosaicism of a novel UBE3A mutation in Angelman syndrome. Am J Med Genet A. 2005;138A(2):187–189. doi:10.1002/ajmg.a.30926
19. Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. doi:10.1146/annurev.genom.2.1.153
20. Beygo J, Buiting K, Ramsden SC, Ellis R, Clayton-Smith J, Kanber D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur J Hum Genet. 2019;27:1326–1340. doi:10.1038/s41431-019-0435-0
21. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi:10.1016/j.ajhg.2010.04.006
22. Levy B, Burnside R. Are all chromosome microarrays the same? What clinicians need to know. Prenat Diagn. 2019;39(3):157–164. doi:10.1002/pd.5422
23. Hoppman N, Rumilla K, Lauer E, Kearney H, Thorland E. Patterns of homozygosity in patients with uniparental disomy: detection rate and suggested reporting thresholds for SNP microarrays. Genet Med. 2018;20(12):1522–1527. doi:10.1038/gim.2018.24
24. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067. doi:10.1093/nar/gkx1153
25. Driscoll DJ, Miller JL, Schwartz S, et al. Prader-Willi Syndrome. Adam MP, Everman DB, Mirzaa GM, et al. editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993.
26. Dagli AI, Mathews J, Williams CA, et al. Angelman Syndrome. In: Adam MP, Everman DB, Mirzaa GM, editors. GeneReviews®. Seattle: University of Washington, Seattle; 1998.
27. Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005;7(14):1–20. doi:10.1017/S1462399405009531
28. Buiting K, Dittrich B, Gross S, et al. Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet. 1998;63(1):170–180. doi:10.1086/301935
29. Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26. doi:10.1038/gim.0b013e31822bead0
30. Keute M, Miller MT, Krishnan ML, et al. Angelman syndrome genotypes manifest varying degrees of clinical severity and developmental impairment. Mol Psychiatry. 2021;26(7):3625–3633. doi:10.1038/s41380-020-0858-6
31. Grootjen LN, Juriaans AF, Kerkhof GF, Hokken-Koelega ACS. Atypical 15q11.2-q13 deletions and the Prader-Willi phenotype. J Clin Med. 2022;11(15):4636. doi:10.3390/jcm11154636
32. Duis J, van Wattum PJ, Scheimann A, et al. A multidisciplinary approach to the clinical management of Prader–Willi syndrome. Mol Genet Genomic Med. 2019;7:e514. doi:10.1002/mgg3.514
33. Butler MG, Miller JL, Forster JL. Prader-Willi syndrome - clinical genetics, diagnosis and treatment approaches: an update. Curr Pediatr Rev. 2019;15(4):207–244. doi:10.2174/1573396315666190716120925
34. Yang-Li D, Fei-Hong L, Hui-Wen Z, et al. Recommendations for the diagnosis and management of childhood Prader-Willi syndrome in China. Orphanet J Rare Dis. 2022;17:221. doi:10.1186/s13023-022-02302-z
35. Shawn E. McCandless; the committee on genetics; health supervision for children with Prader-Willi syndrome. Pediatrics. 2011;127(1):195–204. doi:10.1542/peds.2010-2820
36. Lassi G, Priano L, Maggi S, et al. Deletion of the Snord116/SNORD116 alters sleep in mice and patients with Prader-Willi syndrome. Sleep. 2016;39(3):637–644. doi:10.5665/sleep.5542
37. Miller JL, Tamura R, Butler MG, et al. Oxytocin treatment in children with Prader-Willi syndrome: a double-blind, placebo-controlled, crossover study. Am J Med Genet A. 2017;173(5):1243–1250. doi:10.1002/ajmg.a.38160
38. Kuppens RJ, Donze SH, Hokken-Koelega AC. Promising effects of oxytocin on social and food-related behaviour in young children with Prader-Willi syndrome: a randomized, double-blind, controlled crossover trial. Clin Endocrinol. 2016;85(6):979–987. doi:10.1111/cen.13169
