Back to Journals » Drug Design, Development and Therapy » Volume 15
Potential Anti-Coronavirus Agents and the Pharmacologic Mechanisms
Authors Yang Y, Cui X, Wei H, Guo C, Zhang Y ![]()
Received 20 November 2020
Accepted for publication 6 February 2021
Published 17 March 2021 Volume 2021:15 Pages 1213—1223
DOI https://doi.org/10.2147/DDDT.S293216
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Yang Yang,1,* Xiao Cui,1,* Huaying Wei,1 Caiping Guo,1 Yulin Zhang2
1Department of Infectious Diseases, Beijing You an Hospital, Capital Medical University, Beijing Institute of Hepatology, Beijing, 100069, People’s Republic of China; 2Department of Respiratory and Infectious Diseases, Beijing You an Hospital, Capital Medical University, Beijing Institute of Hepatology, Beijing, 100069, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yulin Zhang
Department of Respiratory and Infectious Diseases, Beijing You an Hospital, Capital Medical University, Beijing Institute of Hepatology, Beijing, 100069, People’s Republic of China
Tel +86 10-83997143
Fax +86 10-63293371
Email [email protected]
Abstract: Severe acute respiratory syndrome coronavirus clade 2 (SARS-CoV-2) is an emerging pathogen, which is similar to previous SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) occurrences. However, we only get few understandings about the pathogenesis of SARS-CoV-2, which need to further be studied. The discovery of an agent that has a treatment efficacy against SARS-CoV-2 is very urgent. In this review, we briefly discuss the virology of this pathogen and focus on the available understanding of the pathogenesis and treatments of this pathogen including the uses of nucleoside analogues, protease inhibitors, interferons, and other small-molecule drugs, on the basis previous comprehensions of SARS and MERS. These reviewed concepts may be beneficial in providing new insights and potential treatments for COVID-19.
Keywords: COVID-19, SARS, MERS, virology, pathogenesis, treatment
Introduction
Currently, coronavirus disease-19 (COVID-19), which is caused by severe acute respiratory syndrome coronavirus clade 2 (SARS-CoV-2), is spreading throughout the world.1 During the past two decades, COVID-19 has become the third epidemic of coronavirus diseases, with the first two epidemics caused by the coronaviruses known as SARS and MERS.2,3 The degrees of homology between SARS-CoV-2 and these human coronaviruses (HCoVs) are high, and the whole genome of SARS-CoV-2 exhibits>85% homology to SARS-CoV.4–6 And the RNA results of them could be detected via samples of nasopharyngeal swabs, sputum and stool.6–8 Although these three HCoVs commonly present with mild influenza-like symptoms (pneumonia, fever, cough and occasional diarrhea), the progression to fatally abnormal features is characterized by acute respiratory distress syndrome (ARDS), acute cardiac injury, and other conditions.9,10 Therefore, the therapeutic conditions that are known to occur with SARS-CoV and MERS-CoV may directly guide and control the emerging infection of SARS-CoV-2.
The Life Cycles and Potential Antiviral Targets
CoVs share conserved genomic factors that are vital to the design of agent targets, in spite of their high species diversity and richness.11,12 Typical CoVs normally contain at least ten open reading frames (ORFs), which encode proteins including nonstructural proteins (nsps), structural proteins, and accessory proteins (Figure 1A).13–15 The first 5ʹ-terminal ORFs (ORF1a/b), which encompass approximately two-thirds of the viral RNA, can be translated into two large replicase proteins: polyprotein 1a (pp1a) and polyprotein 1ab (pp1ab) (Figure 1B).16,17 These two polyproteins are generally cleaved by the papain-like protease (PLpro) and the 3C-like protease (3CLpro) into nsps, including the RNA-dependent RNA polymerase(RdRP), helicase, and other components required for virus replication.18–20 RdRP is a crucial enzyme in the life cycle of coronaviruses, which supports the transcription and replication of their RNA genomes.21 In fact, the RdRp complex represents an Achilles heel for SARS-CoV. Another one-third of ORFs mainly encode structural proteins: spike (S; which are integral membrane proteins), envelope (E), nucleocapsid (N; which binds the viral RNA), and membrane (M) proteins, as well as accessory proteins whose function in virus replication or packaging remains largely uncharacterized (Figure 1C).22,23 Equally, the structure of SARS-CoV-2 also expresses various polyproteins that is similar to those of the typical coronaviruses.24,25
