Back to Journals » Journal of Hepatocellular Carcinoma » Volume 1
Potent efficacy signals from systemically administered oncolytic herpes simplex virus (HSV1716) in hepatocellular carcinoma xenograft models
Authors Braidwood L, Learmonth K, Graham A, Conner J
Received 12 July 2014
Accepted for publication 7 August 2014
Published 16 October 2014 Volume 2014:1 Pages 149—161
DOI https://doi.org/10.2147/JHC.S71019
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ahmed Kaseb
Lynne Braidwood, Kirsty Learmonth, Alex Graham, Joe Conner
Virttu Biologics Ltd, Department of Neurology, Southern General Hospital, Glasgow, UK
Abstract: Oncolytic herpes simplex virus (HSV1716), lacking the neurovirulence factor ICP34.5, has highly selective replication competence for cancer cells and has been used in clinical studies of glioma, melanoma, head and neck squamous cell carcinoma, pediatric non-central nervous system solid tumors, and malignant pleural mesothelioma. To date, 88 patients have received HSV1716 and the virus is well tolerated, with selective replication in tumor cells and no spread to surrounding normal tissue. We assessed the potential value of HSV1716 in preclinical studies with two human hepatocellular carcinoma cell lines, HuH7 and HepG2-luc. HSV1716 displayed excellent replication kinetics in vitro in HepG2-luc cells, a cell line engineered to express luciferase, and virus-mediated cell killing correlated with loss of light emissions from the cells. In vivo, the HepG2-luc cells readily formed light-emitting xenografts that were easily visualized by an in vivo imaging system and efficiently eliminated by HSV1716 oncolysis after intratumoral injection. HSV1716 also demonstrated strong efficacy signals in subcutaneous HuH7 xenografts in nude mice after intravenous administration of virus. In the HuH7 model, the intravenously injected virus replicated prolifically immediately after efficient tumor localization, resulting in highly significant reductions in tumor growth and enhanced survival. Our preclinical results demonstrate excellent tumor uptake of HSV1716, with prolific replication and potent oncolysis. These observations warrant a clinical study of HSV1716 in hepatocellular carcinoma.
Keywords: oncolytic herpes simplex virus, HSV1716, hepatocellular carcinoma, xenografts, efficacy
Introduction
Hepatocellular carcinoma (HCC), a leading cause of cancer-related cell deaths worldwide, is increasing in prevalence.1–3 Early detection of the disease is limited, and many patients present with advanced, inoperable, and aggressive disease, so have an extremely poor prognosis. In addition to surgery or chemotherapy, other treatment options include local ablation by, for example, heat, radiofrequency, or transarterial chemoembolization, although no standard therapy exists for patients who are not suitable for transplantation or surgical resection, or for patients with recurrent HCC, with systemic chemotherapy often considered in these patients albeit in a palliative capacity. Sorafenib is a multikinase inhibitor drug and is the only treatment approved by the US Food and Drug Administration for patients with advanced liver cancer. However, sorafenib is not curative, and patient outcomes remain poor. Monotherapy with sorafenib in HCC reduces the risk of death in year 1 by 31%, and the median survival time in patients treated with sorafenib is typically 3 months longer than for patients treated with placebo.4 New approaches to the treatment of HCC are urgently required, and recent studies with the oncolytic vaccinia virus, JX-594, suggest that oncolytic virotherapy has potential in treating advanced HCC.5
We have investigated the therapeutic potential of the oncolytic herpes simplex virus-1 (HSV-1) HSV1716 in HCC in preclinical studies. The HSV-1 mutant HSV1716 has deletions in both copies of the gene encoding the neurovirulence factor ICP34.5. HSV1716 effectively kills tumor cell lines in vitro and, in a wide range of in vivo cancer models, HSV1716 oncolysis has induced tumor regression and increased survival times in a wide range of solid tumors, including glioma, melanoma, medulloblastoma, mesothelioma, ovarian carcinoma, and teratocarcinoma, supporting translation into clinical studies.6–9 In clinical trials, direct intratumoral injection of HSV1716 has been used to treat patients with recurrent glioma, metastatic melanoma, and squamous cell carcinoma of the head and neck.10–14 A Phase I dose escalation study of intratumoral HSV1716 in pediatric/young adult patients with non-central nervous system solid tumors (http://clinicaltrials.gov/NCT00931931) and a Phase I/IIa study in malignant pleural mesothelioma (http://clinicaltrials.gov/NCT01721018) are currently ongoing. Although the efficacies of other oncolytic HSV have been assessed in various HCC cell lines in tissue culture and xenografts, to date there have been no preclinical studies of HSV1716 in HCC and no xenograft studies that have assessed the potential for its systemic administration.15–23 In this work, we demonstrated strong efficacy signals in two xenograft HCC models, particularly when the virus was administered systemically, which support a clinical study of HSV1716 in advanced HCC.
Materials and methods
Cells
HuH7 (JCRB0403; Japanese Collection of Research Bioresources Cell Bank, Tokyo, Japan) is a well-differentiated, hepatocyte-derived carcinoma cell line that was originally taken from a liver tumor in a 57-year-old Japanese male. HepG2-luc (HT1080-luc2; Perkin-Elmer, Beaconsfield, UK) is a luciferase-expressing cell line stably transfected with the firefly luciferase 2 gene under the control of the human ubiquitin C promoter. The HepG2 cell line was isolated from a liver biopsy of a male Caucasian aged 15 years with a well differentiated HCC. The one58 cell line (10092313; European Collection of Cell Cultures, Salisbury, UK) was derived from the pleural fluid of a patient with malignant mesothelioma, and the U87 cell line (European Collection of Cell Cultures, 89081402) was derived from a female patient with malignant glioma. HuH7, U87, and one58 cells were cultured in Dulbecco’s Modified Eagle’s Medium/F12 medium with 10% newborn calf serum, and HepG2-luc cells were cultured in advanced Roswell Park Memorial Institute 1640 medium supplemented with 20% newborn calf serum (Invitrogen, Paisley, UK) at 37°C in 5% CO2.
Viruses
Stocks of HSV1716 and wild-type HSV-1 17+ were suspended in compound sodium lactate with 10% glycerol. HSV1716gCluc was derived using a site-specific recombination system and has the firefly luciferase gene derived from pGL3 (Promega, Southampton, UK) under control of the HSV-1 gC promoter inserted in both RL1 loci.24,25 Stocks of HSV1716gCluc were prepared with virus suspended in Dulbecco’s Modified Eagle’s Medium/F12 medium. All dilutions from stock to the appropriate titers used compound sodium lactate +10% glycerol and were titrated to confirm pfu/mL prior to use.
