")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Potential for bispecific T-cell engagers: role of blinatumomab in acute lymphoblastic leukemia
Authors Le Jeune C, Thomas X
Received 1 December 2015
Accepted for publication 18 January 2016
Published 18 February 2016 Volume 2016:10 Pages 757—765
DOI https://doi.org/10.2147/DDDT.S83848
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Caroline Le Jeune, Xavier Thomas
Hospices Civils de Lyon, Hematology Department, Lyon-Sud Hospital, Pierre Bénite, France
Abstract: Patients with relapsed/refractory (R/R) B-precursor acute lymphoblastic leukemia (ALL) and patients whose minimal residual disease persists during treatment have a poor leukemia-free survival. Despite improvements in front-line therapy, the outcome in these patients remains poor, especially after relapse. As there are no standard chemotherapeutic regimens for the treatment of patients with R/R B-precursor ALL, T-cell-based therapeutic approaches have recently come to the forefront in ALL therapy. Recently, monoclonal antibodies have been developed to target specific antigens expressed in B-lineage blast cells. In this setting, CD19 is of great interest as this antigen is expressed in B-lineage cells. Therefore, it has been selected as the target antigen for blinatumomab, a new bi-specific T-cell engager antibody. This sophisticated antibody binds sites for both CD19 and CD3, leading to T-cell proliferation and activation and B-cell apoptosis. Owing to its short serum half-life, blinatumomab has been administrated by continuous intravenous infusion with a favorable safety profile. The most significant toxicities were central nervous system events and the cytokine release syndrome. This new therapeutic approach using blinatumomab has been shown to be effective in patients with positive minimal residual disease and in patients with R/R B-precursor ALL leading to a recent approval by the US Food and Drug Administration after an accelerated review process. This review focuses on the profile of blinatumomab and its efficacy and safety.
Keywords: B-cell lineage acute lymphoblastic leukemia, relapsed/refractory, minimal residual disease, BiTE monoclonal antibodies, blinatumomab
Introduction
Patients with relapsed/refractory (R/R) B-precursor acute lymphoblastic leukemia (ALL) and patients whose minimal residual disease (MRD) persists during front-line treatment or who become MRD positive following treatment have a poor leukemia-free survival (LFS). Despite improvements in upfront treatment, the outcome in patients with R/R B-precursor ALL remains dismal. As there are no clear superior chemotherapeutic regimens used in the treatment of patients with R/R B-precursor ALL, T-cell-based therapeutic approaches have recently emerged to the forefront in R/R ALL therapy. Bi-specific T-cell engager (BiTE) antibodies are sophisticated antibodies designed to direct T-effector memory cells toward target cells. Instead of focusing on traditional chemotherapy in R/R ALL, a number of antibody-based therapies have recently been developed to target ALL blast cell surface antigens, particularly CD19 and CD22. This review focuses on the use of blinatumomab, a BiTE antibody binding site for both CD19 and CD3, in B-precursor ALL.
Epidemiology of ALL
ALL is a hematological malignant disease characterized by an uncontrolled proliferation of immature hematopoietic cells. Lymphoid precursors, blocked at an early stage of differentiation, proliferate rapidly and supplant normal hematopoietic cells in the bone marrow. ALL can potentially spread to extramedullary localizations such as central nervous system (CNS), lymph nodes, gonads, spleen, or liver. ALL has a bimodal distribution with an early peak at 4–5 years of age (incidence of 4.5/100,000 per year) followed by a second gradual increase at 50 years (incidence of 2/100,000 per year). The overall incidence of ALL is relatively low, estimated at 1.7/100,000 per year. According to the Surveillance, Epidemiology, and End Result Program, ~6,250 new cases of ALL were expected to be diagnosed in the United States in 2015.1 Although ALL can occur at any age, it comprises <1% of adult cancers but represents the most common childhood malignancy, accounting for ~25% of cancers and 80% of all leukemia in children.2
Advances in the treatment of ALL over the past 3 decades have resulted in a remarkable improvement in terms of survival, especially in childhood ALL with a survival rate approaching 80%–90%. However, standard therapy fails in 10%–20% of newly diagnosed patients, and ALL remains one of the major causes of death from cancer in children.3 In adult ALL, despite a high complete response (CR) rate (80%–90%), most of the patients relapse with chemoresistant disease. Only 40%–50% of adult patients will remain in remission at 5 years. Although response is still high and can reach 50%–60% in R/R childhood ALL, the prognosis of adult patients with R/R ALL is dismal with a 5-year overall survival (OS) of only 10%,4,5 showing a need for different alternative treatment strategies than that required in the pediatric counterpart.