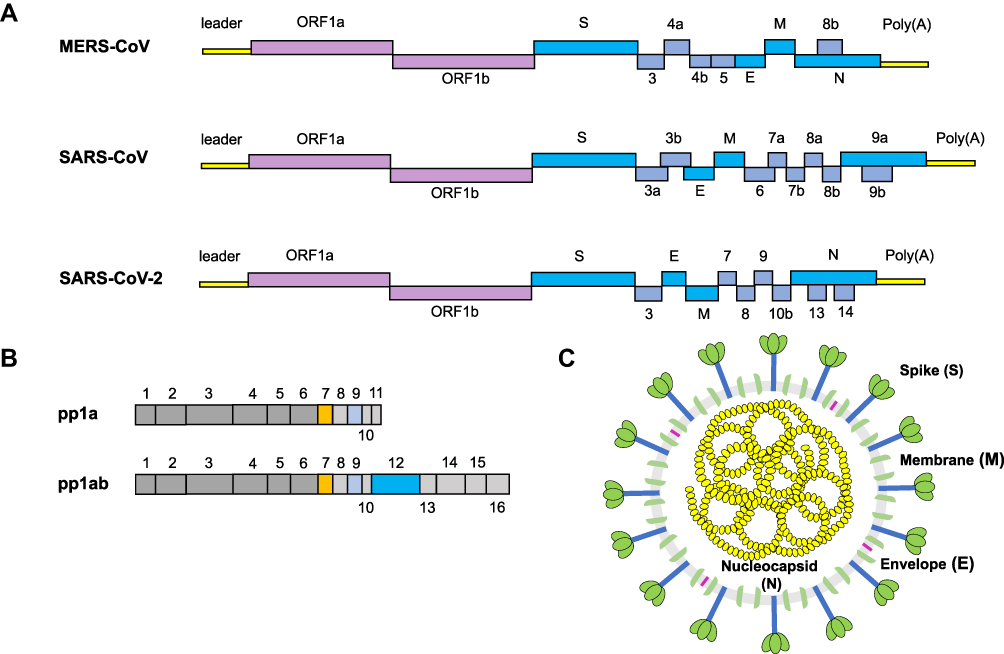 |
Figure 1 The structure of a coronavirus virion and genomic organization. Notes: (A) Coronavirus genome includes 5′ untranslated region (5UTR), such as 5′ leader sequence, open reading frame (ORF) 1a/b encoding non-structural proteins (nsp) for replication, structural proteins such as envelop, membrane and nucleoprotein, accessory proteins. (B) SARS-COV-2 encodes two large polyproteins, pp1a and pp1ab, that are cleaved into 16 non-structural proteins. (C) The coronavirus contains a + sense single stranded RNA (ssRNA) genome encapsidated by the nucleocapsid (N) protein, with the spike (S), membrane (M), and envelope (E) proteins located on the outside of the membrane. |
Viral entry mechanism involves the S protein being split by host proteases into the S1 and the S2 portions to activate virus-host cell interactions and syncytium formation. For instance, cleavage of S generates a C-end rule motif on S1, which binds to cell surface neuropilin-1 and neuropilin-2 receptors. Blocking this interaction by RNA interference can reduce SARS-CoV-2 entry and infectivity.26 Moreover, the combination of the S1 receptor-binding domain (RBD) with the host receptor can alter the conformation of the S2 (the stalk region) for membrane fusion.27 The host proteases of endosomal cysteine protease cathepsin L, transmembrane protease serine 2 (TMPRSS2), and human airway trypsin-like protease could sequentially act on SARS-CoV-S.28 And MERS-CoV-S possesses two cleavage sites for the furin (a type of serine endoprotease), and one of the sites (S1/S2) would be cleaved during the formation of the S protein, whereas another site (S2′) would be cleaved during entry.29 Furthermore, recent studies demonstrated that furin could also active the S protein of SARS-CoV-2 besides TMPRSS2.8,30,31 According to epidemiology, the infectious ability of SARS-CoV-2 is stronger than that of SARS-CoV because of the differences in their S proteins.31 The S protein of SARS-CoV-2 had distinct polybasic amino acids at the cleavage site one digestion region (unlike SARS-CoV), which was easily digested by the furin.31,32 During the viral maturation, furin could drive the RBD structural rearrangements via cutting the S proteins for the adaptation to the receptor, thereby enhancing affinity.30
As is already known, the binding of S proteins with host cell receptors is the meaningful infection triggers. MERS-CoV employs dipeptidyl peptidase 4 (DPP4) as the receptor,33,34 whereas SARS-CoV and SARS-CoV-2 recognizes angiotensin-converting enzyme 2 (ACE2) as the receptor.35,36 To date, strategies were developed to block the ACE2-RBD interaction, further to prevent SARS-CoV-2 entry into the host cell.37 After membrane fusion, the CoVs intracellularly disassemble and release nucleocapsid and RNA into cytoplasm. Then, the ORF1a/b is translated into pp1a and pp1ab with the replication of the genomic RNA. Additionally, numerous nsps are produced by the cleavage of pp1a and pp1ab to form the replication–transcription complex (RTC) in the double-membrane vesicles (DMVs).38,39 Subsequently, the positive-strand genomic RNA would function as the transcription template to synthesize new genomic RNAs (gRNAs).15,39 A nested set of overlapping subgenomic RNAs (sgRNAs) are produced by the RTC via discontinuous transcription, after which the sgRNAs are translated into relevant structural and accessory proteins.40 Finally, proteins and genomic RNAs form the assembled particles in the ER-Golgi intermediate complex (ERGIC) and are transported into the extracellular compartment.41
The involvement of other structural proteins is also implicated in the life cycle. The M protein provides scaffold for virion morphogenesis and combine with the nucleocapsid.42 The E protein contribute to particle assembly and be released by interacting with the M protein.19,43 By serving as a RNA chaperone, the N protein promote template switching and pack the encapsidated genome into virions.19,44,45 Hence, inhibitor compounds against receptors and proteases may be a promising approach to abrogate proteolytic cleavage and block viral transmissibility (Figure 2).
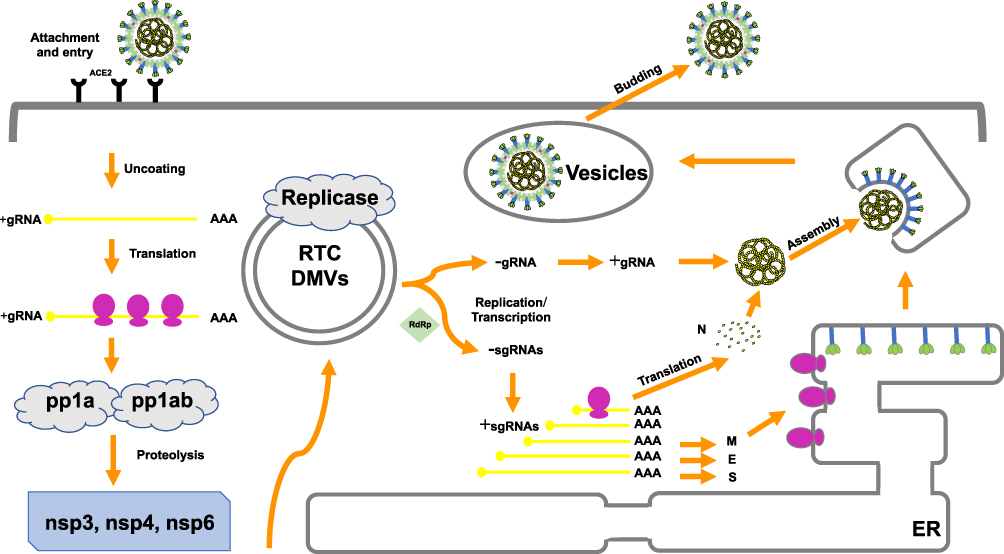 |