Single and multiple step growth kinetics
HuH7 or HepG2-luc cells were plated in 60 mm dishes and after 24 hours were infected with HSV-1 17+ or HSV1716 at various multiplicities of infections (moi). The dilutions of each virus preparation used for these infections were titrated to confirm the amounts of input virus. In each single experiment, virus infection of HepG2-luc and HuH7 were performed in triplicate or quadruplicate, respectively. After 24, 48, or 72 hours of infection, cells and medium were harvested, subjected to one freeze/thaw cycle (−70°C), and titrated. The results are reported in yields of progeny/input infectious virus and the data were analyzed using GraphPad Prism version 4.02.
In vitro toxicity assays for HepG2-luc cells
HSV1716 toxicity in HepG2-luc cells was assessed using loss of light emission and 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) cell survival assays. HepG2-luc cells were plated out in the internal 6×10 grid of a 96-well tissue culture plate (Greiner Bio-One Ltd, Stonehouse, UK) at ~5,000 cells per well. HSV1716 was added at increasing moi after 24 hours in culture in quadruplicate at least and, after a further 72 hours of incubation, the effect of the virus on light emissions and cell survival were determined. Light emission was detected after addition of 0.05 mL luciferase substrate to each well, and after 5 minutes of incubation at 20°C, light output was measured using a 1420 multilabel counter Victor 3 (Perkin-Elmer) in luminometer mode for 0.1 sec/well. Luciferin substrate was prepared by dissolving 1 g of D-luciferin, potassium salt (OZ Biosciences, Marseilles, France) in 66 mL of phosphate-buffered saline. Cell survival was assessed by addition of 0.01 mL of MTT reagent (Promega) to each well and the absorbance at 490 nm was determined after 1 hour of incubation at 37°C in 5% CO2. Killing curves for HSV1716 by loss of light emissions or MTT assay (GraphPad Prism version 4.02) were used to determine the ED50 (effective moi that kills 50% of the cells) for HSV1716.
Animals
All animal procedures were performed under license from the UK Home Office. Female athymic nude mice (6–8 weeks; Charles River Laboratories, Margate, UK) were maintained under specific pathogen-free conditions. Actively growing HuH7 or HepG2-luc cells were harvested, and after resuspension in phosphate-buffered saline, 1×106 cells per mouse were injected subcutaneously. When HuH7 xenografts reached approximately 5 mm in diameter or HepG2-luc xenografts emitted a stable light signal >106 radiance as determined using an in vivo imaging system (IVIS®; Perkin-Elmer), the mice were injected intratumorally or intravenously via the tail vein with phosphate-buffered saline (no virus) or HSV1716, and were inspected daily after treatment.
For IVIS analysis, the mice were subcutaneously injected in the neck with 0.2 mL of luciferin substrate and left for 15 minutes to assure peak bioluminescence. Luciferin substrate was prepared by dissolving 1 g of D-luciferin potassium salt in 66 mL of phosphate-buffered saline, which was filter-sterilized and frozen in small aliquots to avoid repeated freeze/thaw cycles. The mice were anesthetized using isoflurane gas before being moved into the IVIS machine, and each mouse was placed on a nose cone apparatus to maintain anesthesia throughout imaging. Images were acquired using the Living Image (Perkin-Elmer) software. Areas of luminescence were determined in radiance (watts per steradian per square meter). The mice were then returned to their cages and allowed to recover from the anesthetic.
When HuH7 tumor diameters reached 15 mm, the mice were sacrificed and their xenografts and organs were removed for analysis. The viral load in tumors and organs was assessed by plaque-forming assay on Vero cells. Extracted intact tumors/organs were frozen immediately at −70°C, and after thawing, the tissues were mechanically homogenized for 10 seconds in an Omni TH homogenizer in 1 mL of phosphate-buffered saline prior to titration or luciferase assay. Luciferase activity was determined in 96-well enzyme-linked immunosorbent assay plates using D-luciferin (Promega) as substrate added to 0.1 mL of extract with a 1420 multilabel counter Victor 3 in luminometer mode for 0.1 sec/well.
Results
In vitro replication in HuH7 and HepG2-luc cells
In a single experiment, HuH7 cells were plated in 60 mm dishes, and after 24 hours in culture, the cells were infected with HSV1716 or wild-type HSV-1 17+ in quadruplicate at moi of 0.001 or 0.01. After 72 hours of infection, cells were harvested into the medium and total virus was titrated. At moi 0.01, both viruses had similar levels of propagation in HuH7 cells, with 1,298±361 pfu/input virion and 1,871±412 pfu/input virion for HSV1716 and HSV-1 17+, respectively, with no significant differences between the progeny yields (Student’s t-test, P=0.082). However, at moi 0.001, HSV1716 produced approximately five-fold more progeny than HSV-1 17+, with respective yields of 5,283±609 pfu/input virion for HSV1716 compared with 1,183±288 pfu for HSV-1 17+, and the difference was highly significant (Student’s t-test, P=0.0001). Using the same 0.001 moi preparations but in a separate experiment, HSV-1 17+ yielded approximately 43,100±13,988 pfu and 6,850±2,087 pfu/input virion and HSV1716 yielded 8,806±2,713 pfu or 1,573±182 pfu progeny/input virus after 72 hours of infection in U87 and one58 cells, respectively. Thus, HSV1716 replication in U87 and one58 cells was impaired approximately four to five-fold compared with wild-type HSV-1 17+. As an additional control, Vero cells were infected in parallel with HSV1716 or HSV-1 17+ at the same moi, and there were no significant differences in any of the yields (data not shown).
A single multistep growth curve experiment was performed for HuH7 infected in quadruplicate with HSV-1 17+ and HSV1716 at moi 0.002 and 0.02. Cells were harvested into the medium at 24, 48, and 72 hours, total virus was titrated, and yields were determined at the three time points (Figure 1A). At moi 0.02, similar yields were obtained for both HSV-1 17+ and HSV1716 at each time point. At the lower moi 0.002, yields were similar at 24 and 48 hours, but between 48 and 72 hours there was a much larger increase in HSV1716 progeny production compared with that of HSV-1 17+. As with the single-step growth curve experiment, progeny yields for HSV1716 were again approximately five-fold higher compared with HSV-1 17+, and these differences were highly significant (Student’s t-test, P=0.0001). Thus, in two independent experiments with HuH7 cells, HSV1716 at low moi and after 72 hours of infection, replicates with higher efficiency than wild-type HSV-1 17+, with the largest increase in replication occurring between 48 and 72 hours.