39. Tauber M, Boulanouar K, Diene G, et al. The use of oxytocin to improve feeding and social skills in infants with Prader-Willi syndrome. Pediatrics. 2017;139(2):e20162976. doi:10.1542/peds.2016-2976
40. Ng NBH, Low YW, Rajgor DD, et al. The effects of glucagon-like peptide (GLP)-1 receptor agonists on weight and glycaemic control in Prader-Willi syndrome: a systematic review. Clin Endocrinol. 2022;96(2):144–154. doi:10.1111/cen.14583
41. Miller JL, Gevers E, Bridges N, et al. Diazoxide choline extended-release tablet in people with Prader-Willi syndrome: a double-blind, placebo-controlled trial. J Clin Endocrinol Metab;2023. dgad014. doi:10.1210/clinem/dgad014
42. Duis J, Nespeca M, Summers J, et al. A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol Genet Genomic Med. 2022;10(3):e1843. doi:10.1002/mgg3.1843
43. Shaaya EA, Grocott OR, Laing O, Thibert RL. Seizure treatment in Angelman syndrome: a case series from the Angelman syndrome clinic at Massachusetts general hospital. Epilepsy Behav. 2016;60:138–141. doi:10.1016/j.yebeh.2016.04.030
44. Gu B, Zhu M, Glass MR, et al. Cannabidiol attenuates seizures and EEG abnormalities in Angelman syndrome model mice. J Clin Invest. 2019;129(12):5462–5467. doi:10.1172/JCI130419
45. Ascoli M, Elia M, Gasparini S, et al. Therapeutic approach to neurological manifestations of Angelman syndrome. Expert Rev Clin Pharmacol. 2022;15(7):843–850. doi:10.1080/17512433.2022.2109463
46. Bindels-de Heus KG, Mous SE, ten Hooven-Radstaake M, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet Part A. 2020;182:53–63. doi:10.1002/ajmg.a.61382
47. Sewell MD, Wallace C, Gibson A, et al. A retrospective review to assess whether spinal fusion and scoliosis correction improved activity and participation for children with Angelman syndrome. Dev Neurorehabil. 2016;19(5):315–320. doi:10.3109/17518423.2014.980524
48. Peters SU, Bird LM, Kimonis V, et al. Double-blind therapeutic trial in Angelman syndrome using betaine and folic acid. Am J Med Genet A. 2010;152A(8):1994–2001. doi:10.1002/ajmg.a.33509
49. Bird LM, Tan WH, Bacino CA, et al. A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome. Am J Med Genet A. 2011;155A(12):2956–2963. doi:10.1002/ajmg.a.34297
50. Tan WH, Bird LM, Sadhwani A, et al. A randomized controlled trial of levodopa in patients with Angelman syndrome. Am J Med Genet A. 2018;176(5):1099–1107. doi:10.1002/ajmg.a.38457
51. Bird LM, Ochoa-Lubinoff C, Tan WH, et al. The STARS Phase 2 Study: a Randomized Controlled Trial of Gaboxadol in Angelman Syndrome. Neurology. 2021;96(7):e1024–e1035. doi:10.1212/WNL.0000000000011409
52. Grieco JC, Ciarlone SL, Gieron-Korthals M, et al. An open-label pilot trial of minocycline in children as a treatment for Angelman syndrome. BMC Neurol. 2014;14:232. doi:10.1186/s12883-014-0232-x
53. Ruiz-Antoran B, Sancho-López A, Cazorla-Calleja R, et al. A randomized placebo controlled clinical trial to evaluate the efficacy and safety of minocycline in patients with Angelman syndrome (A-MANECE study). Orphanet J Rare Dis. 2018;13(1):144. doi:10.1186/s13023-018-0891-6
54. Carson RP, Herber DL, Pan Z, et al. Nutritional formulation for patients with Angelman syndrome: a randomized, double-blind, placebo-controlled study of exogenous ketones. J Nutr. 2021;151(12):3628–3636. doi:10.1093/jn/nxab284
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.