Back to Journals » OncoTargets and Therapy » Volume 11
PPP5C promotes cell proliferation and survival in human prostate cancer by regulating of the JNK and ERK1/2 phosphorylation
Authors Lv JM, Chen L, Gao Y, Huang H, Pan XW ![]() , Liu X, Chen M, Qu FJ, Li L, Wang JK, Cui XG, Xu DF
, Liu X, Chen M, Qu FJ, Li L, Wang JK, Cui XG, Xu DF
Received 2 January 2018
Accepted for publication 9 June 2018
Published 12 September 2018 Volume 2018:11 Pages 5797—5809
DOI https://doi.org/10.2147/OTT.S161280
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Jianmin Xu
Jian-Min Lv,1–3,* Lu Chen,1,* Yi Gao,1,* Hai Huang,1–3,* Xiu-Wu Pan,2,3,* Xi Liu,1 Ming Chen,2 Fa-Jun Qu,3 Lin Li,3 Jun-Kai Wang,2 Xin-Gang Cui,3,4,* Dan-Feng Xu1,*
1Department of Urinary Surgery, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200025, China; 2Department of Urinary Surgery, Changzheng Hospital, Second Military Medical University, Shanghai 200003, China; 3Department of Urinary Surgery, Third Affiliated Hospital, Second Military Medical University, Shanghai 201805, China; 4Department of Urinary Surgery, Gongli Hospital, Second Military Medical University, Shanghai 200135, China
*These authors contributed equally to this work
Background: Prostate cancer (PCa) is one of the most common malignancies and a major leading cause of cancer-related deaths in males. And it is necessary to explore new molecular targets to enhance diagnosis and treatment level of the PCa. Serine/threonine protein phosphatase 5 (PPP5C) is a vital molecule that Involve in complex cell physiological activity.
Purpose: The objective of this study was to detecte the expression level of PPP5C in the tissue of prostate cancer patients and further discussed the PPP5C biological function and mechanisms on the PCa.
Methods: The expression level of PPP5C was analyzed by immunohistochemistry and ONCOMINE datasets. Lentivirus-mediated short hairpin RNA (shRNA) was constructed to silence the expression of PPP5C in prostate cancer cell. Cell viability and proliferation were measured using MTT and colony formation, and the cell cycle and apoptosis was analyszed by flow cytometry. The changes of downstream protein level and protein phosphorylation level were detected by western blot.
Results: PPP5C was highly expressed in PCa tissue as analyzed by immunohistochemistry and ONCOMINE datasets. PPP5C Knockdown inhibited cell proliferation and colony formation in PCa cells. Flow cytometry analysis showed that DU145, PC3 and 22RV1 PCa cells deprived of PPP5C were arrested in G0/G1 phase and became apoptotic. Western blot analysis indicated that PPP5C knockdown could promote JNK and ERK phosphorylation.
Conclusion: Our study indicated that the PPP5C may become a new potential diagnostic biomarker and therapeutic target for the PCa.
Keywords: PPP5C, prostate cancer, tumorigenesis, JNK, ERK
Introduction
Prostate cancer (PCa) is one of the most common malignancies and a major leading cause of cancer-related deaths in males. Approximately 1,111,700 new cases are diagnosed and roughly 307,500 deaths occur annually, ranking the second incidence and the fifth mortality worldwide.1 Early localized PCa treated with radical prostatectomy or radical radiotherapy can receive an excellent prognosis, with a 5-year survival rate over 99%.2 However, 25%–50% patients will experience biochemical recurrence after radical prostatectomy, which is associated with a high risk of distant metastasis and death.3,4 Androgen deprivation therapy (ADT) is the most significant treatment for new or recurrent metastatic PCa at present.5 However, with continuous ADT, most hormone-sensitive prostate cancer would transform to castration-resistant prostate cancer in several months.6 For which few efficacious therapies are currently available. Thus, it is necessary to explore new molecular targets to enhance diagnosis and treatment level of the PCa.
Serine/threonine protein phosphatase 5 (PPP5C) is a member of the protein serine/threonine phosphatase family that plays an important role in life activities by regulating the phosphorylation of protein serine/threonine residues and activating or inactivating the corresponding substrates.7 PPP5C was found to be expressed in almost all human tissues and involved in numerous cellular processes.8 PPP5C could interfere with cell proliferation and differentiation by influencing the activity of the mitogen-activated protein kinase (MAPK) signaling pathway.9,10 It was also found associating with the DNA damage repair via interacting with DNA-dependent protein kinase catalytic subunit, ataxia telangiectasia mutated, ATM- and RAD3-related, and p53.11–14 Besides, PPP5C could impact the intracellular signaling cascades initiated by hormonal response and cellular stress by regulating several proteins, such as GR, ASK1, and Cdc37.15–17 The latest studies have demonstrated that PPP5C plays an important role in the cellular signaling networks.
To date, there were several evidences indicating that PPP5C is correlated with the malignancy. The study by Jeong et al found that the PPP5C was expressed at elevated levels in proliferating cells of the yeast.18 Several subsequent studies19–21 also reported that PPP5C was upregulated in breast cancer, mantle-cell lymphoma and liver cancer tissues, and ascites. Other studies22–25 reported that downregulation of PPP5C expression could inhibit malignant cells in human glioma, ovarian cancer, hepatocellular carcinoma, and colorectal cancer. The aforementioned findings suggest that PPP5C may play a role in the development and progression of some cancers. However, the association between PPP5C and PCa remains elusive. In this study, we first performed immunohistochemical detection and bioinformatics analysis of Oncomine database between cancer and normal tissues and found that PPP5C was up-expressed in PCa tissue. Then, we employed lentivirus-mediated shRNA to disrupt the PPP5C expression in PCa cells and found that PPP5C silencing inhibited the proliferation and colony formation abilities of PCa cells, arrested them in G0/G1 phase, and increased their apoptosis. Finally, we discovered by Western blot assay that c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated protein kinase (ERK)1/2 phosphorylation were augmented following downregulation of PPP5C. These results demonstrate that PPP5C may play an oncogene role in PCa advancement.
Materials and methods
Immunohistochemistry
Fifty-two pairs of PCa and matched normal prostate tissues were collected from radical prostatectomy specimens at the Department of Urosurgery of Changzheng Hospital (Shanghai, China) between 2012 and 2014. The specimens were formalin fixed, paraffin embedded, sliced into 5-μm thick sections, de-waxed successively from xylene, 100%, 90%, 80%, and 70% ethanol and ddH2O, and incubated overnight with the rabbit anti-PPP5C antibody (#:11715-1-AP, Proteintech, Rosemont, IL, USA) after antigen repair by citrate buffer (0.01M, pH=6.0). On the following day, the tissue sections were interacted with the biotinylated goat anti-rabbit serum and streptavidin-peroxidase conjugate for 15 minutes each at room temperature, stained by diaminobenzidine, and finally, sent to 2 pathologists for judging the positivity and intensity of the PPP5C staining. The staining results were microscopically photographed and assessed as following:
Positivity (A): 0%: 0 score; 0%–20%: 1 score; 20%–60%: 2 score; 60%–100%: 3 score
Intensity (B): Light yellow: 0 score; Yellow: 1 score; Brown yellow: 2 score; Reddish brown: 3 score
Outcome (A+B): 0 score: negative (−); 1 score: weak (+); 2–4 score: medium (++); 4–6 score: strong (+++)
All processes involving human tissues in our study were approved by the ethics committee of the Changzheng Hospital of the Second Military Medical University and written informed consent was obtained from each patient.