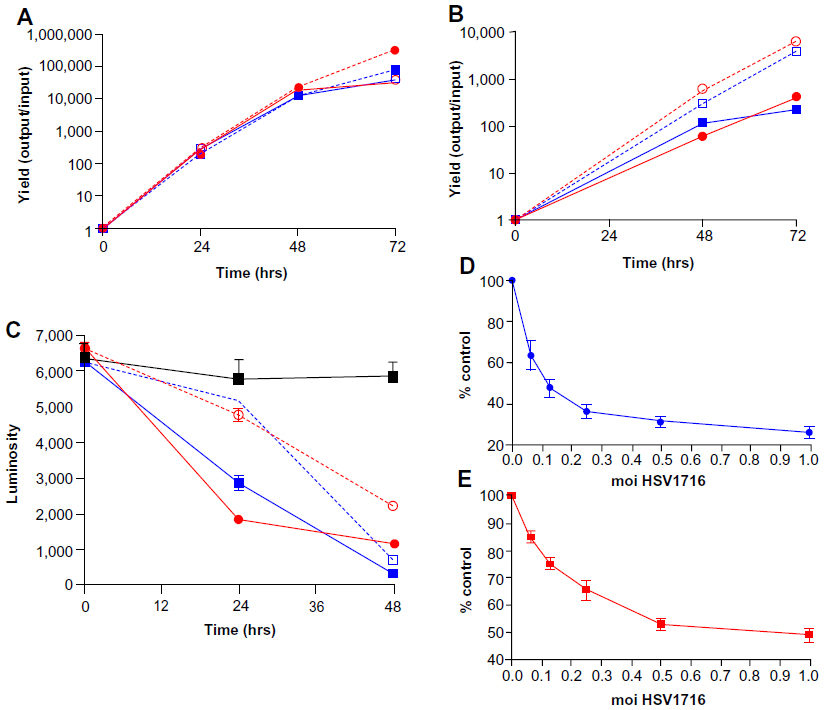 | Figure 1 Replication kinetics of wild-type HSV-1 17+ and HSV1716 in two HCC cell lines, HuH7 (A) and HepG2-luc (B). (A) Mean (± standard deviation) yields (output pfu/input virus) for multistep growth curves from HuH7 infected with HSV-1 17+ (blue) or HSV1716 (red) at moi 0.002 (unbroken line) and 0.02 (dashed line). (B) Mean (± standard deviation) yields (output pfu/input virus) for HepG2-luc cells infected with HSV1716 (red) or HSV-1 17+ (blue) at 0.01 (unbroken line) and 0.001 (dashed line) moi after 48 or 72 hours of infection. (C) Luciferase activity (luminosity) at 24 and 48 hours in HepG2-luc cells infected with HSV1716 (red) or HSV-1 17+ (blue) at 1 (unbroken line) or 0.1 (dashed line) moi. (D and E) HSV1716 moi-dependent HepG2-luc cell killing measured by loss of light emission (D) or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (E) assay. Values are presented as percent mock-infected HepG2-luc cells. |
In a single experiment, HepG2-luc cells were plated in 60 mm dishes, and after 24 hours in culture, cells were infected with HSV1716 or HSV-1 17+ at 0.01 and 0.001 moi in triplicate. After 48 or 72 hours of infection, cells were harvested into the medium and total virus was titrated (Figure 1B). The infected HepG2-luc cells generated high progeny yields of both HSV-1 17+ and HSV1716 at both moi and at both time points, and the yields were similar for both viruses at 0.001 moi. However, after 72 hours of replication at moi 0.01, HSV-1 17+ yielded approximately 4,067±569 pfu progeny/input virus compared with HSV1716 yields of 6,003±987 pfu progeny/input virus, and these differences were significant (Student’s t-test, P=0.0145). Therefore, HSV1716 replication was as at least as efficient and, in some instances more efficient, in both HuH7 and HepG2-luc cells compared with wild-type HSV-1 17+ replication.
HepG2-luc cell death during virus replication was detectable using a loss of light emission assay. Approximately 5,000 HepG2-luc cells/well were plated out in 96-well plates, and after 24 hours in culture, were infected in quadruplicate with HSV1716 or HSV-1 17+ at 1 or 0.1 moi. Luciferase activity (luminosity) was determined at 24 and 48 hours post-infection (Figure 1C). Luciferase activity was constant in uninfected control cells whereas light output decreased in both types of virus-infected cells at the two moi (Figure 1C). Similar levels of moi-dependent and progressive loss of light output were detected for HSV-1 17+ and HSV1716 at 24 and 48 hours, indicating that HSV1716 was as efficient as HSV-1 17+ at killing HepG2-luc cells. Visual inspection of the wells at 48 hours confirmed a virus-mediated cytopathic effect in most cells at this time, and this was confirmed by comparing moi-dependent cell killing for HSV1716 using loss of light emission and an MTT assay. The ED50 of HSV1716 in HepG2-luc cells was equivalent to moi 0.15 as determined using loss of light emission (Figure 1D) and this was very similar to the ED50 of moi 0.12 as determined using the MTT cell survival assay (Figure 1E). Therefore, in HepG2-luc cells, loss of light output correlates with virus replication-mediated HepG2-luc cell death.
Intratumoral injection of nude mice with HepG2-luc xenografts
Fifty nude mice were injected subcutaneously on the flank with the HepG2-luc cells and imaged by IVIS. Luciferase expression in the HepG2-luc cells was visualized within 24–48 hrs as the xenograft formed at the injection site. At day 9 post-cell injection, light-emitting xenografts were formed, with outputs >106 radiance detected in 30 of the 50 mice. These mice were randomized into three groups that received no virus (n=12, injection of phosphate-buffered saline), intratumoral HSV1716 virus at 2×104 pfu (n=6), and intratumoral HSV1716 virus at 2×106 pfu (n=12).
Four of the six mice in the 2×104 pfu HSV1716 group showed a reduction in luciferase levels by day 5 post-injection (Figures 2 and 3A) and light emissions were reduced to background levels in these mice. The other two mice (mice two and three, Figures 2 and 3A) were still positive for light emission on day 32 and were given a further intratumoral injection of HSV1716 2×104 pfu on day 32. One mouse showed a complete loss of light emission at 12 days after the second injection (day 43, Figures 2 and 3A). The second intratumoral injection had no effect on luciferase levels in the xenograft of the remaining mouse (mouse 2, Figures 2 and 3A) and the light-emitting xenograft was still present in this mouse up to day 58 (Figure 3A). This mouse was subsequently euthanized, the tumor was excised and analyzed for the presence of virus, but none was detected, suggesting that HSV1716 failed to establish replication after intratumoral injections in this mouse. Since there was no re-establishment of the HepG2-luc xenografts in five of the six mice receiving HSV1716 2×104 pfu by intratumoral injection, these were designated as cures (Table 1). All control mice maintained light-emitting xenografts throughout this time, as shown for six representative control mice in Figure 2 on the day of treatment and at day 32. Individual radiance outputs are shown in Figure 3B, with the average radiance output for the six control mice on day 0 equal to 5.47×106 radiance, and this had risen to 2.54×107 radiance by day 58, equivalent to an increase of 343,621 radiance/day.