Figure 2 The life cycle of coronavirus. Abbreviations: ACE2, angiotensin-converting enzyme 2; ER, endoplasmic reticulum; +, positive strand RNA; –, negative strand RNA; RTC, replication–transcription complex; DMVs, double-membrane vesicles (DMVs); gRNA, genomic RNAs; sgRNA, sub genomic RNA; nsp, nonstructural protein; pp1a, polyprotein 1a; pp1ab, polyprotein 1ab. Notes: The coronavirus replication cycle in the host cell encompasses four steps, including attachment and entry, translation of the viral replicase, genome transcription and replication, translation of the structural proteins, and virion assembly and release. Potential therapeutic targets are noted in red. |
Pathogenesis
The pathogenesis of SARS-CoV-2 is possibly resembles those of SARS-CoV and MERS-CoV. SARS-CoV-2 generally spread via respiratory droplets, close contact, aerosol transmission, and (potentially) in fecal-oral transmission (Figure 3A).46 Zou et al discovered that the viral loads of COVID-19 patients (detected in nasal swabs) are higher than those that are detected in throat swabs.47 And viral replication is assumed to likely begin in the upper respiratory tract, with proliferation occurring in the lower respiratory tract to cause pneumonia.48
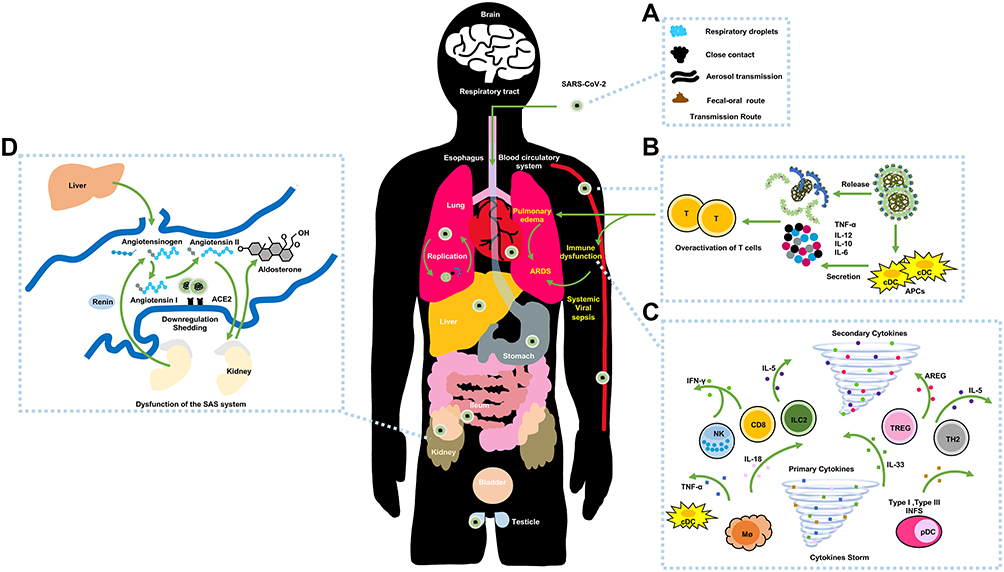 |
Figure 3 The possible pathogenesis of SARS-CoV-2. Notes: (A) The transmission routes. (B) Active innate immunity. (C) Cytokine storm. (D) Dysfunction of the renin-angiotensin system (RAS). |
Histopathology
One of the major pathogenesis features of SARS-CoV-2 is severe pneumonia.49–51 Histological cases from severe COVID-19 patients revealed that their bilateral lung tissue samples were consistent with the exhibition of early-phase ARDS.52 And interstitial lymphocytic inflammatory infiltrates and viral cytopathic-like changes were observed in the intra-alveolar spaces. In addition to the viral tropism for pulmonary involvement, extrapulmonary histopathology was also observed. Because these organ cells broadly express ACE2 receptors (which are vulnerable to S proteins),53 they have a high risk for infection. Liver biopsies demonstrated microvesicular steatosis, lobular and portal activity, which were associated with either SARS-CoV-2 infection or drug-induced liver injury.52 Notably, whereas COVID-19 patients could develop acute cardiac injury and acute heart failure, few interstitial mononuclear inflammatory infiltrates were observed in histological heart changes.52,54 Therefore, it is not clear as to whether heart diseases were influenced by SARS-CoV-2 or if they were complications of abnormal cardiometabolic requirements.54
Possible Mechanism of Inflammatory Responses
The aggressive inflammation features of COVID-19 patients maybe the outcome of direct virus-induced cytopathic effects and immunopathology that is induced by the cytokine storm.55,56 ARDS has proven to be deadly in critical COVID-19 patients, which is a similar feature of deterioration that occur in the later stages of SARS and MERS patients or during the period of recovery.10,51,57,58 Some fatal cases would suddenly exhibit grievous dyspnea that requires mechanical ventilation, and the histology also support the development of ARDS.6,51,57,58
The chief mechanism for ARDS is the cytokine storm, which is an uncontrollably violent immune disorder and cause the inevitable failure of multiple organs during aggravation. At the original onset of viral replication, massive respiratory epithelial cells, dendritic cells (DCs) and macrophages induce delayed (but elevated) levels of proinflammatory cytokines/chemokines.18 Afterwards, mononuclear macrophages and DCs (or plasmacytoid dendritic cells) continue to secrete low levels of antiviral factor interferons (IFNs), and induce the increased production of proinflammatory cytokines (Figure 3B).59 The delayed release of type I IFNs at the early stages of virus replication would attract the accumulation of inflammatory cells, thus contributing to excessive lung cytokine/chemokine levels, lung injury, and defective virus-specific T-cell responses.60 The expressions of serum cytokine and chemokine levels in patients with severe MERS and SARS are higher than those levels in mild to moderate infections. Additionally, the upregulation of serum cytokine and chemokine levels are connected with elevated numbers of neutrophils and monocytes in lung tissues, thus implying that they are involved in lung pathology.