Bioinformatic analysis of the ONCOMINE microarray datasets
A set of public and published microarray data were used to analyze the PPP5C mRNA level between the PCa and normal prostate tissue or other through the Oncomine microarray datasets (www.onocomine.org), using “prostate cancer”, “PPP5C”, and “mRNA” as terms for searching the objective data. Then, we further selected the microarray datasets that contained the information about the “Cancer vs Normal tissue” or the “distant metastasis vs the primary lesion”, and we further extracted the mRNA expression value in each corresponding tissue samples. In this way, we finally acquired several microarray datasets for depth analysis. The 4 datasets, “Wallace Prostate”, “Vanaja Prostate”, “Welsh Prostate”, and “Arredouani Prostate”, were used to evaluate the PPP5C mRNA expression between level cancer and normal tissues, using “Yu Prostate”, “Holzbeierlein Prostate”, “Ramaswamy Prostate”, and “Ramaswamy multi-cancer 2 Prostate” for analyzing the PPP5C level between the metastatic and primary PCa tissues.
Cell culture
We purchased the human PCa LNCaP, 22RV1, DU145 and PC3 cells, and human embryonic kidney cell line 293T from the cell bank of the Chinese Academy of Science (Shanghai, China). The DU145 and PC3 cells were cultured in Ham’s F-12 Nutrient Mixture (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS, Thermo Fisher Scientific). The 22RV1 and LNCaP cells were maintain in Roswell Park Memorial Institute-1640 (HyClone, Logan, UT, USA) supplemented 10% FBS (Thermo Fisher Scientific). And, the 293 T cells were held in DMEM medium (Hyclone) added with 10% FBS (Thermo Fisher Scientific). Finally, all the cells were cultured under the recommended conditions: 5% CO2 at 37°C.
Lentivirus construction
siRNA targeting PPP5C (NC_000019.10) and non-silencing siRNA were transformed into shRNA and cloned into pFH-L green fluorescent protein (GFP) vector (Shanghai Hollybio, China) by double digestion with NheI and PacI. The generated plasmid shPPP5C-pFH-L or shCon-pFH-L, together with 2 pHelper plasmids pVSVG-I and pCMVΔR8.92 was transfected into 293 T cells. After 72-hour transfection, the lentivirus (Lv-shPPP5C or Lv-shCon) in the culture medium was collected and purified by ultracentrifugation, and the titer of each lentivirus was determined. The sequences of the shRNA are as follows: 5′-GAGACAGAGAAGATTACAGTACTCGAGTACTGTAATCTTCTCTGTCTCTTTTT-3′ (shPPP5C) and 5′-TTCTCCGAACGTGTCACGTATCTCGAGATACGTGACACGTTCGGAGAATTTTT-3′ (shcon).
Cell infection with lentivirus
DU145, PC3, or 22RV1 cells at a density of 1 × 105/well were placed in 6-cm dishes for 6 hours. Then, cells in each group were infected with the lentivirus Lv-shPPP5C, lentivirus Lv-shCon, and no lentivirus medium. The multiplicity of infection (MOI) of DU145, PC3, and 22RV1 was 45, 40, and 15, respectively. The virus volume was calculated using the following equation: viral particle = MOI*5 × 104, viral volume = viral particle/TU·mL−1. The infection efficiency was evaluated by the percentage of GFP-positive cells after 3–5 days.
Quantitative real-time polymerase chain reaction (PCR)
DU145, PC3, and 22RV1 cells were collected. Five days after infection with lentivirus, total RNA was extracted by using the Trizol reagent (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer’s instructions, and the total RNA concentration was tested by using ultraviolet analysis. The purification quality was estimated by the ratio of 260/280. Total RNA (2 μg) was reverse transcribed to cDNA with the M-MLV Reverse Transcriptase (Promega, Madison, WI, USA) and Oligo primer as the standard processes. The resulting cDNA was subjected to qPCR (SYBR™ Green, Bio-Rad Laboratories Inc., Hercules, CA, USA) using CFX Connect™ Real-Time PCR Detection System (Takara, Dalian, China). Primers used in real-time PCR are as follows: PPP5C forward, GGTGAGGTGAAGGCCAAGTA; PPP5C reverse, TGTGGATCTGACCAGAGCAG; β-actin forward, GTGGACATCCGCAAAGAC; β-actin reverse, AAAGGGTGTAACGCAACTA.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot assay
The SDS-PAGE was confected by H2O, 30% PAGE, 1.0 mol/L Tris (pH 6.8) or 1.5 mol/L Tris (pH 8.8), 10% SDS, 10% ammonium persulfate, and TEMED as the standard processes. In this way, 10% separation and 5% concentrated gels were acquired for next electrophoresis. Cells for Western blot assay were collected and lysed in the ice-cold lysate (100 mM Tris-HCl, pH 6.8, 10 mM EDTA, 4% SDS, and 10% Glycine). Then, the protein of varying molecular weight was separated via electrophoresis on the SDS-PAGE after centrifugation and protein quantification of the lysate. Next, the protein on the SDS-PAGE was transferred into the polyvinylidene difluoride (PVDF) membranes for 60–120 minutes. Following, the PVDF membranes were blocked in the 40–50 mL tris-buffered saline with Tween 20 (TBST) with 5% milk for 1 hour and incubated with the following primary antibodies overnight at 4°C: glyceraldehyde-3-phosphate dehydrogenase (#:10494-1-AP, Proteintech); PPP5C (#:11715-1-AP, Proteintech), JNK (#:9252, Cell Signaling, Beverly, MA, USA), P-JNK (#:4668, Cell Signaling), P38 (#:14064-1-AP, Proteintech), P-P38 (#:11581, Cell Signaling), ERK1/2 (#:11257-1-AP, Proteintech), and P-ERK1/2 (#:4370, Cell Signaling). Afterward, the PVDF membranes were washed 3 times in the TBST and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (#:Sc-2054, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1–2 hour at the room temperature. Finally, the PVDF membranes were imaged by the enhanced chemiluminescence plus TM Western blotting system kit.
Cell proliferation assay
DU145, PC-3, and 22RV1 cells at a density of 3 × 103/well were grown in 96-well plates for 5 repetitions and at 5 time points of day 1, 2, 3, 4, and 5. Then, each well was added with MTT (Sigma-Aldrich Co., St Louis, MO, USA) 20 μL (5 mg/mL) for 4 hours and acidic isopropanol (10% SDS, 5% isopropanol, and 0.01 mol/L HCl) for 12 hours at 37°C at the same time every 5 days. On the following day, the absorbance of each well was determined at 570 nm.