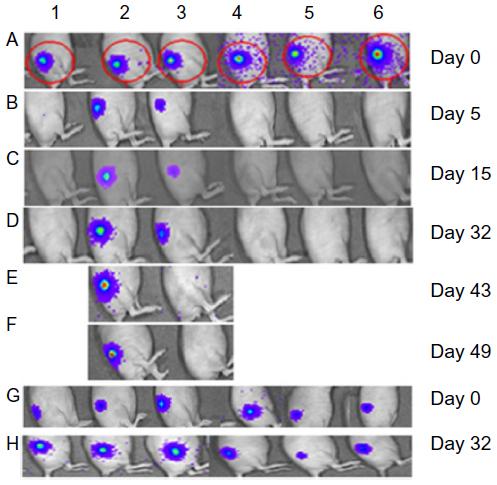 | Figure 2 (A–D) IVIS images of nude mice with HepG2-luc xenografts prior to intratumoral treatment with 2×104 pfu HSV1716 and at 5, 15, and 32 days post-injection. The remaining two mice with an IVIS signal on day 32 (D) are shown 11 (E) and 17 (F) days after a second intratumoral injection of HSV1716 at 2×104 pfu. (G and H) Six representative control mice on days 0 and 32. |
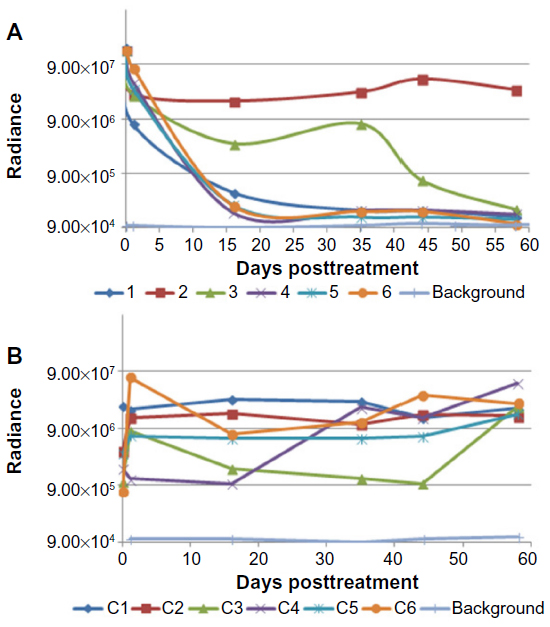 | Figure 3 Individual light output from HepG2-luc xenografts as determined by IVIS imaging of nude mice treated with an intratumoral injection of 2×104 pfu HSV1716 (A) and six representative mice treated with 0.1 mL of phosphate-buffered saline (B). The IVIS background readings for each determination are also shown. |
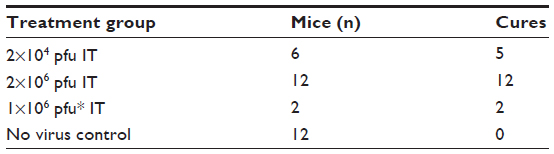 | Table 1 Overall summary of HepG2-luc in vivo results |
For mice treated with HSV1716 2×106 pfu or with phosphate-buffered saline, there was an increase in light output between days 3 and 9 posttreatment, which returned to almost pretreatment levels by day 10 (Figure 4). The reason for this transient increase is unknown. Thereafter, all 12 of the mice treated with HSV1716 2×106 pfu showed a reduction in luciferase levels by day 12 post-injection, and light emissions were reduced to background levels by day 20 (Figure 4). During this time, none of the 12 mice injected with phosphate-buffered saline showed a reduction in luciferase activity levels, which increased steadily from an average of 2.44×106 radiance to 2.2×107 radiance on day 42, equivalent to an increase of 464,782 radiance/day and indicating xenograft growth (Figure 4). The radiance values of the two groups (HSV1716 2×106 and no virus) were significantly different (by Student’s t-test), with P-values of less than 0.05 from day 16 onwards.
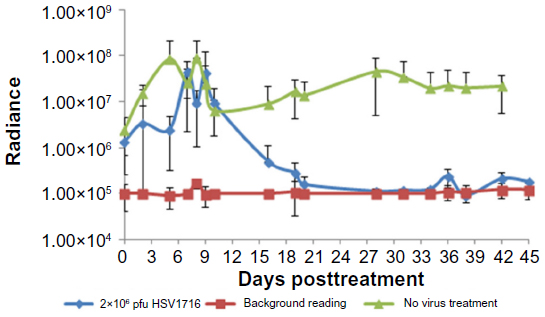 | Figure 4 Average light output (± standard deviation) from HepG2-luc xenografts as determined by IVIS imaging of nude mice treated with an intratumoral injection of 2×106 pfu HSV1716 or phosphate-buffered saline. The IVIS background readings for each determination are also shown. |
The ability of HSV1716 to treat long-established HepG2-luc xenografts was investigated using two mice from one of the control groups (Figure 5A and B). At day 73, these two mice had strong light-emitting xenografts and received HSV1716 1×106 pfu by intratumoral injection. By 17 (Figure 5A) and 11 (Figure 5B) days post-injection, all light emissions from the xenografts were completely lost. The results of the HepG2-luc studies are summarized in Table 1 and clearly demonstrate complete remission of the HepG2-luc xenografts, in most instances by HSV1716 administered via intratumoral injection. Skin from the xenograft site was collected at the time of sacrifice from a number of these cured mice, but no HSV1716 was detected by titration of tissue extracts (data not shown).
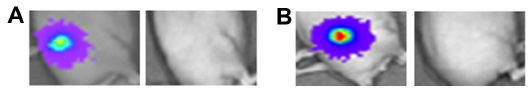 | Figure 5 (A) IVIS image from a mouse with a 73-day-old HepG2-luc xenograft before and 17 days after receiving 1×106 pfu HSV1716 by intratumoral injection. (B) IVIS image from a mouse with a 73-day-old HepG2-luc xenograft before and 11 days after receiving 1×106 pfu HSV1716 by intratumoral injection. |
Intravenous injection of nude mice with HuH7 xenografts
HuH7 cells were injected into the flanks of nude mice and subcutaneous tumors were allowed to develop. Twenty-two mice with appropriately sized tumors were divided into three groups, comprising no virus (phosphate-buffered saline control, n=7), 1×107 pfu (n=8), or 1×106 pfu (n=7) HSV1716 administered by tail vein injection on days 1 and 4. HuH7 tumor xenografts treated with HSV1716 at both 1×106 pfu and 1×107 pfu by intravenous injection on days 1 and 4 had greatly reduced rates of growth compared with controls that received intravenous phosphate-buffered saline (Figure 6A). The difference was highly significant by one-way analysis of variance (P<0.0001) with Tukey’s multiple comparison test for each of the three groups, indicating that both doses of HSV1716 significantly reduced tumor growth compared with the no virus controls (P<0.001 for both). There was no significant difference between the 1×107 and 1×106 pfu doses (P>0.05). By day 13, all of the no virus control mice had been sacrificed due to unrestricted tumor growth (Figure 6A), and the Kaplan–Meier survival plot for each group of mice indicated highly significant improved survival (log-rank test, P=0.0008) in both groups compared with the no virus controls (Figure 6B). Tumor xenografts were completely eradicated by administration of HSV1716 in four of 15 long-term survivors, including one from the 1×107-treated group and the other three from the 1×106-treated group. After sacrifice of the other mice with tumors in the virus-treated groups, tumors and organs were removed and extracts were titrated. In most tumors, large amounts of virus (>1×105 pfu/mL) were present even at 20 days after administration (Table 2), indicating excellent and persistent replication of HSV1716 in the HuH7 xenografts. No virus was detected by titration in the liver, lung, spleen, kidney, gut, skin, brain, or heart from any of these mice, indicating highly specific tumor localization and replication of HSV1716 (data not shown).