Few factors have been associated with an increased risk of ALL. Some unusual genetic conditions, such as Down syndrome, neurofibromatosis,6 Shwachman syndrome,7 Bloom syndrome,8 and ataxia telangiectasia,9 have been recognized at an increased risk of leukemia. Genome-wide association studies showed that some inherited genetic polymorphisms are associated with the development of childhood ALL.10 Some endogenous or exogenous exposures have been found to contribute to childhood ALL, but this topic remains contentious. Thus, ionizing radiation is an established causal exposure for childhood ALL.11 Medical radiation is one of the most common sources of radiation exposure. ALL may also occur as a side effect of chemotherapy. Some studies suggest that exposure to infections (viral or bacterial) may also promote ALL by an abnormal or dysregulated immune response.12
The World Health Organization classifies ALL as either B-cell lineage ALL or T-cell lineage ALL.13 The major subsets of precursor B-cell ALL include common precursor B-cell ALL (CD10 positive and no surface or cytoplasmic immunoglobulin [Ig]), pro-B ALL (CD10 negative and no surface or cytoplasmic Ig), and pre-B ALL (presence of cytoplasmic Ig). Cytogenetic analyses and molecular marker determinations are important to quickly distinguish patients whose leukemic cells harbor the Philadelphia chromosome (Ph) [t(9;22)] and/or B-cell receptor (BCR)–ABL rearrangement, leading to tyrosine kinase inhibitor-based treatment schedules.
Classical stratification into risk groups is based on a range of clinical, biological, and genetic features, such as age and sex; white blood cell count and CNS involvement at diagnosis; immunophenotypic, cytogenetic, and molecular characteristics; and early response to induction chemotherapy.14,15 A major breakthrough was the systematic study of the MRD. Methods for determining MRD are based on the detection of leukemia-specific aberrant immunophenotypes by flow cytometry, the evaluation of leukemia-specific rearranged Ig or T-cell receptor (TCR) sequences by quantitative real-time polymerase chain reaction, or the detection of fusion genes associated with chromosomal abnormalities. The detection limit with these methods is 10−3–10−5 (0.1%–0.001%).16 Persistence or reappearance of MRD is now the most important adverse prognostic factor allowing the implementation of a risk-adapted therapy of ALL and has erased most of the classical prognostic factors.17,18
Current and emerging therapy for ALL and outcomes
The currently accepted rules for the front-line treatment of B-cell lineage ALL generally involve the use of a multiagent chemotherapy combination that includes glucocorticoid (prednisone or dexamethasone), vincristine, an anthracycline, and asparaginase. Some regimens also add other drugs, such as high-dose cytarabine or cyclophosphamide.19–21 A 7-day corticosteroid treatment can precede the induction phase in order to assess the corticosteroid sensitivity of leukemia cells and decrease the leukemia burden. With these induction regimens, >90% of children and 60%–80% of adults achieve CR.3,21,22 Based on the retrospective reports in adolescents with ALL, pediatric-inspired regimens have recently been applied in adult patients with newly diagnosed B-cell lineage ALL, resulting in a significant decreased incidence of relapse (37% at 5 years).23 The consolidation phase starts immediately after the induction phase to eradicate residual leukemia cells and continue the CNS prophylaxis therapy. This phase includes the sequential administration of high-dose chemotherapy, commonly methotrexate, asparaginase, cytarabine, and cyclophosphamide. For patients with no indication for allogeneic hematopoietic stem-cell transplantation (HSCT), re-induction (or delayed intensification) typically includes the same agents used during the induction and initial consolidation phases. Maintenance therapy lasts for 2 years and typically combines 6-mercaptopurine and methotrexate, with eventually vincristine/steroid pulses.