Similarly, SARS-CoV-2 employs ACE2 as critical receptors, which indicates the likelihood of the same types of cells being hijacked and attacked. Yang et al demonstrated that COVID-19 patient exhibited pyroptosis in macrophages and lymphocyte, and most of these patients developed peripheral blood lymphopenia.55 Furthermore, researchers also detected high concentrations of proinflammatory CD4 T-cells and cytotoxic granules of CD8 T-cells, thus indicating that T-cells may become overactivated. And viral replication and pulmonary vascular leakage may trigger the robust secretion of proinflammatory cytokines/chemokines (Figure 3C). Overall, the abnormal levels of cytokine/chemokine levels in COVID-19 patients are correlated with disease severity,9,56,61 and these factors could activate the event of specific immunity, such as the T-helper type 1 (Th1) cell response. Study reported that COVID-19 patients in intensive care units (ICU) displayed higher serum levels of granulocyte colony-stimulating factor, interferon-inducible protein-10, monocyte chemotactic protein 1, macrophage inflammatory protein-1A, and TNF-α than those levels in non-ICU patients,62 thus indicating that a cytokine storm is connected with disease severity. Additionally, the downregulation and shedding of pulmonary ACE2 receptors could cause the dysfunction of the renin-angiotensin system (RAS) (Figure 3D). Correspondingly, the compensatory overproduction of angiotensin II via ACE leads to the excessive binding to the type 1a receptor that aggravate lung vascular permeability and acute lung injury.
Potential Anti-Coronavirus Agents
Protease Inhibitor
Most of the enzymes and proteins that are critical to coronaviral replication are promising agent targets. Therefore, protease inhibitors maybe an attractive option in the treatment of COVID-19.63 Shortly after the SARS epidemic in 2003, protease inhibitors were screened and developed. Lopinavir (LPV) is a type of HIV type 1 aspartate protease inhibitor, with ritonavir (RV) used as an accelerator, and their combination act against the viral 3CL protease in in vitro, MERS-CoV-infected NHPs and SARS patients.64 The reason why LPV/RV are universally accepted is because they not only have a reliable safety profile but are also easily available for immediate usage. For the recent emergence of SARS-CoV-2, LPV/RV (administered in combination with IFN-α) are recommended for clinical treatments.65–67 Nevertheless, it is unknown if LPV/RV could be powerfully against SARE-CoV-2 because the HIV protease and the 3CLpro are classified into different protease families. And LPV/RV is specifically designed to match catalytic site of the HIV protease dimer, whereas the viral proteases do not possess the C2-symmetry pocket. Clinical experiments have been launched to test the effects of LPV/RV in COVID-19 patients. In a randomized open-label trial of LPV/RV involving severe COVID-19 patients, no benefits were observed in terms of clinical improvement, mortality, or viral RNA levels, compared to standard care.67 Another randomized trial of combing LPV/RV with ITF-β-1b plus ribavirin in mild to moderate COVID-19 patients, the combination therapy showed significantly accelerate clinical improvement and reduce viral RNA loads than the use of LPV/RV alone.66
Antiviral Targeting the RdRp
Nucleoside analogues that target viral nucleic acids could universally resist ranges of CoVs via specific or nonspecific chain stops, lethal mutations, and retarded nucleotide biosynthesis.9 Ribavirin is one of the common options to treat COVID-19 patients.68 Mechanistically, ribavirin (in the form of a guanine derivative) competitively inhibits mRNA translation by RdRp to block the 5ʹ end of mRNA capping. Although SARS-CoV and MERS-CoV are susceptible to ribavirin in in vitro, its adverse reactions are severe at high doses in patients. Paradoxically, doses of ribavirin that inhibit viral proliferation in vitro exceed the concentrations that are accessible by classical human regimens.11 Present treatment experiences implied that ribavirin should be used at early stages of COVID-19, in case strains have developed drug resistance.66 Meta-analyses of cases demonstrated that the impact of ribavirin in the treatment of highly pathogenic coronavirus respiratory syndromes was inadequate (if there were any impacts at all), despite it being used as a part of treatment regimens for HCoVs.
Remdesivir is an adenosine analogue that also specifically target the viral RdRp and affect RNA chain synthesis. Previous studies demonstrated that remdesivir has strong broad-spectrum activities against SARS-CoV and MERS-COV in in vitro and in vivo.69 Wang et al detected that remdesivir could effectively inhibit SARS-CoV-2 in Vero E6 cells (EC50=0.77 μM, EC90=1.76 μM).68 Williamson et al observed that remdesivir could potently alleviate disease symptoms and lung injury in rhesus macaque model of SARS-CoV-2 infection that simulate mild to moderate disease progression. And their investigation data supported that remdesivir treatment in COVID-19 patients should be initiated as fast as possible, in order to reduce virus replication and to achieve clinical improvements at early stages of the drug course regimens. To rapidly estimate as to whether remdesivir has in-vivo antiviral activity against SARS-CoV-2, researchers also engineered chimeric mice-adapted SARS-CoV variants encoding the viral target of remdesivir (the RdRp, of SARS-CoV-2). Based on these mice infections with the chimeric virus, they concluded that the administration of remdesivir could decrease viral loads and improve pulmonary tissue damage. All of these data supported the further performance of clinical trials in COVID-19 patients. A clinical report demonstrated encouraging outcomes in a small cohort of COVID-19 patients with severe symptoms who were treated with remdesivir. The result showed that the oxygen-support statuses of 68% (53 cases) of patients were improved after receiving remdesivir.70 In contrast, a recent randomized, double-blind controlled trial in severe COVID-19 patients revealed that remdesivir could not ameliorate the time to clinical improvements, the clearance of the virus and the mortality rate. And there were higher proportions of toxicity and adverse events in recipients than in the placebo group.71 Overall, the efficacy, safety, and optimal dosing of remdesivir in different severity statuses of COVID-19 patients still need to be confirmed globally.