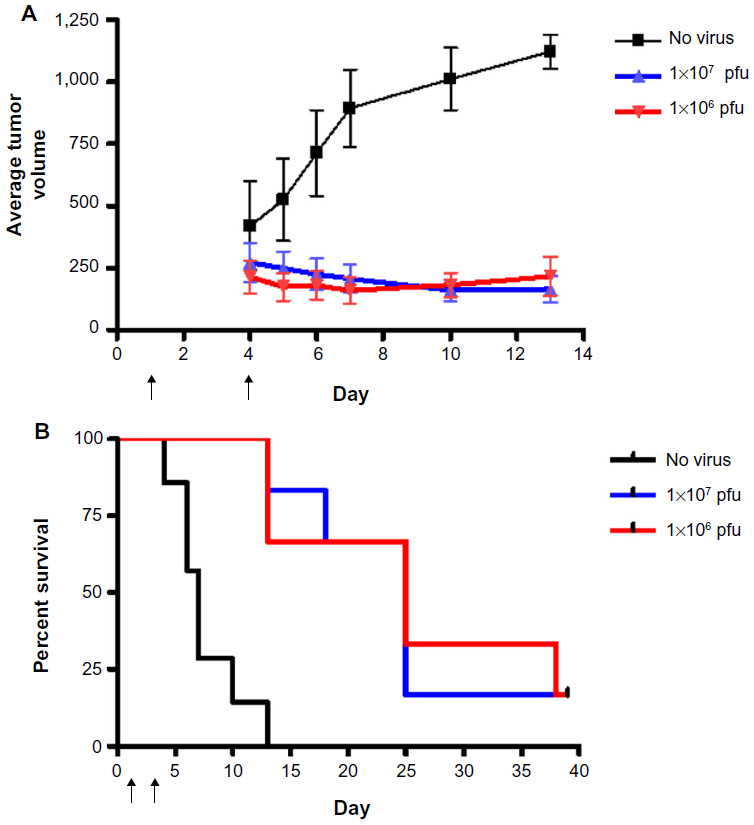 | Figure 6 Growth (A) and survival (B) for mice with HuH7 xenografts treated intravenously with 1×106 (red line) or 1×107 pfu (blue line) HSV1716 on days 1 and 4. |
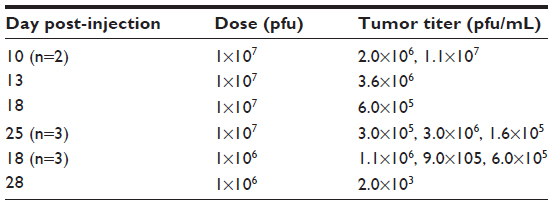 | Table 2 Titers from excised HuH7 xenografts after intravenous administration of HSV1716 on days 1 and 4 |
In a second HuH7 xenograft experiment, 17 mice were randomly assigned into three groups to receive no virus (n=5), HSV1716 1×105 (n=6), or HSV1716 1×106 pfu (n=6) on days 1, 14, and 29 by tail vein injection. Again, HSV1716 by intravenous injection had profound inhibitory effects on tumor growth (Figure 7A) and the differences between treated and control mice were highly significant by analysis of variance (P<0.0001). Tukey’s multiple comparison test for each of the three groups indicated that both doses of HSV1716 significantly reduced tumor growth compared with no virus controls (P<0.005 for both); however, there was no significant difference between the 1×106 and 1×105 pfu doses (P>0.05). All control mice had been sacrificed by day 13 posttreatment due to their tumor burden. The Kaplan–Meier survival plots for each group of mice indicated significantly improved survival (log-rank test, P=0.0157) in both virus-treated groups compared with the no virus controls (Figure 7B). When the experiment was terminated on day 66, four of six mice in the 1×106 pfu group and two of six mice in the 1×105 pfu group had no visible tumor. After sacrifice, if tumors were present, they were removed, extracted, and virus-titrated. In most tumors, large amounts of virus (>1×106 pfu/mL) were present even at 35 days after administration (Table 3). No virus was detected in a sample of skin removed from the xenograft sites in cured mice on day 66 after treatment. Thus, intravenously administered HSV1716 was highly effective at reducing growth and extending survival in HuH7-xenografted mice, with highly efficient replication kinetics within the tumors.