Plate colony formation assay
After lentivirus infection, DU145, PC3, and 22RV1 cells at a density of 6 × 102/well in the logarithmic growth phase were trypsinized, resuspended, and plated in 6-well plates, with the culture medium replaced every 3 days during cell clonal growth at 37°C with 5% CO2. After culture for 9–12 days, cells were fixed in 4% paraformaldehyde and stained with purple crystals. Then, the number of colonies in the 6-well plate were counted under a microscope and photographed with a digital camera.
Cell cycle analysis
After stable transduction with the lentivirus, each group of DU145, PC3, and 22RV1 cells was cultured in 6-cm plates for 3 days, washed with PBS, fixed in 75% ethanol at 4°C overnight, washed again, and stained with 20 mg/mL propidium iodide (PI; Sigma). The DNA content was recorded by a flow cytometer (BD Biosciences, Bedford, MA, USA). Every experiment was completed in triplicate.
Cell apoptosis analysis
DU145, PC3, and 22RV1 cells at a density of 1 × 105/well were infected with the lentivirus and cultured in 6-cm dishes. Three days later, each group cells were harvested and treated with annexin V/PI (BD Biosciences) for cell apoptosis analysis by flow cytometry (BD Biosciences). Living cells (annexin V negative, PI negative), early apoptotic cells (annexin V positive, PI negative), late apoptotic cells (annexin V positive, PI positive), and necrotic cells (annexin V negative, PI positive) were identified by FlowJo 7.6 Software (TreeStar Inc, Ashland, OR, USA). Each experiment was performed in triplicate.
Statistical analysis
All the experiments were repeated for at least 3 times. The value was presented as mean ± SD. The Wilcoxon signed-rank test was used to analyze the PPP5C level of the Immunohistochemistry score and microarray datasets. Statistical comparisons between the Lv-shCon and Lv-shPPP5C of the biological experiments were performed using Student’s t-test. The SPSS version 20 (IBM Corporation, Armonk, NY, USA) was used to calculate the statistics, and P-values<0.05 were considered statistically significant.
Results
PPP5C is overexpressed in PCa tissue
The PCa and the corresponding adjacent tissues were obtained from the patients for immunohistochemical analysis of the PPP5C protein level. As shown in Figure 1A, PPP5C staining of the PCa tissue was deeper and wider than that of the adjacent tissue. The negative, weakly positive, positive, and strongly positive rates in PCa group were 25%, 11.5%, 50%, and 13.5%, respectively vs 50%, 30.8%, 19.2%, and 0% in adjacent group (Table 1), indicating that PPP5C protein level was highly expressed in PCa tissue compared with the adjacent normal tissue (P<0.001). Subsequently, differences in PPP5C expression between PCa and normal prostate tissues were further validated by public online ONCOMINE microarray database, showing that the PPP5C mRNA level in PCa tissue was overexpressed in multiple databases vs the normal tissue (Figure 1B). Further analysis revealed that the PPP5C expression was even higher in the metastatic tissue than that in the primary PCa tissue (Figure 1C).
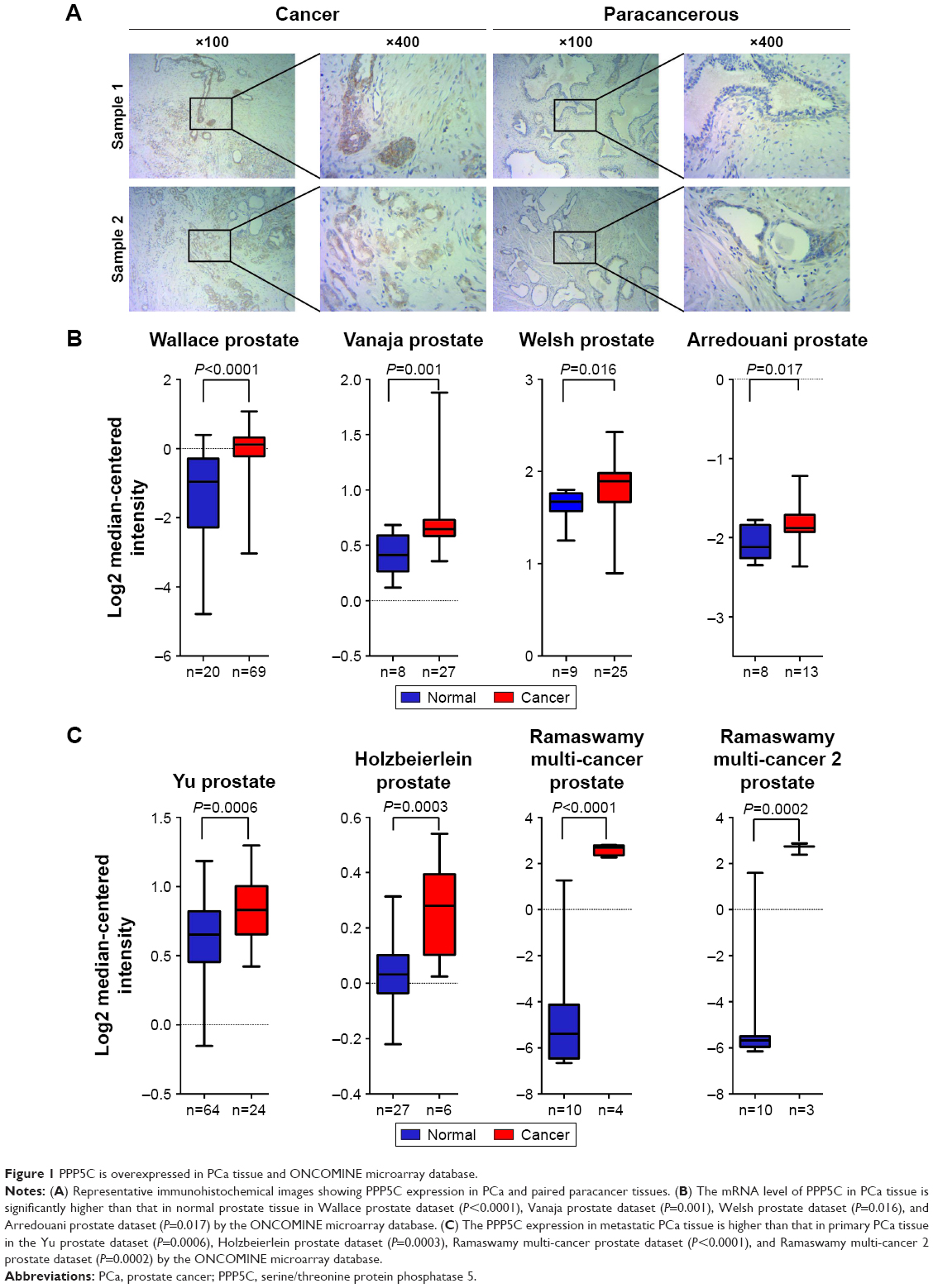 | Figure 1 PPP5C is overexpressed in PCa tissue and ONCOMINE microarray database. |
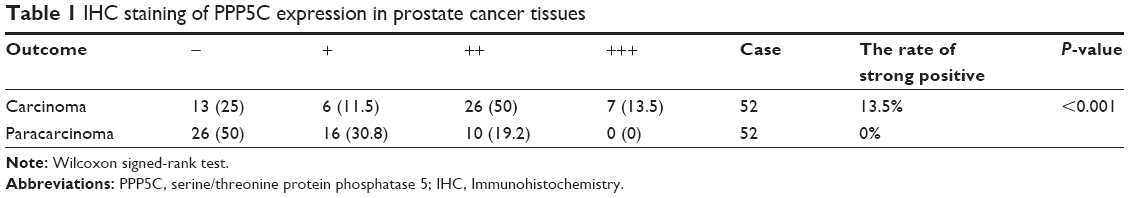 | Table 1 IHC staining of PPP5C expression in prostate cancer tissues |
The expression level and lentivirus-mediated interference of PPP5C in PCa cell lines
To explore the role of PPP5C, the baseline expression level of PPP5C in PCa cell lines was first examined. qPCR and Western blot assay results showed that PPP5C was highly expressed in common PCa cell lines LNCap, 22RV1, PC3, and DU145, and the expression level of PPP5C in DU145, PC3, and 22RV1 cells were more highly expressed than that in LNCap cells (Figure 2A and B). Thus, DU145, PC3, and 22RV1 were chosen for founding the PPP5C down-expressed PCa cell line model in lentivirus-mediated transduction. As shown in Figure 2C, the transfection efficiency of the LV-shPPP5C lentivirus to PCa DU145, PC3, and 22RV1 cells was over 90% via the GFP-expression analysis of the fluorescence microscope. The results of qPCR and Western blot assays showed that the mRNA and protein levels of PPP5C were fully knocked down in Lv-shPPP5C-infected DU145, PC3, and 22RV1 cell lines 3–5-days post-transduction (Figure 2D and E), indicating that the PPP5C low-expression PCa cell lines were successfully established by lentivirus-mediated RNAi.