Similar to remdesivir, clinical trials are in progress for the treatment of COVID-19 by using favipiravir, the RdRP inhibitor.21 Favipiravir could insert into viral RNA, provoking C-to-U and G-to-A transitions, and subsequent disrupt viral replication and fidelity, leading to lethal mutagenesis.72,73 In SARS-CoV-2-infected hamsters, a high dose of favipiravir reduced infectious virus titers in the lungs and improved lung histopathology. Moreover, it markedly decreased virus transmission by direct contact.74 In May 2020, the Russian Ministry of Health granted favipiravir for the treatment of COVID-19 patients, based on the results of Phase II/III clinical trial.75
Sofosbuvir and daclatasvir are well tolerated and effective antivirals against HCV. Evidence from in silico and in vitro studies suggests the potential for sofosbuvir/daclatasvir as a therapeutic option for COVID-19.76–78 Clinical trials were conducted to evaluate the effectiveness and safety of the combination of sofosbuvir/daclatasvir (Sovodak). A randomized active-controlled trial observed that after 7 days of treatment, compared with control, Sovodak did not significantly alleviate symptoms of COVID-19. One month use of Sovodak reduced the number of patients with fatigue and dyspnoea.79 Another study found the addition of sofosbuvir and daclatasvir to standard care markedly reduced the duration of hospital stay.80 In open-label study, treatment with Sovodak in patients with severe COVID-19 improved clinical symptoms decreased length of hospital stays.81 Given the small sample size, larger randomized controlled trials were needed to establish the evidence to support approval of Sovodak.
Interferon
IFNs are a kind of immunomodulator that are secreted by the host innate immune response.55 IFNs could strengthen the phagocytic activity, recruitment of innate immune cells and facilitate the antiviral states of host cells.82 More precisely, IFNs promote the expressions of antiviral proteins (2,5-oligoadenylate synthetase, protein kinase, and phosphodiesterase) by JAK/STAK (mainly), IRF-1 signal transduction pathways.
Researchers have already studied IFNs with other antiviral drugs, such as ribavirin and/or LPV/RV, in the treatment of HCoV infections, but the efficacy outcomes were not consistent, which were due to the various intervention regimens. Although previous studies reported that IFN had a modest protective function in SARS and MERS mice and NPHs, the combination regimens (comprised of IFN and ribavirin or LPV/RV) only displayed limited beneficial effects in clinical trials. Currently, the common antiviral therapies in COVID-19 patients include ribavirin, LPV/RV, IFN, or their combinations. Hung et al observed that the early administration of the combination of INFβ-1b, LPV/RV, and ribavirin was superior to the use of LPV/RV alone in reducing initial viral loads and relieving symptoms in mild to moderate SARS-CoV-2 infection patients.82 Another report demonstrated that COVID-19 patients exhibited insufficient production of endogenous IFNs based on blood results, and most of these patients later possess worse prognosis. And it may explain the fact that the host innate IFN response had a positive impact on the immune system for the control of COVID-19, which could be enhanced by the use of IFNs or INF inducers. Most importantly, a high virus titer and the excessive immune response are associated with high risks of morbidity and mortality in HCoVs-infected patients. IFN is reliably beneficial in diminishing viral loads and in suppressing imbalanced inflammatory responses at the early phases of disease based on the experiences of curing SARS and MERS. In contrast, IFN could not exhibit additional beneficial immunomodulatory properties during other stages. Hence, the timing of medication is significant for a curative effect.
Monoclonal Antibody
Elevated interleukin-6 was reported to be associated with cases of severe COVID-19 and the need for mechanical ventilation.83–85 Tocilizumab, an anti-interleukin-6 receptor monoclonal antibody, appeared to improve outcomes of COVID-19 pneumonia in observational studies,86–88 while randomized trials have shown mixed results.89–91 A global, Phase 3 clinical trial investigated the safety and efficacy of tocilizumab in hospitalized patients with COVID-19 pneumonia. The likelihood of progression to mechanical ventilation or death was significantly reduced by the use of tocilizumab, while no benefit with respect to mortality was observed.89 To date, available data on the use of tocilizumab showed variable effects.
Other Small-Molecule Drugs
Small-molecule immune modulator (chloroquine [CQ] and/or its derivative hydroxychloroquine [HCQ]) has been considered as a potential therapeutic agent against SARS-CoV-2,68,92 which affect different steps in viral replication (the glycosylation of host receptors, the termination of membrane fusion, and the inhibition of pH-dependent lysosomal events).93
CQ and HCQ affect the biosynthesis of sialic acid by inhibiting quinone reductases, thereby interfering with the binding of the virus to host receptors.94 Because they are weakly alkaline and easily accumulate in the acidic lysosome, the Golgi network (TGN) vesicles, and the endosome, to disturb the acid-base in the organelles and the function of several enzymes (acid hydrolase, glycosylase, and other enzymes).93,95 To date, no studies on the binding of SARS-CoV-2 (like other CoVs) with sialic acids have been reported, but this possibility cannot be entirely dismissed. CQ and HCQ also have the ability to suppress mitogen-activated protein kinase activation and to block the viral molecular crosstalk with target cells.94 Moreover, they could impair the infectivity of the pH-dependent endosome-mediated CoVs, hamper the lysosome-triggered membrane fusion, as well as interfere with the posttranslational modifications and glycosylation of synthesized glycoproteins within the endoplasmic reticulum or the TGN vesicles.93,95 CQ and HCQ could also alter M protein maturation, thus suggesting a possible intervention of SARS-CoV-2 assembly and budding.94,95
Moreover, CQ and HCQ could synergistically modulate cell signaling and pro-inflammatory cytokines,96 and impede the expression of the major histocompatibility complex (MHC) class II, as well as antigen presentation and immune activation, via Toll-like receptor (TLR) signaling and cGAS stimulation of INF genes.97 Thereby, they could downregulate the secretion of pro-inflammatory cytokines in the cytokine storm; particularly, the secretions of tumor necrosis factor (TNF)-α, IL-2, and IL-6,95,96 Hence, immunomodulatory options for controlling the cytokine storm maybe a potential selection.
Although both CQ and HCQ have promising results in in vitro, the anti-CoV activity remains to be fully explored. A preliminary trial reported that the repurposing combination of HCQ and azithromycin (AZT) reduced SARS-CoV-2 shedding and the time to clinical recovery,98 whereas most of other studies did not indicate any beneficial effects in infected patients.99,100 Given the present controversies, clinicians should rationally use CQ and HCQ in COVID-19 patients until high-quality randomized clinical trials verify their functions in patients.