CNS prophylaxis includes generally intrathecal chemotherapy (methotrexate, cytarabine, and hydrocortisone) throughout the different phases of the treatment and high-dose methotrexate and high-dose cytarabine during the consolidation phase.24 To minimize neurotoxicity, cranial irradiation may be reserved to those with active meningeal leukemia or at high risk of CNS relapse.25
Patients with high-risk features at diagnosis are typically recommended to undergo allogeneic HSCT in first CR, given the availability of a human leukocyte antigen-compatible donor. The indications for allogeneic HSCT are reassessed continuously. The previously existing criteria are now questioned in view of MRD information. The prognostic relevance of MRD for predicting relapse risk and survival has been reported in several clinical trials in pediatric and adult ALL.26 Several studies have reported a better OS and LFS in patients with positive MRD treated with allogeneic HSCT than in those with positive MRD not undergoing allogeneic HSCT.27
Alternative stem cell sources for transplantation (cord blood units, mismatched unrelated donor, haplo-identical donor) can be considered in clinical trials or in case of second CR. Autologous HSCT has been shown less effective than consolidation and maintenance chemotherapy and is not currently recommended in the treatment of non-Ph ALL.28
Adult patients with R/R B-cell lineage ALL have a poor prognosis. R/R B-cell lineage ALL represents a challenging situation. There are no established standards of care in this setting. The use of chemotherapeutic agents similar to those administered in first-line therapy allows a second CR achievement of <50% and the 5-year OS does not exceed 10%.29 The ultimate goal for these patients is to proceed to allogeneic HSCT. However, only a fraction of patients can be bridged to allogeneic HSCT after a first relapse, and further efforts must be developed to speed the process of donor selection, including the identification of potential unrelated donors at the time of initial disease presentation rather than at relapse. Survival rates after HSCT are 16%–23% versus only 4% with chemotherapy alone.29–31 Consequently, the development of effective and relatively nontoxic rescue therapies is needed. The nucleoside analog clofarabine aims to achieve CR in ~30% when used as a single agent.32 A remission rate of ~50% has been reported with a combination chemotherapy regimen consisting of clofarabine, etoposide, and cyclophosphamide.33 The poor response rates with standard chemotherapy have led to trials using newer targeted approaches, especially monoclonal antibodies or T-cell engaging strategies.34 Chimeric antigen receptor T-cells are T-cells that have been genetically modified to express anti-CD19. This novel cellular therapeutic approach is now being developed by several programs to target CD19-positive malignancies including pediatric and adult ALL.35 With this approach, CR rates of 80% have been observed in patients with R/R ALL.35 However, there are concerns about the high incidence of severe cytokine release syndrome (CRS). Recent data show that antibody-based therapy is a highly promising treatment approach. ALL leukemic cells express several surface antigens amenable to target therapies, including CD19, CD20, CD22, and CD52. Monoclonal antibodies work through a number of mechanisms, including antibody-dependent cytotoxicity, complement-dependent cytotoxicity, and direct induction of apoptosis. Monoclonal antibodies target leukemic surface antigens selectively and minimize off-target toxicity.36,37 These include unconjugated monoclonal antibodies (ofatumumab, alemtuzumab, epratuzumab), monoclonal antibodies conjugated to cytotoxic agents (inotuzumab ozogamycin, SAR3419) or to toxins (BL22, moxetumomab pasudotox), and the recently developed class of T-cell engaging bi-specific single-chain antibodies (BiTE antibodies). Blinatumomab is a bi-specific antibody targeting CD19 and CD3.
Pharmacology, mode of action, and pharmacokinetics of blinatumomab
BiTE antibodies represent a novel class of monoclonal antibodies that bind to surface antigens on target cells and activate TCRs, linking the cells to directly recruit effector T-cells and to augment the antineoplastic effect. Blinatumomab is a bi-specific CD19-directed CD3 T-cell engager that has been shown to induce durable responses in patients with various B-cell malignancies.
CD19 is a 95 kDa transmembrane B-cell-specific coreceptor. It belongs to the Ig superfamily. With the exception of plasma cells, CD19 is continuously expressed during B-cell development, including all B-cell lineage-derived leukemia, making it an attractive target in malignancies of B-cell origin.38 CD19 is presumed to play important roles in the BCR signaling, being an adaptor and activator of PI3 kinase, thereby regulating B-cell survival and differentiation. In this pathway, CD19 is thought to sustain the malignant B-cell phenotype via mechanisms of proliferation, cell survival, and self-renewal.39,40
Blinatumomab, derived from murine B-cell antibodies, is a 55 kDa single-chain antibody that contains both an anti-CD3 arm and an anti-CD19 arm that are joined by a nonimmunogenic linker. This structure allows a high degree of flexibility needed for simultaneous binding of two cells.41,42 Once the CD19+ B-cell and the CD3+ T-cell have been linked via blinatumomab, a cytolytic synapse between the T-cell and the B-cell occurs. Engaged-cytotoxic T-cells release granzymes and perforin via exocytosis into the target cells resulting in the B-cell apoptosis (Figure 1). Blinatumomab-activated T-cells also release inflammatory cytokines that lead to T-cell proliferation and activation.43 This therapeutic approach is independent from the TCR specificity and from the peptide antigen presentation with major histocompatibility complex class I molecules.44 This allows a polyclonal T-cell recruitment and a lower susceptibility to major immune escape mechanisms of tumor cells through downregulation of major histocompatibility complex class molecules.