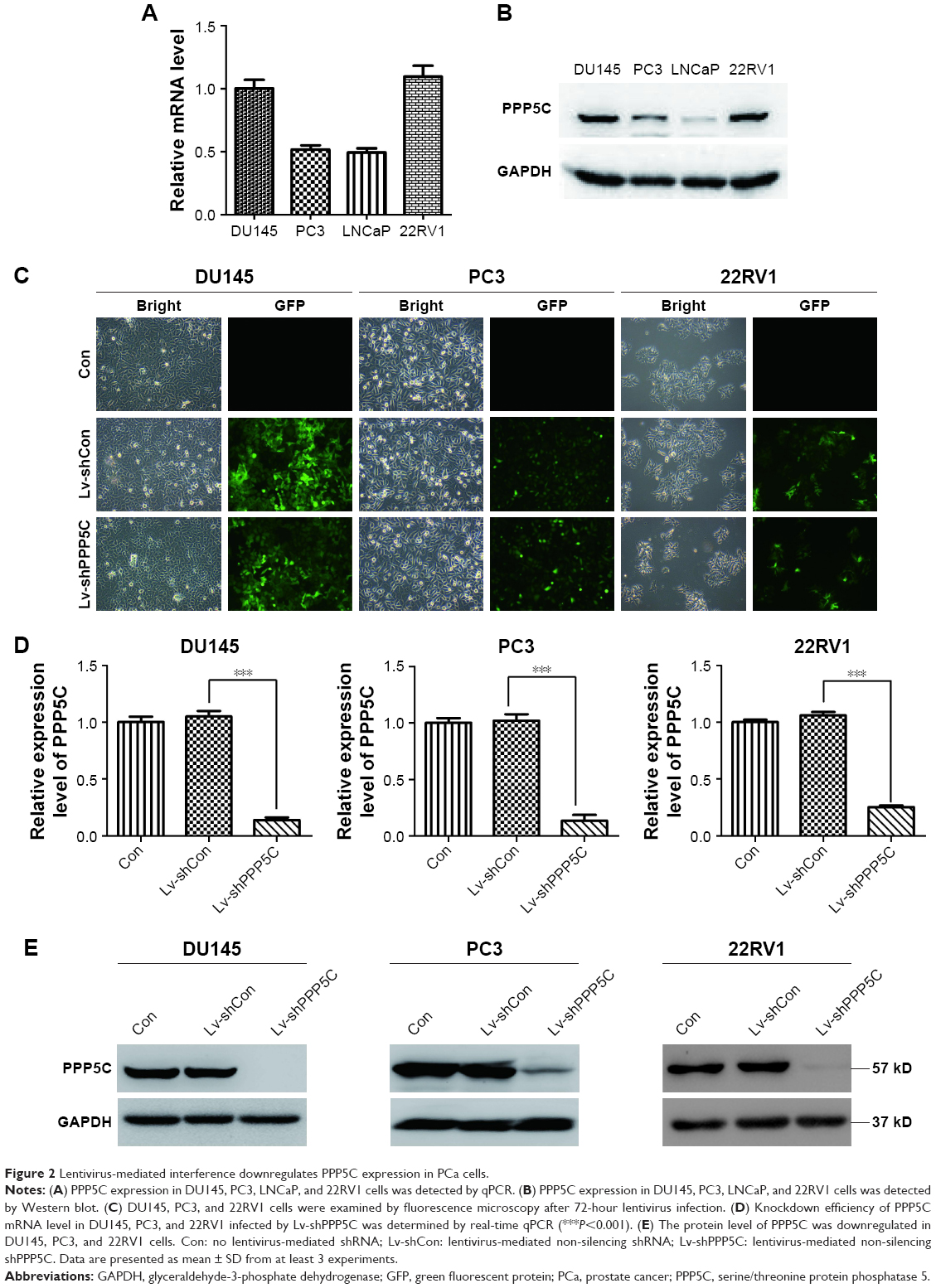 | Figure 2 Lentivirus-mediated interference downregulates PPP5C expression in PCa cells. |
Knockdown of the PPP5C expression inhibits the proliferation and colony ability of DU145, PC3, and 22RV1 cells
MTT assay was performed to evaluate the impact of PPP5C on the proliferation ability of PCa cells. As shown in Figure 3A, the growth curves in PPP5C knockdown groups of DU145, PC3, and 22RV1 cells were remarkably flatter in a time-dependent manner.