Conclusion
Taken together, SARS-CoV-2, the pathogen that is responsible for the coronavirus infectious disease-19 (COVID-19), is currently spreading throughout the world. Unfortunately, the major treatment regimen against SARS-CoV-2 consist of supportive treatments, and the clinical development of specific antiviral agents is still a primary concern. There are still numerous limitations in the current pandemic. First, the lack of evaluation approaches and ideal animal models that are used for testing drug activity among the different labs around the world is one of the obstacles to overcome, in order to discover effective drug treatments. Particularly, the absence of reproducible HCoV diseases has never been resolved in NHP models, which reduces the operability and reliability of results in vivo. Established suitable models require strict laboratory conditions and superior technology, which is also a problem. Therefore, few therapeutic options that have been applied in COVID-19 patients have been based on clear in vivo evidence of efficacy thus far, and meta-analyses of treatments have not indicated valid therapies. Specifically, current experimental systems for the study of the CoVs are not adequate to support the further development of antiviral agents. Second, the replication of CoVs can periodically produce progeny viruses with an abundance of genetic diversity. Recombination also frequently appears between viral genomes, and these gene level changes can lead to medication resistance if the mutations can alter the agents’ target domains. Third, one of the urgent strategies for treatment regimens mainly depends on combinations with licensed drugs that are known to possess receivable safety profiles and broad-spectrum antiviral activities. Although many of these drugs have exhibited anti-coronavirus effects in vitro, their pharmacokinetic, pharmacodynamic, and side effect aspects may not meet in vivo standards. Finally, virologic and patient-related elements may pose challenges in the clinical study of COVID-19. For example, the sample size and quality of the subjects may not reach the standard value, which results in the low reliability and operability of the test data.
Acknowledgments
We would like to thank Professor Yingmin Ma (Department of Respiratory and Infectious Diseases, Beijing You An Hospital, Capital Medical University, Beijing Institute of Hepatology, Beijing 100069, China) for his advices about the manuscript framework and the interpretation of the data.
Author Contributions
Yulin Zhang supervised the entire study, including the procedures, conception, design and revision. Caiping Guo and Huaying Wei were responsible for the collection of data. Yang Yang and Xiao Cui contributed the data analysis and drafted the article. Yang Yang and Xiao Cui equally contributed to the manuscript. All authors made a significant contribution to the conception, study design, execution, acquisition of data, analysis and interpretation of manuscript; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; agreed to submit to the current journal; and agree to be accountable for all aspects of the work.
Funding
Supported by the National Natural Science Foundation of China (81873761, 81571178, 81371399) and the 13th Key Science and Technology Five Year Plan of China (2018ZX10302104, 2018ZX10715-005-002-002).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gorbalenya AE, Baker SC, Baric RS, et al. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536–544.
2. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523–534. doi:10.1038/nrmicro.2016.81
3. Tu HL, Tu S, Gao SQ, Shao AW, Sheng JF. Current epidemiological and clinical features of COVID-19; a global perspective from China. J Infect. 2020;81(1):1–9. doi:10.1016/j.jinf.2020.04.011
4. Chan JFW, Zhu K-HK, Zhu Z. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan (vol 23, pg 2511, 2019). Emerg Microbes Infect. 2020;9(1):540.
5. Skipper CP, Pastick KA, Engen NW, et al. Hydroxychloroquine in nonhospitalized adults with early COVID-19: a randomized trial. Ann Intern Med. 2020. doi:10.7326/M20-4207
6. Zhu N, Zhang DY, Wang WL, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. doi:10.1056/NEJMoa2001017
7. Lai CC, Liu YH, Wang CY, et al. Asymptomatic carrier state, acute respiratory disease, and pneumonia due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): facts and myths. J Microbiol Immunol Infect. 2020;53(3):404–412. doi:10.1016/j.jmii.2020.02.012
8. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271. doi:10.1016/j.cell.2020.02.052
9. Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. 2020;109:102433. doi:10.1016/j.jaut.2020.102433
10. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:10.1016/S0140-6736(20)30183-5
11. Zumla A, Chan JFW, Azhar EI, Hui DSC, Yuen KY. Coronaviruses - drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15(5):327–347.
12. Belshaw R, Gardner A, RarnbaUt A, Pybus OG. Pacing a small cage: mutation and RNA viruses. Trends Ecol Evol. 2008;23(4):188–193. doi:10.1016/j.tree.2007.11.010
13. Frieman MB, Yount B, Sims AC, et al. SARS coronavirus accessory ORFs encode luxury functions. Adv Exp Med Biol. 2006;581:149–152.
14. Menachery VD, Mitchell HD, Cockrell AS, et al. MERS-CoV accessory ORFs play key role for infection and pathogenesis. mBio. 2017;8(4). doi:10.1128/mBio.00665-17.
15. Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res. 2014;109:97–109. doi:10.1016/j.antiviral.2014.06.013
16. Snijder EJ, van der Meer Y, Zevenhoven-Dobbe J, et al. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol. 2006;80(12):5927–5940. doi:10.1128/JVI.02501-05
17. van Boheemen S, de Graaf M, Lauber C, et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio. 2012;3(6). doi:10.1128/mBio.00473-12.
18. Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81(Pt 4):853–879. doi:10.1099/0022-1317-81-4-853
19. Sturman LS, Holmes KV. The molecular biology of coronaviruses. Adv Virus Res. 1983;28:35–112.
20. da Silva SJR. Role of nonstructural proteins in the pathogenesis of SARS-CoV-2. J Med Virol. 2020;92(9):1427–1429. doi:10.1016/j.jmii.2020.03.022
21. Wang Q, Wu J, Wang H, et al. Structural basis for RNA Replication by the SARS-CoV-2 polymerase. Cell. 2020;182(2):417–428.e13. doi:10.1016/j.cell.2020.05.034
22. Chen L, Zhong L. Genomics functional analysis and drug screening of SARS-CoV-2. Genes Dis. 2020;7(4):542–550. doi:10.1016/j.gendis.2020.04.002
23. Finkel Y, Mizrahi O, Nachshon A, et al. The coding capacity of SARS-CoV-2. Nature. 2020. doi:10.1038/s41586-020-2739-1
24. Chan JF, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–236. doi:10.1080/22221751.2020.1719902
25. Masters PS. The molecular biology of coronaviruses. In: Maramorosch K, Shatkin AJ, editors. Advances in Virus Research. Vol. 66. Elsevier Academic Press Inc; 2006:193.