 | Figure 1 Generation, structure, and mode of action of blinatumomab. |
Pharmacokinetics from preclinical and early clinical studies on blinatumomab has shown a relatively short serum half-life of 2–3 hours.45 Therefore, blinatumomab is administered over a 28-day continuous infusion using a portable mini-pump in order to maintain steady drug concentration, followed by a 14-day rest period before starting the next cycle. Steady-state concentrations were achieved within 24 hours and persisted over the entire treatment course. Pharmacokinetics assessments demonstrated a mean volume of distribution at the terminal phase of 4.52 (±2.89) L, a mean half-life of 2.11 (±1.42) hours, and an estimated mean clearance of 2.92 (±2.83) L/h.46 Blinatumomab pharmacokinetic parameters were not affected by age, sex, weight, body surface area, disease status, and creatinine clearance. The metabolic pathway of blinatumomab has not yet been characterized. Renal excretion is limited, but pharmacokinetic studies in patients with renal impairment have not been conducted. Thus, recommendations for patients with severe renal impairment or those receiving dialysis cannot be made at this time. However, blinatumomab clearance in the case of mild or moderate renal impairment approximates blinatumomab clearance with normal renal function.47 Blinatumomab clearance is not affected by hepatic dysfunction. For all patients treated with a minimal dose of 0.005 mg/m2/d, a rapid and lasting eradication of B-cells from peripheral blood has been observed. B-cell counts remained below the level of detection for the entire cycle and did not recover between cycles.48 Peripheral CD8+ and CD4+ T-cell counts fell within a few hours after blinatumomab infusion due to redistribution of T-cells into the tissue. T-cells returned to baseline by day 1 to day 2. In many patients, T-cell counts exceeded baseline levels during treatment. Inflammatory cytokines associated with the activation of T-cells increased within 1 day and were detectable for 2 days. This increase was transient, and the cytokine levels normalized after 2 days with no recurrence in future cycles.49
Comparative efficacy, safety, and tolerability of blinatumomab
Patients with MRD-positive B-lineage ALL after multiagent chemotherapy regimen have a poor outcome. A multicenter, single-arm, Phase II clinical trial by the German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia assessed blinatumomab in adult patients with B-cell lineage ALL with MRD persistence or relapse after induction and consolidation therapy.50 Twenty-one patients received blinatumomab at 15 μg/m2/d by continuous intravenous infusion for a 4-week period followed by a 14-day rest period before starting the next cycle. Patients received an average of three additional consolidation cycles. Twenty patients were evaluable. The primary objective was MRD response rate determined by a quantitative real-time polymerase chain reaction. Sixteen patients (80%) became MRD negative after the first cycle. Among these MRD responders, 12 patients were molecularly refractory to previous chemotherapy. Three of the five patients with a BCR–ABL translocation, which in all cases was refractory to tyrosine kinase inhibitors (imatinib and/or dasatinib), turned MRD negative. At a median follow-up of 33 months, LFS was 61% for the 20 evaluable patients. Nine patients underwent allogeneic HSCT after blinatumomab therapy. Their estimated LFS was 65%.51
One hundred and sixteen patients were included into the BLAST study, a single-arm, Phase II clinical trial that evaluated efficacy, safety, and tolerability of blinatumomab in patients with MRD-positive ALL.52 Median age was 45 years (range: 18–76 years). At the time of enrollment, 65% of the patients were in first CR. As of February 2014, 106 patients had ended treatment: 74 had completed treatment (four cycles or one cycle followed by HSCT) and 32 had discontinued treatment for various reasons; 79 patients were still alive and being followed. Complete MRD response after the first cycle of blinatumomab was achieved in 88 patients (78%) and two additional patients achieved a complete MRD response after more than one cycle. Overall, the complete MRD response rate was 80%. MRD response did not differ significantly across baseline age, sex, line of treatment, and MRD burden categories.
Based on the positive experience in adult patients with MRD-positive B-cell lineage ALL, a clinical Phase II trial was started in 2010 in adult patients with R/R B-cell lineage ALL.53 The initial dose was 5 μg/m2/d and then increased to 15 μg/m2/d. A total of 36 patients were included and treated. The median age was 32 years. Fifteen patients (42%) had a prior history of allogeneic HSCT. Twenty-five patients (69%) achieved CR or CR with partial hematological recovery (CRh), of whom 88% obtained an MRD CR. Median LFS was 7.6 months with a follow-up of 9.7 months. Median OS was 9.8 months with a median follow-up of 12.1 months. Thirteen responders (52%) underwent allogeneic HSCT, of whom six died from transplant-related toxicity and two relapsed.
Topp et al54 confirmed these results in a large, multicenter, single-arm, Phase II clinical trial that included adult patients with R/R B-cell lineage ALL. The median age was 38 years. Approximately one-third of the patients had undergone allogeneic HSCT. Blinatumomab was administrated by continuous intravenous infusion for a 4-week period followed by a 14-day rest period before starting the next cycle. The dose was 9 μg/kg/d for the first 7 days of cycle 1 and 28 μg/kg/d thereafter. A total of 189 patients were included and treated. Eighty-two patients (43%) achieved CR or CRh within two cycles of treatment. Response rate was higher in patients with <50% of blasts in bone marrow at baseline. With a median follow-up of 8.9 months, 37 of the 82 patients who achieved CR or CRh (45%) were still in remission and 32 (40%) underwent allogeneic HSCT. The median LFS was 6.9 months for those with CR and 5.0 months for those with CRh. The median OS for the entire cohort was 6.1 months. Median OS was longer for patients achieving an MRD response (11.5 months versus 6.7 months for patients with positive MRD).