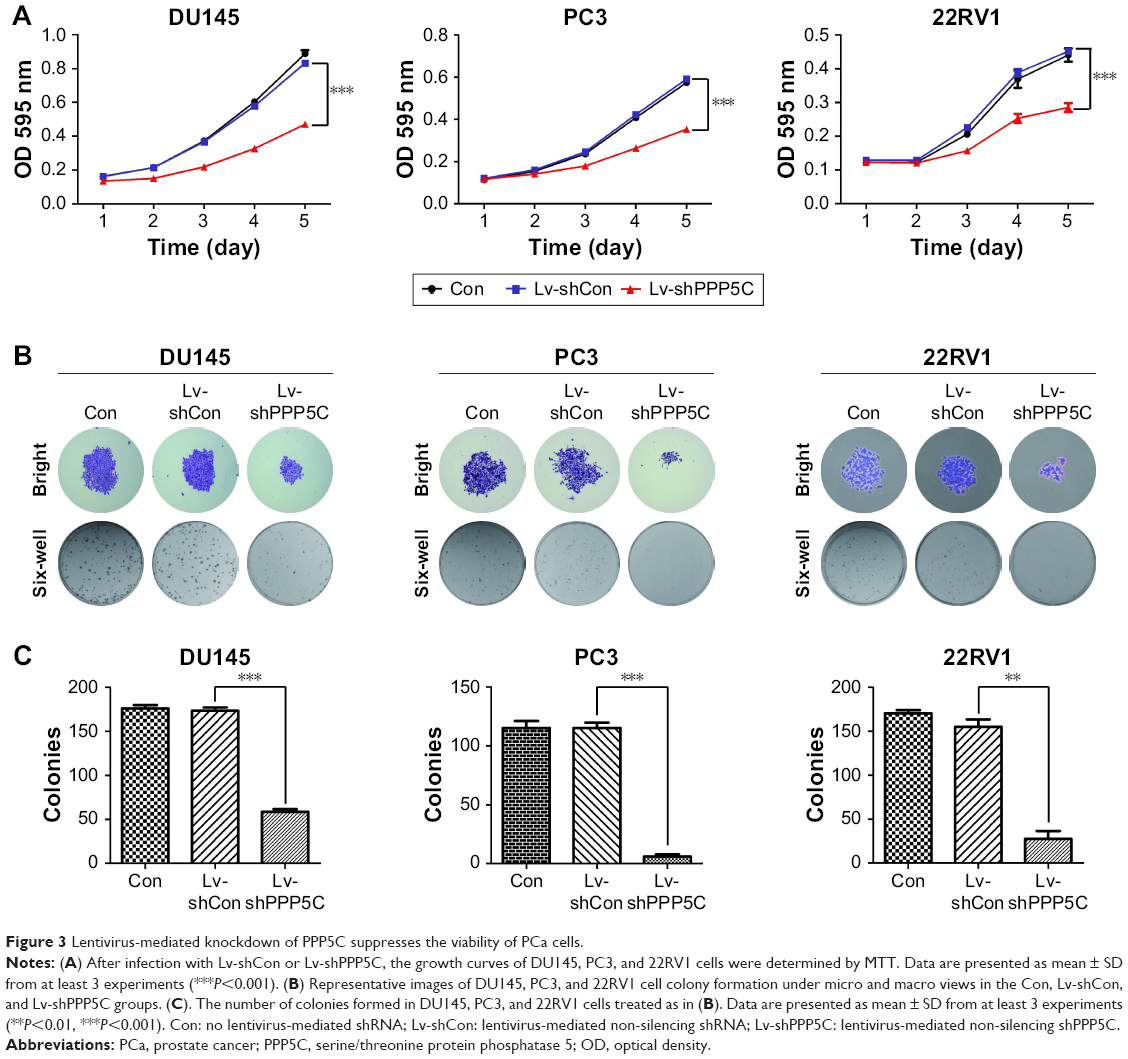 | Figure 3 Lentivirus-mediated knockdown of PPP5C suppresses the viability of PCa cells. |
The plate colony formation assay was used to examine the colony formation ability of the cells. It was found that the size and quantity of colony formation were reduced markedly in LV-shPPP5C transduction group (Figure 3B and C), indicating that PPP5C played an important role in the growth and colony formation of PCa cells.
Disruption of the PPP5C level induces G0/G1 arrest in PCa cells
Knowing that abnormal cell cycle is a significant characteristic during carcinogenesis, flow cytometry was performed to analyze the transform of each cell cycle phase via PI staining. As shown in Figure 4A, DU145, PC3, and 22RV1 cells underwent significant difference changes in cell cycle between Lv-shPPP5C and Lv-shCon groups. Further statistical analysis showed that PPP5C knockdown increased the number of cells arrested in G0/G1 phase, and the number of cells in S and G2/M phase decreased in varying degrees (Figure 4B), indicating that disruption of the PPP5C expression would result in G0/G1 arrest in PCa DU145, PC3, and 22RV1 cells.
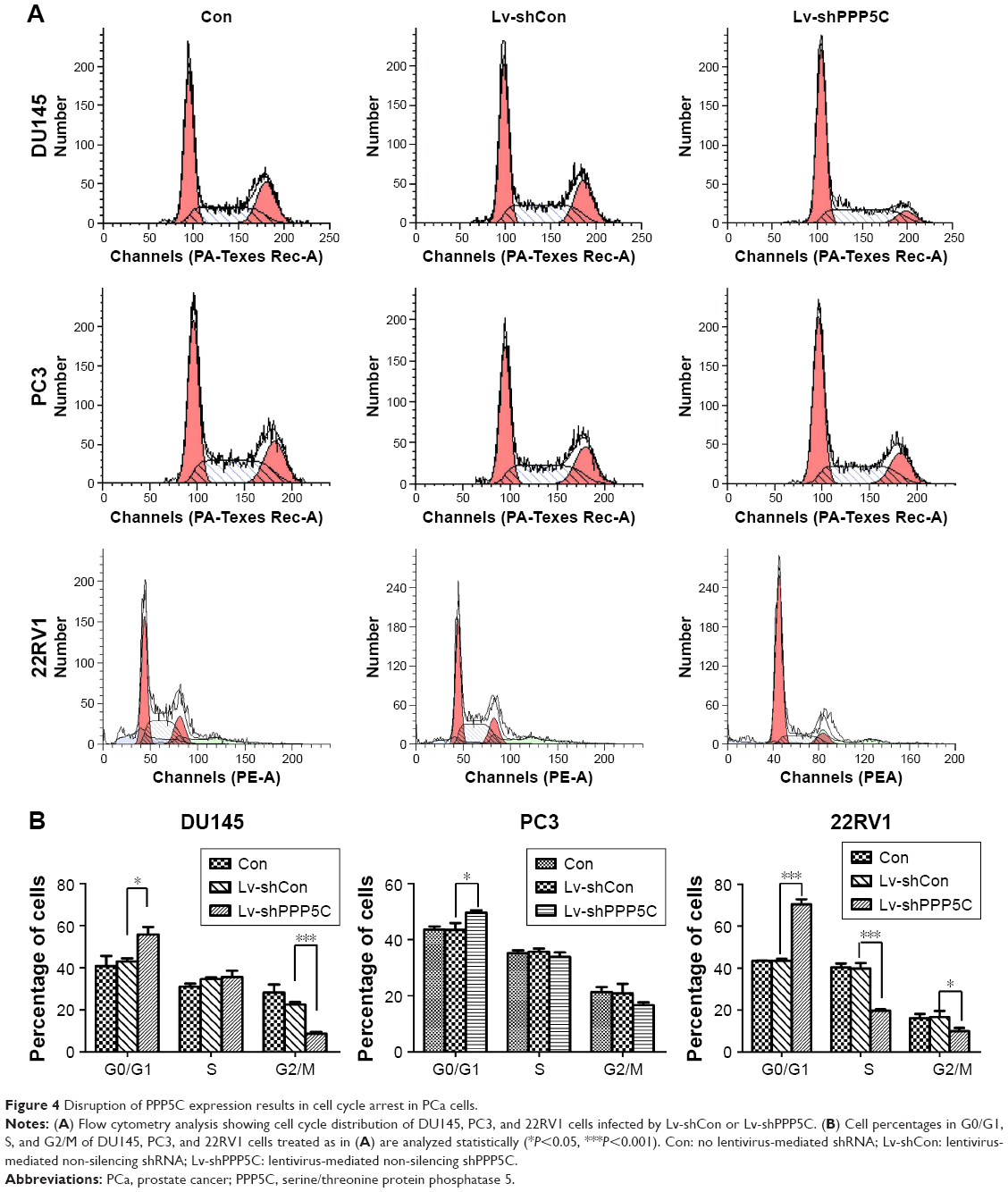 | Figure 4 Disruption of PPP5C expression results in cell cycle arrest in PCa cells. |
Downregulation of the PPP5C expression increases apoptosis of PCa cells
Knowing that abnormal apoptosis is the most important biological characteristic of malignant cells, flow cytometry and annexin V/PI staining were performed to detect apoptosis to further evaluate the impact of PPP5C on PCa cells. As shown in Figure 5A, the number of early apoptotic cells (annexin V+/PI−) and late apoptotic cells (annexin V+/PI+) were slightly elevated in PPP5C knockdown group of PCa DU145, PC3, and 22RV1 cells. Then, we performed statistical analysis of the proportion of each phase according to the annexin V and PI staining (Figure 5B).