26. Daly JL, Simonetti B, Klein K, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–865. doi:10.1126/science.abd3072
27. Fung TS, Liu DX. Human coronavirus: host-pathogen interaction. In: Gottesman S, editor. Annual Review of Microbiology. Vol. 73. 2019: 529–557.
28. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc Natl Acad Sci U S A. 2004;101(12):4240–4245. doi:10.1073/pnas.0306446101
29. Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A. 2014;111(42):15214–15219. doi:10.1073/pnas.1407087111
30. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176(5):104742. doi:10.1016/j.antiviral.2020.104742
31. Chen Y, Guo Y, Pan YH, Zhao ZJ. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020;525(1):135–140. doi:10.1016/j.bbrc.2020.02.071
32. Millet JK, Whittaker GR. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134. doi:10.1016/j.virusres.2014.11.021
33. Alagaili AN, Briese T, Mishra N, et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. mBio. 2014;5(2):e00884–14. doi:10.1128/mBio.01002-14
34. Mackay IM, Arden KE. MERS coronavirus: diagnostics, epidemiology and transmission. Virol J. 2015;12(1):222. doi:10.1186/s12985-015-0439-5
35. Ge XY, Li JL, Yang XL, et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503(7477):535–538. doi:10.1038/nature12711
36. Menachery VD, Yount BL
37. Schoof M, Faust B, Saunders RA, et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science. 2020;370(6523):1473–1479.
38. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ, Moscona A. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio. 2013;4(4). doi:10.1128/mBio.00524-13
39. Maier HJ, Hawes PC, Cottam EM, et al. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio. 2013;4(5):e00801–13. doi:10.1128/mBio.00801-13
40. Lundin A, Dijkman R, Bergstrom T, et al. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the Middle East respiratory syndrome virus. PLoS Pathog. 2014;10(5):e1004166. doi:10.1371/journal.ppat.1004166
41. Lai MM, Cavanagh D. The molecular biology of coronaviruses. Adv Virus Res. 1997;48:1–100.
42. Klumperman J, Locker JK, Meijer A, Horzinek MC, Geuze HJ, Rottier PJ. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J Virol. 1994;68(10):6523–6534. doi:10.1128/JVI.68.10.6523-6534.1994
43. Ruch TR, Machamer CE. The coronavirus E protein: assembly and beyond. Viruses. 2012;4(3):363–382. doi:10.3390/v4030363
44. Eleouet JF, Slee EA, Saurini F, et al. The viral nucleocapsid protein of transmissible gastroenteritis coronavirus (TGEV) is cleaved by caspase-6 and −7 during TGEV-induced apoptosis. J Virol. 2000;74(9):3975–3983. doi:10.1128/JVI.74.9.3975-3983.2000
45. Zuniga S, Sola I, Moreno JL, Sabella P, Plana-Duran J, Enjuanes L. Coronavirus nucleocapsid protein is an RNA chaperone. Virology. 2007;357(2):215–227. doi:10.1016/j.virol.2006.07.046
46. Wang J, Du G. COVID-19 may transmit through aerosol. Ir J Med Sci. 2020. doi:10.1007/s11845-020-02218-2
47. Zou L, Ruan F, Huang M, et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med. 2020;382(12):1177–1179. doi:10.1056/NEJMc2001737
48. Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology. 2020;158(6):1831–1833 e3. doi:10.1053/j.gastro.2020.02.055
49. Ding Y, Wang H, Shen H, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200(3):282–289. doi:10.1002/path.1440
50. Ng DL, Al Hosani F, Keating MK, et al. Clinicopathologic, immunohistochemical, and ultrastructural findings of a fatal case of middle east respiratory syndrome coronavirus infection in the united arab emirates, April 2014. Am J Pathol. 2016;186(3):652–658. doi:10.1016/j.ajpath.2015.10.024
51. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–1720. doi:10.1056/NEJMoa2002032
52. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420–422. doi:10.1016/S2213-2600(20)30076-X
53. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020;14(2):185–192. doi:10.1007/s11684-020-0754-0
54. Rizzo P, Vieceli Dalla Sega F, Fortini F, Marracino L, Rapezzi C, Ferrari R. COVID-19 in the heart and the lungs: could we “Notch” the inflammatory storm? Basic Res Cardiol. 2020;115(3). doi:10.1007/s00395-020-0791-5
55. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J Infect. 2020;80(6):607–613. doi:10.1016/j.jinf.2020.03.037
56. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418–423. doi:10.1002/jmv.25681
57. Chan JF, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523. doi:10.1016/S0140-6736(20)30154-9
58. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–1069. doi:10.1001/jama.2020.1585
59. Liu J, Zheng X, Tong Q, et al. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J Med Virol. 2020;92(5):491–494. doi:10.1002/jmv.25709
60. Jin Y, Yang H, Ji W, et al. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses. 2020;12(4):4. doi:10.3390/v12040372
61. Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect. 2020.
62. Nikolich-Zugich J, Knox KS, Rios CT, Natt B, Bhattacharya D, Fain MJ. SARS-CoV-2 and COVID-19 in older adults: what we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience. 2020;42(2):505–514. doi:10.1007/s11357-020-00186-0
63. Baden LR, Rubin EJ. Covid-19 - the search for effective therapy. N Engl J Med. 2020;382(19):1851–1852. doi:10.1056/NEJMe2005477
64. Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov. 2020;19(3):149–150. doi:10.1038/d41573-020-00016-0
65. Dalerba P, Levin B, Thompson JL. A trial of lopinavir–ritonavir in Covid-19. N Engl J Med. 2020.
66. Hung IF-N, Lung K-C, Tso EY-K, et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, Phase 2 trial. Lancet. 2020. doi:10.1016/S0140-6736(20)31042-4
67. Cao B, Wang Y, Wen D, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. 2020;382(19):1787–1799. doi:10.1056/NEJMoa2001282
68. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. doi:10.1038/s41422-020-0282-0
69. Agostini ML, Andres EL, Sims AC, et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9(2). doi:10.1128/mBio.00221-18.
70. Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med. 2020. doi:10.1056/NEJMoa2007016
71. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569–1578. doi:10.1016/S0140-6736(20)31022-9
72. Shannon A, Selisko B, Le NT, et al. Rapid incorporation of favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat Commun. 2020;11(1):4682. doi:10.1038/s41467-020-18463-z
73. Robson F, Khan KS, Le TK, et al. Coronavirus RNA proofreading: molecular basis and therapeutic targeting. Mol Cell. 2020;79(5):710–727. doi:10.1016/j.molcel.2020.07.027
74. Kaptein SJF, Jacobs S, Langendries L, et al. Favipiravir at high doses has potent antiviral activity in SARS-CoV-2-infected hamsters, whereas hydroxychloroquine lacks activity. Proc Natl Acad Sci U S A. 2020;117(43):26955–26965. doi:10.1073/pnas.2014441117
75. Ivashchenko AA, Dmitriev KA, Vostokova NV, et al. AVIFAVIR for treatment of patients with moderate COVID-19: interim results of a phase II/III multicenter randomized clinical trial. Clin Infect Dis. 2020.
76. Jácome R, Campillo-Balderas JA, Ponce de León S, Becerra A, Lazcano A. Sofosbuvir as a potential alternative to treat the SARS-CoV-2 epidemic. Sci Rep. 2020;10(1):9294. doi:10.1038/s41598-020-66440-9
77. Chien M, Anderson TK, Jockusch S, et al. Nucleotide analogues as inhibitors of SARS-CoV-2 polymerase, a key drug target for COVID-19. J Proteome Res. 2020;19(11):4690–4697. doi:10.1021/acs.jproteome.0c00392
78. Beck BR, Shin B, Choi Y, Park S, Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. 2020;18:784–790. doi:10.1016/j.csbj.2020.03.025
79. Roozbeh F, Saeedi M, Alizadeh-Navaei R, et al. Sofosbuvir and daclatasvir for the treatment of COVID-19 outpatients: a double-blind, randomized controlled trial. J Antimicrob Chemother. 2020.
80. Sadeghi A, Ali Asgari A, Norouzi A, et al. Sofosbuvir and daclatasvir compared with standard of care in the treatment of patients admitted to hospital with moderate or severe coronavirus infection (COVID-19): a randomized controlled trial. J Antimicrob Chemother. 2020;75(11):3379–3385. doi:10.1093/jac/dkaa334
81. Eslami G, Mousaviasl S, Radmanesh E, et al. The impact of sofosbuvir/daclatasvir or ribavirin in patients with severe COVID-19. J Antimicrob Chemother. 2020;75(11):3366–3372. doi:10.1093/jac/dkaa331
82. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. 2020;35(3):266–271. doi:10.1007/s12250-020-00207-4
83. Aziz M, Fatima R, Assaly R. Elevated interleukin-6 and severe COVID-19: a meta-analysis. J Med Virol. 2020;92(11):2283–2285. doi:10.1002/jmv.25948
84. Zhu J, Pang J, Ji P, et al. Elevated interleukin-6 is associated with severity of COVID-19: a meta-analysis. J Med Virol. 2020.
85. Herold T, Jurinovic V, Arnreich C, et al. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J Allergy Clin Immunol. 2020;146(1):128–136.e4. doi:10.1016/j.jaci.2020.05.008
86. Xu X, Han M, Li T, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A. 2020;117(20):10970–10975. doi:10.1073/pnas.2005615117
87. Morrison AR, Johnson JM, Griebe KM, et al. Clinical characteristics and predictors of survival in adults with coronavirus disease 2019 receiving tocilizumab. J Autoimmun. 2020;114:102512. doi:10.1016/j.jaut.2020.102512
88. Gupta S, Wang W, Hayek SS, et al. Association between early treatment with tocilizumab and mortality among critically ill patients with COVID-19. JAMA Intern Med. 2020.
89. Salama C, Han J, Yau L, et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med. 2020. doi:10.1056/NEJMoa2030340
90. Salvarani C, Dolci G, Massari M, et al. Effect of tocilizumab vs standard care on clinical worsening in patients hospitalized with COVID-19 pneumonia: a randomized clinical trial. JAMA Intern Med. 2020.
91. Hermine O, Mariette X, Tharaux PL, Resche-Rigon M, Porcher R, Ravaud P. Effect of tocilizumab vs usual care in adults hospitalized with COVID-19 and moderate or severe pneumonia: a randomized clinical trial. JAMA Intern Med. 2020.
92. Kono M, Tatsumi K, Imai AM, Saito K, Kuriyama T, Shirasawa H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: involvement of p38 MAPK and ERK. Antiviral Res. 2008;77(2):150–152. doi:10.1016/j.antiviral.2007.10.011
93. Hashem AM, Alghamdi BS, Algaissi AA, et al. Therapeutic use of chloroquine and hydroxychloroquine in COVID-19 and other viral infections: a narrative review. Travel Med Infect Dis. 2020. doi:10.1016/j.tmaid.2020.101735
94. Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today’s diseases. Lancet Infect Dis. 2003;3(11):722–727. doi:10.1016/S1473-3099(03)00806-5
95. Devaux CA, Rolain JM, Colson P, Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int J Antimicrob Agents. 2020;105938.
96. Zhao M. Cytokine storm and immunomodulatory therapy in COVID-19: role of chloroquine and anti-IL-6 monoclonal antibodies. Int J Antimicrob Agents. 2020. doi:10.1016/j.ijantimicag.2020.105982
97. Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. 2020;10(2):102–108. doi:10.1016/j.jpha.2020.03.001
98. Gautret P, Lagier JC, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;56(1):105949. doi:10.1016/j.ijantimicag.2020.105949
99. Molina JM, Delaugerre C, Le Goff J, et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med Mal Infect. 2020;50(4):384. doi:10.1016/j.medmal.2020.03.006
100. Sharma A. Chloroquine paradox may cause more damage than help fight COVID-19. Microb Infect. 2020;22(4–5):154–156. doi:10.1016/j.micinf.2020.04.004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.