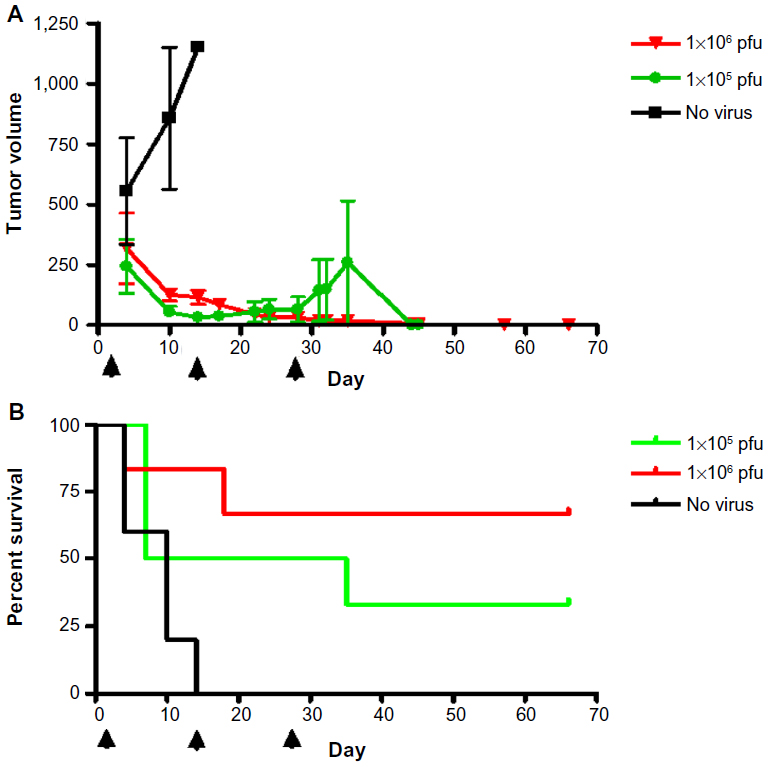 | Figure 7 Growth (A) and survival (B) of mice with HuH7 xenografts treated intravenously with 1×105 (green line) or 1×106 pfu (red line) HSV1716 on days 1, 14, and 29. Control mice (black line) received phosphate-buffered saline injections. Arrows indicate days of treatment. |
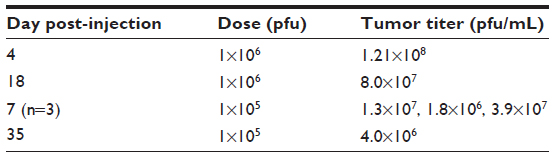 | Table 3 Titers from excised HuH7 xenografts after intravenous administration of HSV1716 on days 1, 14, and 29 |
Tumor localization of intravenously injected HSV1716 in nude mice with HuH7 xenografts
The potent efficacy signals in the HuH7 xenograft studies suggested effective tumor localization of systemically administered HSV1716 followed by highly efficient replication. This was confirmed in biodistribution studies. HSV1716gCluc has the firefly luciferase gene under control of the HSV-1 gC promoter and was used in several tumor localization studies. Luciferase is expressed in the HSV1716gCluc-infected cell as a late gene approximately 8–10 hours after the lytic cycle is initiated (data not shown). Subcutaneous flank HuH7 tumors developed in ten nude mice, and HSV1716gCluc 1×107 pfu was administered to all mice via single tail vein injection, with tumor and organs harvested from two mice on days 1, 4, 7, 14, and 21 post-injection. Biodistribution of virus was analyzed by both titration and luciferase assay, and the average results from the two mice at each time point are presented in Figure 8. Exceptionally high titers of HSV1716, even early on after administration, were detected in the tumors of all mice at all times post-injection (Figure 8A), indicating very efficient viral replication in the HuH7 xenografts. Apart from 1,000 pfu detected transiently in the liver of one mouse on day 4 and in the lungs of another mouse on day 7, HSV1716 was absent from all other tissues at any of the time points. High levels of luciferase activity were detected only in tumor extracts (Figure 8B), with luciferase activity completely absent from all tissue extracts. In particular, no luciferase activity was detected in the liver extract on day 4 or the lung extract on day 7, despite the presence of titratable virus in these tissues at this time. Since luciferase will only be expressed during active HSV1716gCluc infection, this suggests non-replicating HSV1716gCluc in the liver on day 4 and in the lungs on day 7. The presence of virus in the tissues of these two mice was possibly due to leakage of virus into the circulation from the tumor, as extremely high levels of replicating virus were present in the tumor extract.
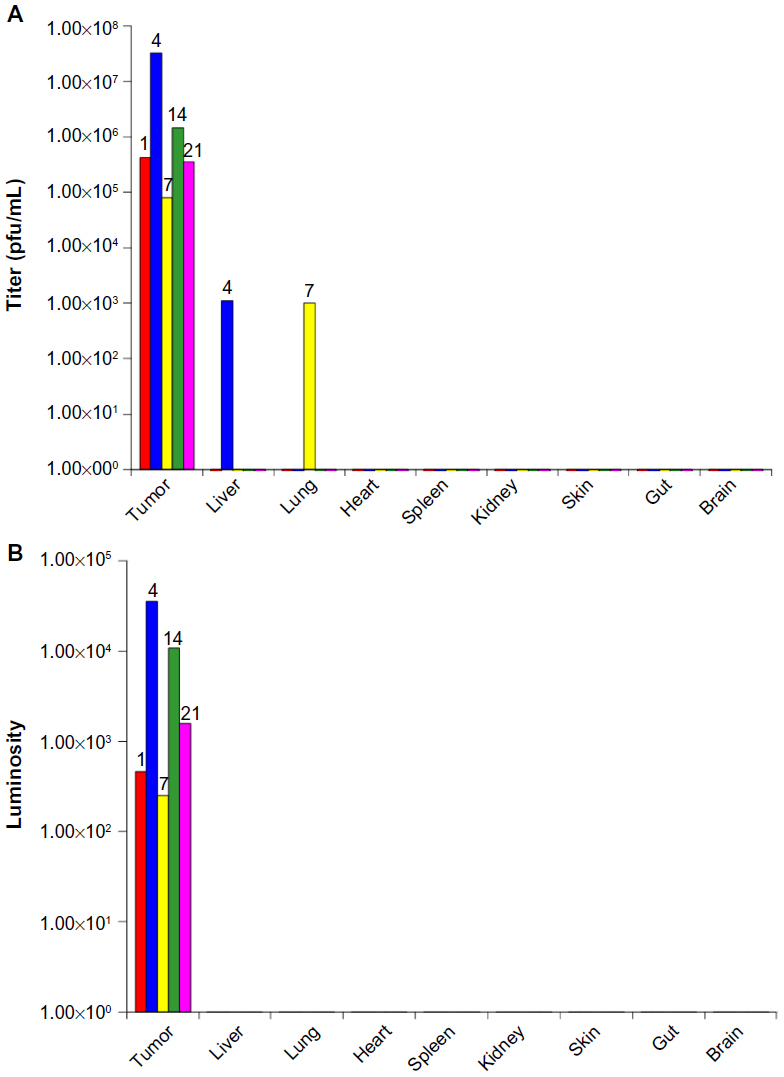 | Figure 8 Biodistribution of HSV1716gCluc analyzed by both titration (A) and luciferase (B) assay in organs from nude mice with HuH7 xenografts harvested at one (red), 4 (blue), 7 (yellow), 14 (green), and 21 (pink) days after receiving a single intravenous injection of 1×107 pfu HSV1716gCluc. Day of sacrifice is indicated at the top of each bar. |
Four nude mice with HuH7 xenografts were injected via the tail vein with HSV1716gCluc 1×107 pfu and tumors were harvested and titrated at 16, 24, 48, and 72 hours post-injection. At 16 hours post-injection, 9×104 pfu/mL were titrated within the tumor indicating that, as the HSV-1 replication cycle is between 18–24 hours, tumor localization of systemically administered virus is extremely rapid and highly efficient. Titratable HSV1716 progeny increased thereafter, with 2.41×105 pfu/mL detected at 24 hours, 1.7×107 pfu/mL at 48 hours, and 8.8×107 pfu/mL by 72 hours (Figure 9A). Virus levels within the tumor were approximately ten-fold greater than the input dose within 72 hours, indicating rapidly established highly efficient replication of HSV1716gCluc in the HuH7 xenografts. The luciferase activity in the extracted tumors was also determined and, presented alongside the titers in Figure 9A, demonstrated a good correlation between luciferase activity and virus titers in the tumor extracts.