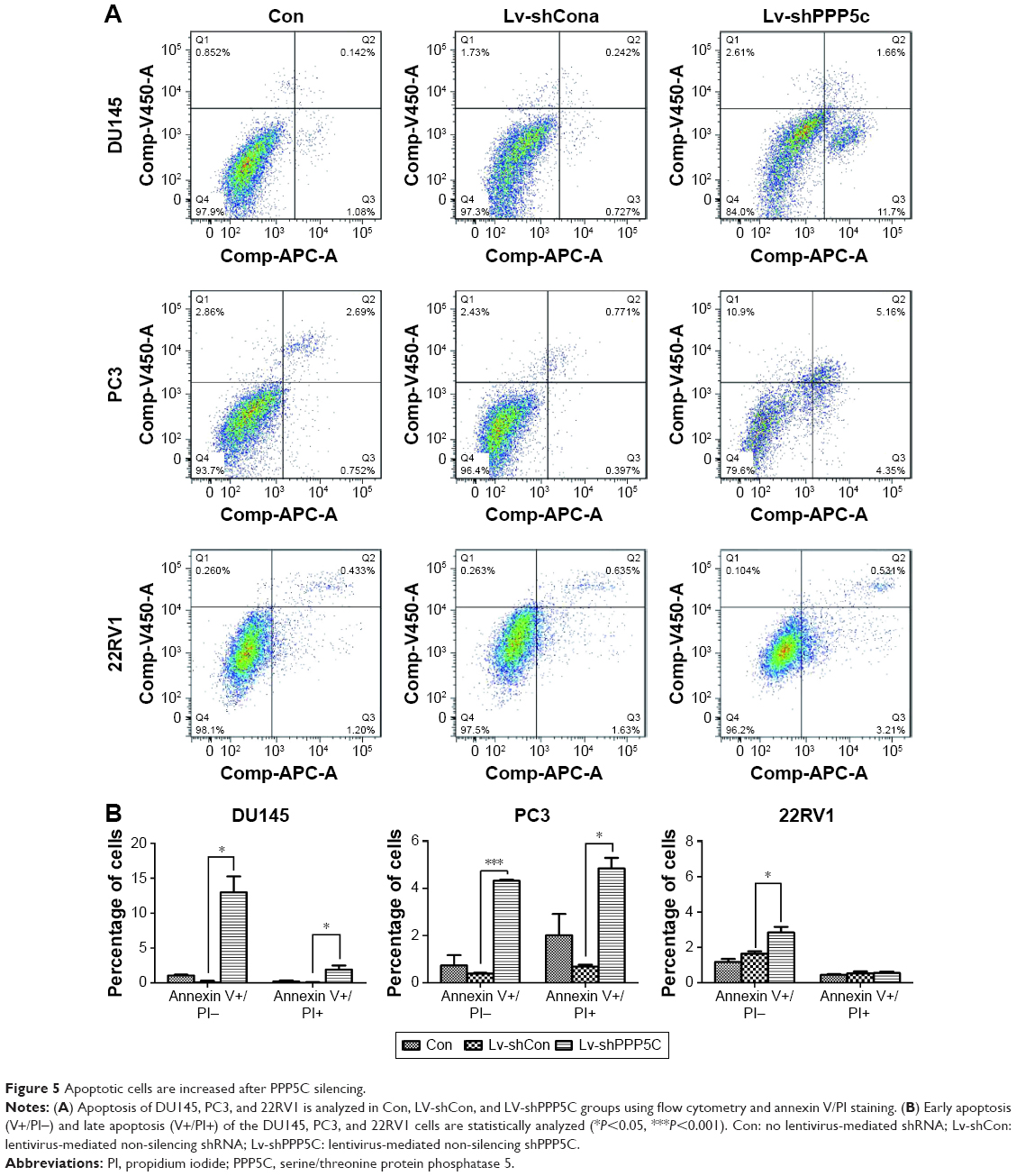 | Figure 5 Apoptotic cells are increased after PPP5C silencing. |
PPP5C knockdown augments phosphorylation of JNK and ERK1/2 in PCa cells
To explore the underlying mechanism of PPP5C in regulating the biological characteristics of the PCa cells, Western blot was performed to detect changes in protein expression. As shown in Figure 6A, the phosphorylation level of JNK and ERK1/2 was markedly increased with invariable non-phosphorylation. At the same time, the P38 protein level was always consistent with phosphorylation or non-phosphorylation after PPP5C knockdown.