In childhood ALL, blinatumomab was initially evaluated as compassionate use in three children with R/R ALL after allogeneic HSCT. All three patients achieved a complete molecular remission. Later, a Phase I/II study included 41 pediatric patients with R/R B-cell lineage ALL.55 These patients were previously heavily treated. Among the 13 patients (32%) who achieved CR, ten obtained a complete molecular response and nine further underwent allogeneic HSCT. With a median follow-up of 12.4 months, median LFS was 8.3 months and median OS was 5.7 months for patients who achieved CR.
Several clinical trials are currently ongoing for adult and pediatric patients with B-cell lineage ALL. The results of a Phase III study comparing blinatumomab with chemotherapy (TOWER study) in patients with ALL in first or second relapse (NCT 02013167) and those of a Phase II study in patients with relapsed Ph+ ALL (ALCANTARA study) (NCT 02000427) are awaited. Seven other trials are currently recruiting (Table 1).
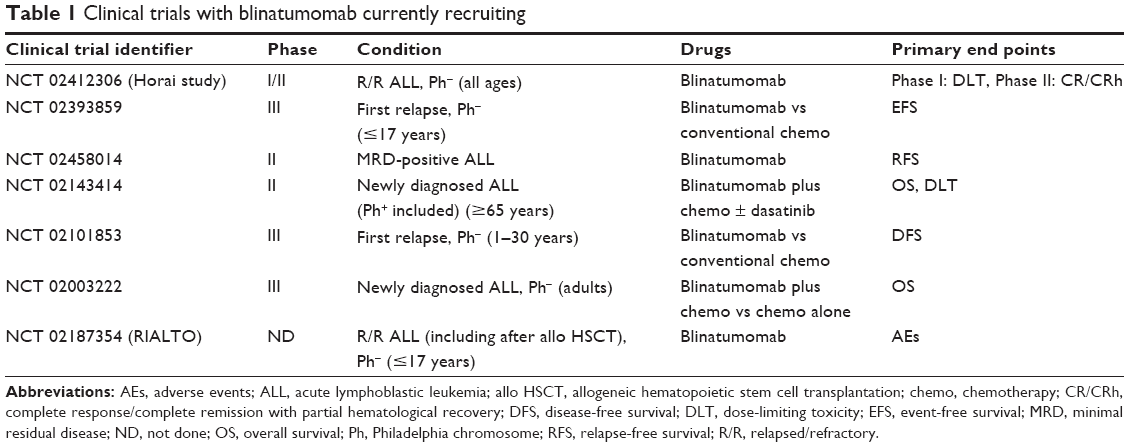 | Table 1 Clinical trials with blinatumomab currently recruiting |
Safety and tolerability
The safety profile of blinatumomab treatment is acceptable. However, some adverse events (AEs) have been reported in clinical trials (Table 2). The most common AEs (>20% of cases) were pyrexia, fatigue, headache, tremor, chills, peripheral edema, nausea, vomiting, and diarrhea.50,53,54 Almost all AEs were transient and occurred early during cycle 1. Changes in laboratory parameters were observed as expected because of blinatumomab mechanisms of action: leukopenia with neutropenia, B-cell lymphopenia, thrombocytopenia, hypogammaglobulinemia, elevation of gamma-glutamyltransferase, and transaminases. CRS and neurologic events were the most severe observed toxicities. Grade 3 and higher neurological AEs occurred in 11%–28% of patients across clinical trials.50,53,54 These AEs included encephalopathy, seizure, aphasia, and disorientation, leading to discontinuation of blinatumomab treatment. Although not fully explained, causes of blinatumomab-related CNS toxicity were thought to be in relation to the adherence of activated T-cells to the endothelium.56 Grade 3 neurological AEs required the interruption of blinatumomab and were managed by dexamethasone administration. After resolution, blinatumomab could be reintroduced at a lower dose. However, in case of grade 4 neurological AEs or more than one seizure, permanent discontinuation of blinatumomab was mandatory.57 CRS is due to rapid lysis of malignant cells by T-lymphocytes during the first infusion. CRS is mediated by transient release of inflammatory cytokines, interleukin (IL)-2, IL-6, IL-10, tumor necrosis factor, and interferon-γ, from blinatumomab-engaged T-cell effectors. These cytokines increased within 1 day, declined rapidly, and were detectable for 2 days. In most patients, symptoms related to CRS were moderate, such as flu-like syndrome with chills, pyrexia, and myalgia. In the most severe cases, patients presented hypotension, capillary leak, pulmonary edema, coagulopathy, and multiorgan failures.58 Dexamethasone was required for all patients as a premedication to prevent CRS events associated with blinatumomab treatment.57
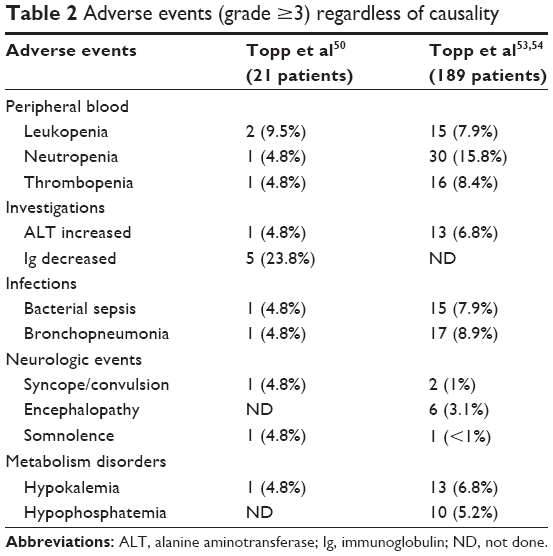 | Table 2 Adverse events (grade ≥3) regardless of causality |
Conclusion and future directions