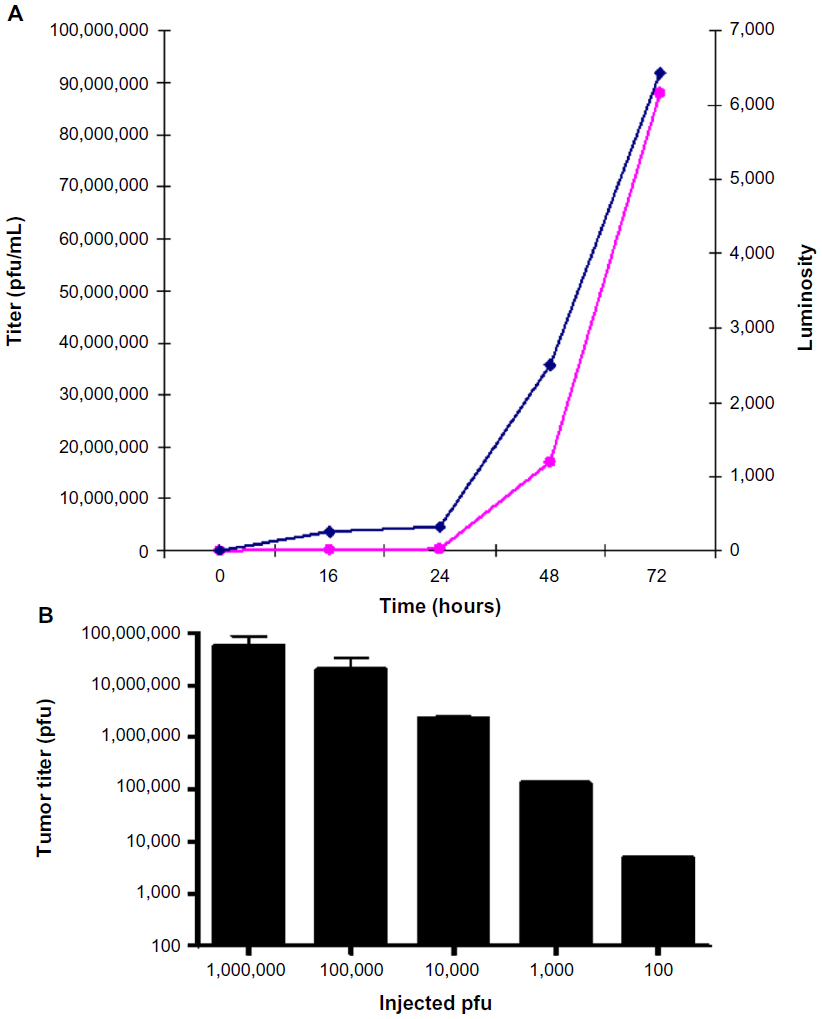 | Figure 9 (A) Titers (dark blue) and luciferase activity (pink) in tumor extracts from nude mice with HuH7 xenografts injected via the tail vein with 1×107 pfu HSV1716gCluc. (B) Titers from HuH7 xenografts 72 hours after intravenous injection of 100, 1×103, 1×104, 1×105, or 1×106 pfu HSV1716. |
In a further tumor localization study, ten nude mice with HuH7 xenografts received increasing amounts of HSV1716 by tail vein injection and the virus was titrated 72 hours after administration. Two mice each received HSV1716 1×106, 1×105, 1×104, 1×103, and 100 pfu, and in all cases, the tumor titers at 72 hours had exceeded the input dose by at least ten-fold and in some instances by 100-fold (Figure 9B). Systemic administration of HSV1716 as little as 100 pfu was sufficient to establish xenograft replication, and intravenously injected HSV1716 is therefore able to locate rapidly to HuH7 xenografts and establish highly efficient replication, leading to inhibition of growth, prolonged survival, and, in many instances, a cure.
Discussion
In vitro, HSV1716 demonstrated excellent replication kinetics in two well characterized HCC cell lines, ie, HuH7 and a luciferase-expressing variant of HepG2 cells. In both cell lines, HSV1716 was better at replicating and produced significantly more progeny than the wild-type virus from which it was derived, in HuH7 in two separate experiments at lower moi and in HepG2-luc in a single experiment at higher moi. This superior replication competence in both HCC cell lines is unusual. We observed the reverse, ie, a lower replication competence of HSV1716 compared with HSV-1 17+, in U87 and one58 cells and in many different human cancer cell lines.24 This was also observed in vivo, with infection of HuH7 xenografts rapidly generating exceptionally high titers of HSV1716 within 72 hours. It is possible that in wild-type HSV-1-infected HuH7 or HepG2-luc, ICP34.5 has a negative influence on replication efficiency and this is lost in the ICP34.5-deleted HSV1716, but more extensive studies are required to confirm this.
In vitro, HSV1716 infection reduced the light output from HepG2-luc cells in culture and was correlated with reduced cell survival; we used this to investigate HSV1716 efficacy in a HepG2-luc xenograft model. IVIS analysis was used to assess the success of the virus in eliminating the rapidly formed light-emitting HepG2-luc xenografts in the flanks of nude mice. Loss of light emission following virus treatment was permanent, and these animals were therefore scored as cures. In control animals, light emissions were consistently at least 100-fold higher than background (>106 radiance), and were often greater than 108 radiance, with light output increasing at a constant rate for over 100 days. Single intratumoral injections of HSV1716 were highly effective at eliminating the HepG2-luc xenografts, even at low doses of 2×104 pfu, with most mice cured. Even at later times after implantation of HepG2-luc cells, single intratumoral injections of HSV1716 were highly effective. Additionally, a repeat intratumoral injection was usually sufficient to eliminate xenografts that persisted after initial virus administration. A small number of mice with HepG2-luc xenografts received HSV1716 by intravenous injection, and although the xenografts were still present after the initial intravenous injection, they were eliminated in four of five mice after two subsequent intravenous injections. The xenograft persisting in one mouse despite these three injections suggests that intravenously administered HSV1716 most probably failed to localize to this tumor, and no virus was detected by titration in the xenograft of this mouse at the time of sacrifice (data not shown).
Monitoring light emission from the HepG2-luc xenografts was a useful and convenient method for monitoring the success of oncolytic virus treatment. Loss of light emission as visualized by IVIS correlated well with xenograft regression. Nonresponding mice were also readily identifiable, and further virus treatments could be administered. However, the method is limited by the availability of luciferase-expressing cell lines. We also considered the usefulness of an HSV1716 variant that expressed luciferase in a replication-dependent manner via an HSV-1 late promoter. HSV1716gCluc replication was readily visualized by IVIS in HuH7 xenografts, and light emission correlated with virus replication. Rapid and specific tumor localization of systemically administered HSV1716gCluc was readily observed by IVIS 72 hours after intravenous injection of HSV1716gCluc. These strong light emissions from the replication of HSV1716gCluc within the HuH7 xenografts persisted for at least 21 days after a single administration, indicating the continuous presence of replicating virus within the xenograft (data not shown). Further, in biodistribution studies, although some virus was detected sporadically in normal organs (liver and lung) by titration, luciferase assays demonstrated that it was nonreplicating and therefore most likely a result of tumor-derived, blood-born virus contaminating the tissues. Studies with HepG2-luc and HSV1716gCluc therefore demonstrate the utility of light-based methodologies to monitor oncolytic virus efficacy in xenograft models.