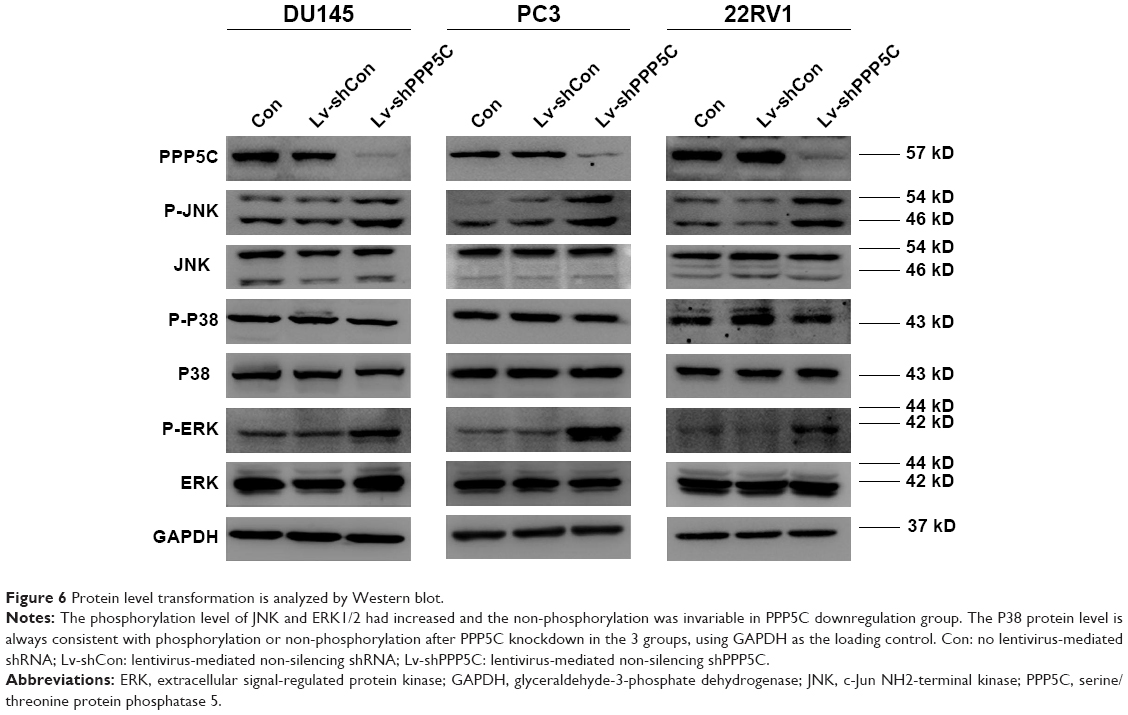 | Figure 6 Protein level transformation is analyzed by Western blot. |
Discussion
Reversible phosphorylation of protein is an important regulatory mechanism in life by changing the substrate activity through phosphorylation and dephosphorylation of protein for regulating the physiological function and pathological changes of cells. It has been demonstrated to play important roles in various physiological activities, such as cell growth, senescence, and apoptosis by influencing numerous signaling networks.26 PPP5C is one of the most common molecules in the phosphoprotein phosphatase (PPP) family of Ser/Thr phosphatases. Previous studies have demonstrated that PPP5C plays numerous physiological functions in life. It was found in our study that PPP5C worked as an oncogene in PCa, though the underlying mechanism needs to be further explored.
The MAPK is one of the most important regulators of cellular physiopathological activities, especially in cell growth and survival both in normal and cancer cells.27,28 There are mainly 3 groups of MAPKs in mammalian species: JNK, ERK1/2, and p38 MAP kinase (P38).
Knowing that PPP5C has certain relevance with MAPK pathways via ERK1/2, JNK, and P38,29–31 we detected ERK, JNK, and P38 levels after PPP5C downregulation and found that the phosphorylation of ERK1/2 and JNK was significantly increased with the non-phosphorylation was fixed (Figures 6 and 7). But whether the phosphorylation or the non-phosphorylation level of the P38 molecule was immovable when the PPP5C changed. These results suggest that PPP5C promoted cell proliferation and survival, probably by regulating the phosphorylation of JNK and ERK1/2 in human PCa. JNK is reported to suppress carcinogenesis by regulating the pro-apoptotic activity. Several studies demonstrated that JNK activation participated in initiating apoptosis of many types of cancer cells, including gastric, lung, and breast cancer cells via different stimulation.32–34 We therefore infer that regulation of JNK phosphorylation is a potential mechanism underlying the oncogene role of PPP5C. ERK1/2 pathway is one of the vital parts of the MAPK pathways. Study35 showed that aberrant ERK1/2 activation was implicated in one-third of all human cancers. Many researchers36,37 considered that the activation of ERK phosphorylation could lead to DNA damage in cells, leading to cell cycle arrest and apoptosis. However, some other researchers38,39 argued that increased activation of ERK could resist the apoptosis induced by endoplasmic reticulum stress and therefore, was beneficial to cell survival, which is similar to the finding in the present study. We consider that increased ERK1/2 phosphorylation is likely the reason leading to cell growth inhibition, cell cycle arrest, and increased apoptosis after PPP5C knockdown. Nevertheless, we had demonstrated the biological function of PPP5C and the JNK and ERK1/2 transformation in PCa cells, the exact molecular mechanism and accurate modulatory relation of JNK and ERK1/2 between the PPP5C abnormality and the tumor biological function need to be further explored.
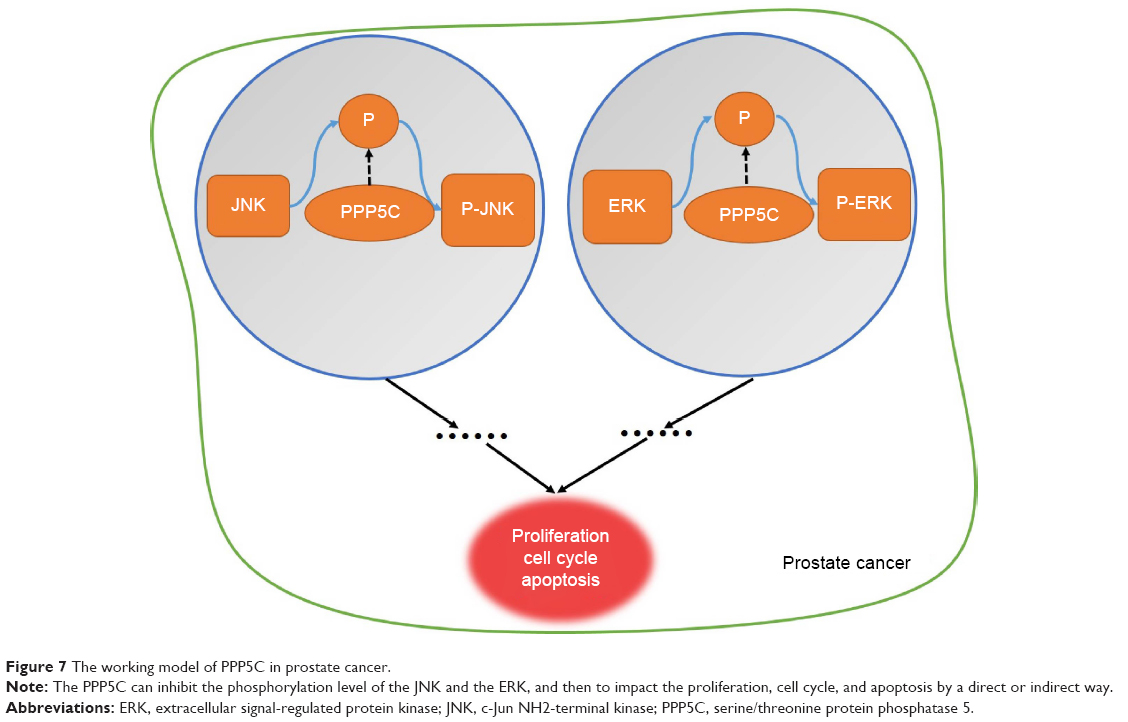 | Figure 7 The working model of PPP5C in prostate cancer. |
In summary, we demonstrated that PPP5C played a vital role in viability, proliferation, and apoptosis a of PCa cells, possibly by regulating JNK and ERK1/2 phosphorylation. PPP5C will likely to become a new potential therapeutic target for the treatment of PCa.
Conclusion
By using immunohistochemistry and ONCOMINE datasets analysis, we discovered that PPP5C was high-expression in PCa. Then, we successfully proved that knockdown of the PPP5C can disturb the cell proliferation, colony formation, and induce the cell apoptosis as well as G0/G1 arrest. Besides, we found that it can regulate the JNK and ERK1/2 phosphorylation of the PCa cell by Western blot assay. Our study indicated that the PPP5C may become a new potential diagnostic biomarker and therapeutic target for the PCa.