The role of monoclonal antibodies and other novel targeted approaches in ALL continues to be defined. Blinatumomab is a novel BiTE antibody that has demonstrated promising activity and a relatively favorable safety profile in patients with MRD-positive ALL and in patients with R/R ALL. The first data support that blinatumomab has efficacy on MRD, is applicable to all cases of B-cell lineage ALL, including Ph-positive ALL even with T315I mutation, since the first results of the ALCANTARA trial showed 86% of complete MRD response in patients achieving CR,59 and is non-cross-resistant with chemotherapy and allogeneic HSCT. However, blinatumomab also needs to be compared with other monoclonal antibodies and chimeric antigen receptor T-cells in terms of efficacy and toxicity as all these agents have evidence of efficacy but concerns regarding toxicity. In patients with poor prognosis, the main goal remains to bridge to allogeneic HSCT.
An advantage with blinatumomab therapy is that management is relatively simple. However, it is strongly recommended that patients are hospitalized for the first 9 days of the first induction cycle and the first 2 days of the following cycles, as well as after any additional dose step in order to prevent the high-grade AEs. But after the initial hospitalization, blinatumomab infusion may be continued on an outpatient setting. Continuous IV infusion through a portable mini-pump is generally well accepted by patients, and this should be further confirmed by the analysis of quality-of-life questionnaires. However, this could represent challenges in frail patients or in children.
Safety profile appears as favorable, but life-threatening reactions can result from massive release of cytokines. Blinatumomab can also cause B-cell depletion, leading to a decrease in Ig levels and therefore an increased risk of severe infections. Which patients will benefit from blinatumomab has yet to be defined. Despite promising overall initial results, some patients do not respond to blinatumomab or experience disease progression after an initial response. The causes of this resistance remain unknown. A CD19-negative escape may be a contributing factor by CD19 internalization or selection of CD19-negative subclones.60 Another hypothesis sustained by a short duration of effect is that blinatumomab can target bulk leukemic cells but not leukemia stem cells. What has also yet to be defined is whether there is a way to predict which patients will respond to blinatumomab and whether the degree of pretreatment or prior therapies affects the potential for response.
The ability to target sanctuary sites remains a major challenge. Indeed, some relapses after blinatumomab are extramedullary, suggesting an insufficient activity of blinatumomab in sanctuary sites, especially in CNS.
Blinatumomab, as all active agents, will ultimately need to be incorporated into the front-line regimens to optimize treatment efficacy. Incorporating active monoclonal antibodies into front-line ALL therapy may likely induce higher rates of MRD negativity. However, the optimal placement of blinatumomab in earlier phases of ALL treatment has yet to be determined. The promising results may lead to combining blinatumomab with standard chemotherapy in ALL salvage and frontline regimens. Combination with other monoclonal antibodies may also be a potential option. Integration into combination regimens will need to account for the unique toxicity profile to ensure optimal safety for patients and the best efficacy potential.
Combinations might reduce the risk of resistant clonal outgrowth. However, it is not clear if blinatumomab should be combined simultaneously or sequentially with other agents and what would be the optimal sequence. The dependence of blinatumomab on circulating immune cells seems to limit concomitant use with myelosuppressive therapies. But concurrent use of targeted treatment, including l-asparaginase, should be plausible.