Acknowledgments
This work is supported by grants from the National Natural Science Foundation of China (No 81772747 and 81572525); the Medical Guidance (Chinese and Western Medicine) Science and Technology Support Project (No 17411960200); the National Natural Science Foundation of China (No 81602238, No 81702515, No 81702501); the General Program of Shanghai Municipal Commission of Health and Family Planning (No 201440511); Shanghai Sailing Program (No 17YF1425400); and the Youth Foundation of Second Military Medical University (No 2016QN17).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Chen R, Ren S; Chinese Prostate Cancer Consortium, et al. Prostate cancer in Asia: a collaborative report. Asian J Urol. 2014;1(1):15–29. | ||
Molitierno J, Evans A, Mohler JL, et al. Characterization of biochemical recurrence after radical prostatectomy. Urol Int. 2006;77(2):130–134. | ||
Roehl KA, Han M, Ramos CG, Antenor JA, Catalona WJ. Cancer progression and survival rates following anatomical radical retropubic prostatectomy in 3,478 consecutive patients: long-term results. J Urol. 2004;172(3):910–914. | ||
Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA. 2005;294(2):238–244. | ||
Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6(2):76–85. | ||
Shenolikar S, Nairn AC. Protein phosphatases: recent progress. Adv Second Messenger Phosphoprotein Res. 1991;23:1–121. | ||
Xu X, Lagercrantz J, Zickert P, Bajalica-Lagercrantz S, Zetterberg A. Chromosomal localization and 5′ sequence of the human protein serine/threonine phosphatase 5′ gene. Biochem Biophys Res Commun. 1996;218(2):514–517. | ||
von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat Cell Biol. 2006;8(9):1011–1016. | ||
Shah BH, Catt KJ. Protein phosphatase 5 as a negative key regulator of Raf-1 activation. Trends Endocrinol Metab. 2006;17(10):382–384. | ||
Wechsler T, Chen BP, Harper R, et al. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci U S A. 2004;101(5):1247–1252. | ||
Ali A, Zhang J, Bao S, et al. Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 2004;18(3):249–254. | ||
Zhang J, Bao S, Furumai R, et al. Protein phosphatase 5 is required for ATR-mediated checkpoint activation. Mol Cell Biol. 2005;25(22):9910–9919. | ||
Zuo Z, Dean NM, Honkanen RE. Serine/threonine protein phosphatase type 5 acts upstream of p53 to regulate the induction of p21 (WAF1/Cip1) and mediate growth arrest. J Biol Chem. 1998;273(20):12250–12258. | ||
Chen MS, Silverstein AM, Pratt WB, Chinkers M. The tetratricopeptide repeat domain of protein phosphatase 5 mediates binding to glucocorticoid receptor heterocomplexes and acts as a dominant negative mutant. J Biol Chem. 1996;271(50):32315–32320. | ||
Morita K, Saitoh M, Tobiume K, et al. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001;20(21):6028–6036. | ||
Vaughan CK, Mollapour M, Smith JR, et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol Cell. 2008;31(6):886–895. | ||
Jeong JY, Johns J, Sinclair C, Park JM, Rossie S. Characterization of Saccharomyces cerevisiae protein Ser/Thr phosphatase T1 and comparison to its mammalian homolog PP5. BMC Cell Biol. 2003;4:3. | ||
Golden T, Aragon IV, Rutland B, et al. Elevated levels of Ser/Thr protein phosphatase 5 (PP5) in human breast cancer. Biochim Biophys Acta. 1782;2008(4):259–270. | ||
Ghobrial IM, Mccormick DJ, Kaufmann SH, et al. Proteomic analysis of mantle-cell lymphoma by protein microarray. Blood. 2005;105(9):3722–3730. | ||
Shirato H, Shima H, Nakagama H, et al. Expression in hepatomas and chromosomal localization of rat protein phosphatase 5 gene. Int J Oncol. 2000;17(5):909–912. | ||
Feng L, Sun P, Li Z, Liu M, Sun S. Knockdown of PPP5C inhibits growth of hepatocellular carcinoma cells in vitro. Appl Biochem Biotechnol. 2015;175(1):526–534. | ||
Wang J, Zhu J, Dong M, et al. Inhibition of protein phosphatase 5 (PP5) suppresses survival and growth of colorectal cancer cells. Biotechnol Appl Biochem. 2015;62(5):621–627. | ||
Zhi X, Zhang H, He C, et al. Serine/threonine protein phosphatase-5 accelerates cell growth and migration in human glioma. Cell Mol Neurobiol. 2015;35(5):669–677. | ||
Zheng X, Zhang L, Jin B, et al. Knockdown of protein phosphatase 5 inhibits ovarian cancer growth in vitro. Oncol Lett. 2016;11(1):168–172. | ||
Golden T, Swingle M, Honkanen RE. The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer Metastasis Rev. 2008;27(2):169–178. | ||
Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119(Pt 22):4607–4615. | ||
Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–549. | ||
Liu C, Zhang R, Sun C, et al. Resveratrol prevents cadmium activation of Erk1/2 and JNK pathways from neuronal cell death via protein phosphatases 2A and 5. J Neurochem. 2015;135(3):466–478. | ||
Han X, Xu B, Beevers CS, et al. Curcumin inhibits protein phosphatases 2A and 5, leading to activation of mitogen-activated protein kinases and death in tumor cells. Carcinogenesis. 2012;33(4):868–875. | ||
Zhou G, Golden T, Aragon IV, Honkanen RE. Ser/Thr protein phosphatase 5 inactivates hypoxia-induced activation of an apoptosis signal-regulating kinase 1/MKK-4/JNK signaling cascade. J Biol Chem. 2004;279(45):46595–46605. | ||
Lin HH, Chen JH, Kuo WH, Wang CJ. Chemopreventive properties of Hibiscus sabdariffa L. on human gastric carcinoma cells through apoptosis induction and JNK/p38 MAPK signaling activation. Chem Biol Interact. 2007;165(1):59–75. | ||
Su JC, Lin KL, Chien CM, et al. Novel indoloquinoline derivative, IQDMA, induces G(2)/M phase arrest and apoptosis in A549 cells through JNK/p38 MAPK signaling activation. Life Sci. 2009;85(13–14):505–516. | ||
Brosseau CM, Pirianov G, Colston KW. Involvement of stress activated protein kinases (JNK and p38) in 1,25 dihydroxyvitamin D3-induced breast cell death. Steroids. 2010;75(13–14):1082–1088. | ||
Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. | ||
Bacus SS, Gudkov AV, Lowe M, et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20(2):147–155. | ||
Tang D, Wu D, Hirao A, et al. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277(15):12710–12717. | ||
Arai K, Lee SR, van Leyen K, Kurose H, Lo EH. Involvement of ERK MAP kinase in endoplasmic reticulum stress in SH-SY5Y human neuroblastoma cells. J Neurochem. 2004;89(1):232–239. | ||
Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J Biol Chem. 2004;279(47):49420–49429. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.