Blinatumomab could find a place in a window period and serve as a bridge to allogeneic HSCT. It could certainly find a place for reaching MRD negativity before HSCT. Its efficacy has been related to a low leukemia burden, which could be explained by an adequate ratio between T-cell effectors and targeted cells. Blinatumomab could also play a role in molecular relapses after HSCT and potentiate the effect of donor lymphocyte infusions.
Although there are limited data available in the elderly, blinatumomab should be useful in this setting, in which neither intensive chemotherapy nor HSCT is an optimal choice because of a lack of efficacy and a high risk of severe toxicity.
In conclusion, the use of monoclonal antibodies is now entering common practice in the treatment of adults with ALL. Blinatumomab is a BiTE antibody recognizing both CD3 and CD19, leading to adequate T-cell activation that works in a human leukocyte antigen-independent pathway. Single-agent blinatumomab treatment has shown a reduced relapse incidence when administered to patients with B-cell lineage ALL in morphologic remission but detectable MRD and a high rate of morphological response with achievement of an MRD-negative status in R/R ALL. These results suggest that blinatumomab has the potential to improve CR duration and OS in patients with B-cell lineage ALL.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
SEER Cancer Statistics Review [webpage on the Internet]. Previous Version: SEER Cancer Statistics Review, 1975–2010. 2015. Available from: http://seer.cancer.gov/archive/csr/1975_2010/. Accessed October 4, 2015. | ||
Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012; 30(14):1663–1669. | ||
Stock W, Johnson JL, Stone RM, et al. Dose intensification of daunorubicin and cytarabine during treatment of adult acute lymphoblastic leukemia: results of Cancer and Leukemia Group B Study 19802. Cancer. 2013;119(1):90–98. | ||
Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28(24):3880–3889. | ||
Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer. 1994;70(5):969–972. | ||
Strevens MJ, Lilleyman JS, Williams RB. Shwachman’s syndrome and acute lymphoblastic leukaemia. Br Med J. 1978;2(6129):18. | ||
Passarge E. Bloom’s syndrome: the German experience. Ann Genet. 1991;34(3–4):179–197. | ||
Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood. 1996;87(2):423–438. | ||
De Jonge R, Tissing WJE, Hooijberg JH, et al. Polymorphisms in folate-related genes and risk of pediatric acute lymphoblastic leukemia. Blood. 2009;113(10):2284–2289. | ||
Preston DL, Kusumi S, Tomonaga M, et al. Cancer incidence in atomic bomb survivors. Part III. Leukemia, lymphoma and multiple myeloma, 1950–1987. Radiat Res. 1994;137(2 suppl):S68–S97. Erratum in: Radiat Res. 1994;139(1):129. | ||
Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6(3):193–203. | ||
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. | ||
Hoelzer D, Thiel E, Löffler H, et al. Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood. 1988;71(1):123–131. | ||
Wetzler M, Dodge RK, Mrózek K, et al. Prospective karyotype analysis in adult acute lymphoblastic leukemia: the cancer and leukemia Group B experience. Blood. 1999;93(11):3983–3993. | ||
Hoelzer D. Personalized medicine in adult acute lymphoblastic leukemia. Haematologica. 2015;100(7):855–858. | ||
Campana D. Minimal residual disease in acute lymphoblastic leukemia. Hematol Hematology Am Soc Hematol Educ Program. 2010;2010:7–12. | ||
Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk-specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153–4162. | ||
Larson RA, Dodge RK, Burns CP, et al. A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: cancer and leukemia group B study 8811. Blood. 1995;85(8):2025–2037. | ||
Linker CA, Levitt LJ, O’Donnell M, Forman SJ, Ries CA. Treatment of adult acute lymphoblastic leukemia with intensive cyclical chemotherapy: a follow-up report. Blood. 1991;78(11):2814–2822. | ||
Kantarjian H, Thomas D, O’Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 2004;101(12):2788–2801. | ||
Sive JI, Buck G, Fielding A, et al. Outcomes in older adults with acute lymphoblastic leukaemia (ALL): results from the international MRC UKALL XII/ECOG2993 trial. Br J Haematol. 2012;157(4):463–471. | ||
Huguet F, Leguay T, Raffoux E, et al. Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol. 2009;27(6):911–918. | ||
Bleyer WA, Poplack DG. Prophylaxis and treatment of leukemia in the central nervous system and other sanctuaries. Semin Oncol. 1985;12(2): 131–148. | ||
Cortes J, O’Brien SM, Pierce S, Keating MJ, Freireich EJ, Kantarjian HM. The value of high-dose systemic chemotherapy and intrathecal therapy for central nervous system prophylaxis in different risk groups of adult acute lymphoblastic leukemia. Blood. 1995;86(6):2091–2097. | ||
Gökbuget N, Kneba M, Raff T, et al; German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120(9):1868–1876. | ||
Litzow MR. Finally moving forward in adult ALL. Blood. 2015;125(16):2453–2454. | ||
Gupta V, Richards S, Rowe J. Acute Leukemia Stem Cell Transplantation Trialists’ Collaborative Group. Allogeneic, but not autologous, hematopoietic cell transplantation improves survival only among younger adults with acute lymphoblastic leukemia in first remission: an individual patient data meta-analysis. Blood. 2013;121(2):339–350. | ||
Fielding AK, Richards SM, Chopra R, et al; Medical Research Council of the United Kingdom Adult ALL Working Party; Eastern Cooperative Oncology Group. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109(3):944–950. | ||
Gökbuget N, Stanze D, Beck J, et al; German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032–2041. | ||
Tavernier E, Boiron JM, Huguet F, et al; GET-LALA Group; Swiss Group for Clinical Cancer Research SAKK; Australasian Leukaemia and Lymphoma Group. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia. 2007;21:1907–1914. | ||
Kantarjian H, Gandhi V, Cortes J, et al. Phase 2 clinical and pharmacologic study of clofarabine in patients with refractory or relapsed acute leukemia. Blood. 2003;102(7):2379–2386. | ||
Abbi KKS, Rybka W, Ehmann WC, Claxton DF. Phase I/II study of clofarabine, etoposide, and mitoxantrone in patients with refractory or relapsed acute leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(1): 41–46. | ||
Le Jeune C, Thomas X. Antibody-based therapies in B-cell lineage acute lymphoblastic leukaemia. Eur J Haematol. 2015;94:99–108. | ||
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. | ||
Kantarjian H, Thomas D, Wayne AS, O’Brien S. Monoclonal antibody-based therapies: a new dawn in the treatment of acute lymphoblastic leukemia. J Clin Oncol. 2012;30(31):3876–3883. | ||
Ai J, Advani A. Current status of antibody therapy in ALL. Br J Haematol. 2015;168(4):471–480. | ||
Raponi S, De Propris MS, Intoppa S, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52(6):1098–1107. | ||
Fujimoto M, Poe JC, Inaoki M, Tedder TF. CD19 regulates B lymphocyte responses to transmembrane signals. Semin Immunol. 1998;10(4): 267–277. | ||
Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol. 2011;23(2):178–183. | ||
Dreier T, Lorenczewski G, Brandl C, et al. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002; 100(6):690–697. | ||
Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136(3):334–342. | ||
Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res. 2011; 317(9):1255–1260. | ||
Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006; 43(6):763–771. | ||
Blincyto® (blinatumomab). Available from: http://pi.amgen.com/united_states/blincyto/blincyto_pi_hcp_english.pdf. Accessed October 11, 2015. | ||
Wu B, Hijazi Y, Wolf A, Brandl C, Sun YN, Zhu M. Pharmacokinetics (PK) of blinatumomab and its clinical implications. J Clin Oncol. 2013; 31(15):abstract 3048. | ||
Kaplan JB, Grischenko M, Giles FJ. Blinatumomab for the treatment of acute lymphoblastic leukemia. Invest New Drugs. 2015;33(6):1271–1279. | ||
Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321(5891):974–977. | ||
Klinger M, Brandl C, Zugmaier G, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119(26):6226–6233. | ||
Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29(18):2493–2498. | ||
Topp MS, Gökbuget N, Zugmaier G, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120(26):5185–5187. | ||
Gökbuget N, Dombret H, Bonifacio M, et al. BLAST: a confirmatory, single-arm, phase 2 study of blinatumomab, a bispecific T-cell engager (BiTE®) antibody construct, in patients with minimal residual disease B-precursor acute lymphoblastic leukemia (ALL). Poster presented at: American Society of Hematology (ASH) Annual Meeting; December 6–9, 2014; San Franscisco, CA. Abstract 379. | ||
Topp MS, Gökbuget N, Zugmaier G, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134–4140. | ||
Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. | ||
Schlegel P, Lang P, Zugmaier G, et al. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica. 2014;99(7):1212–1219. | ||
Ribera J-M, Ferrer A, Ribera J, Genescà E. Profile of blinatumomab and its potential in the treatment of relapsed/refractory acute lymphoblastic leukemia. Onco Targets Ther. 2015;8:1567–1574. | ||
Barrett DM, Teachey DT, Grupp SA. Toxicity management for patients receiving novel T-cell engaging therapies. Curr Opin Pediatr. 2014; 26(1):43–49. | ||
Teachey DT, Rheingold SR, Maude SL, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–5157. | ||
Martinelli G, Dombret H, Chevallier P, et al. Complete molecular and hematologic response in adult patients with relapsed/refractory (R/R) Philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia (ALL) following treatment with blinatumomab: results from a phase 2 single-arm, multicenter study (ALCANTARA). Blood. 2015;126(issue 23):abstract 679. | ||
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.