HuH7 cells formed rapidly growing aggressive xenografts, and we assessed the efficacy of intravenously administered HSV1716 in mice with HuH7 xenografts. Mice received either two injections close together or three injections at 2-weekly intervals. Although both treatment schemes were highly effective at restricting tumor growth, the three injections at 2-weekly intervals were better at effecting cures when compared with the two injections on days 1 and 4. In both experiments, all control mice had to be sacrificed by 13 days after treatment was started due to their tumor burdens. Approximately 25% of the mice treated with two doses were cured, compared with 50% of mice receiving the three doses, and interestingly, most of the mice receiving three doses of HSV1716 were sacrificed within the 14-day interval between the first and second injections (n=4), with only two mice sacrificed thereafter, one after the second injection and one after the third. These results suggest that a more effective dosing scheme may comprise two initial injections within the first 4 days followed by two subsequent injections 14 and 28 days later.
In the HuH7 xenograft model, there was extremely rapid tumor localization of intravenously administered HSV1716, which quickly established highly efficient replication, with levels of intratumoral virus exceeding the input doses. Surprisingly, uptake of HSV1716 from the circulation by HuH7 cells was highly efficient, as at least one virion from an intravenous injection of 100 pfu (ie, at least 1%) was sufficient to establish replicating virus in the xenograft. Thus, HSV1716 is highly effective at reducing growth and extending survival in HuH7 and HepG2-luc xenografts in nude mice as a result of highly efficient uptake of virus by cells and prolific replication kinetics within the tumors.
HCC is the third most common cause of cancer-related deaths.2 Early detection is limited, and given that the majority of patients present with advanced inoperable disease, chemotherapy is the only option. There are several internationally recognized staging systems that combine liver function and tumor extension for the prognosis and management of therapeutic options in patients with HCC, including Cancer of the Liver Italian Program, Barcelona Clinic Liver Cancer, and Japanese Integrated Staging, with the Barcelona Clinic Liver Cancer system most widely accepted in clinical practice.3,26 Doxorubicin is routinely used as a single agent, but shows insufficient response rates of 15%–20%.27 Sorafenib is the only systemic therapy universally approved for advanced liver cancer. Sorafenib blocks RAF protein kinase, a critical component of the RAF/MEK/ERK signaling pathway that controls cell division and proliferation. In addition, sorafenib inhibits the vascular endothelial growth factor receptor-2/platelet-derived growth factor receptor-beta signaling cascade, thereby blocking tumor angiogenesis. Phase III clinical trials have indicated that single-agent sorafenib might have a beneficial therapeutic effect, adding about 3 months to the lifespan of late-stage HCC patients with well-preserved liver function.4,28 However, in these trials, objective tumor responses were rare, and toxicities, including rash, diarrhea, and fatigue, often led to dose reductions and/or discontinuation of treatment.29 Recently, impressive results with the oncolytic vaccinia virus JX-594 in advanced HCC have been reported in a Phase II study.5 JX-594 has selective replication competence for cancer cells via inactivation of the viral thymidine kinase gene, and additionally expresses immunostimulatory human granulocyte-macrophage colony-stimulating factor and beta-galactosidase.30–32 In a randomized Phase II dose-finding study including 30 patients, JX-594 was infused intratumorally at 108 pfu or 109 pfu on days 1, 15, and 29, and demonstrated significantly improved survival between the 108 pfu and 109 pfu cohorts with median survivals of 14.1 and 6.7 months, respectively.5 Although both oncolytic and immunostimulatory mechanisms of action were demonstrated, their relative contribution to survival could not be determined, but the authors did note that the acute peak JX-594 concentration in blood correlated with duration of overall survival, suggesting that control of tumor growth and improved survival may be achievable via high-dose intravenous administration.
In other relevant clinical studies, NV1020, which is a highly attenuated, multimutated replication competent derivative of HSV-1, has been delivered by hepatic artery infusion in patients with metastatic colorectal carcinoma to the liver in Phase I and Phase I/II studies.33–35 Patients received four NV1020 doses via weekly hepatic artery infusion, followed by two or more cycles of conventional chemotherapy. Phase I included cohorts receiving 3×106, 1×107, 3×107, or 1×108 pfu/dose, and the 1×108 pfu/dose was identified as the optimal biological dose for Phase II. All of the 22 Phase I/II patients who received the optimal biological dose had previously received 5-fluorouracil, most had received oxaliplatin or irinotecan (50% had both), and many had received at least one targeted agent. After administration of NV1020, 50% showed stable disease and the best overall tumor control rate after chemotherapy was 68% (one with a partial response, 14 with stable disease). Therefore, the study suggested that NV1020 extended overall survival by stabilizing colorectal cancer liver metastases with minimal toxicity and potentially resensitizing them to salvage chemotharapy.
In our preclinical HCC studies, HSV1716 was highly efficacious by intravenous administration in an aggressive HuH7 model, and although there was some dose effect, scheduling of administration was potentially more important. Excellent uptake of HSV1716 from the circulation combined with prolific replication are the most likely key determinants of inhibition of tumor growth and enhanced survival. Our preclinical results in two HCC models therefore support clinical translation of systemic HSV1716.
Disclosure
All authors are employees of Virttu Biologics Ltd but have no other conflicts of interest in this work.
References
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. | |
Llovet JM, Bruix J. Novel advancements in the management of hepatocellular carcinoma in 2008. J Hepatol. 2008;48 Suppl 1:S20–S37. | |
Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–1022. | |
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. | |
Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. | |
Kesari S, Randazzo BP, Valyi-Nagy T, et al. Therapy of experimental human brain tumors using neuroattenuated herpes simplex virus mutant. Lab Invest. 1995;73:636–648. | |
Kucharczuk JC, Randazzo B, Chang MY, et al. Use of a “replication restricted” herpes virus to treat experimental human malignant mesothelioma. Cancer Res. 1997;57:466–471. | |
Randazzo BP, Bhat MG, Kesari S, Fraser NW, Brown SM. Treatment of experimental subcutaneous human melanoma with a replication-restricted herpes simplex virus mutant. J Invest Dermatol. 1997;108:933–977. | |
Coukos G, Makrigiannakis A, Kang EH, et al. Use of carrier cells to deliver a replication-selective herpes simplex virus-1 mutant for the intraperitoneal therapy of epithelial ovarian cancer. Clin Cancer Res. 1999;5:1523–1537. | |
Rampling R, Cruikshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859–866. | |
Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoral injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9:398–406. | |
Harrow S, Papanastassiou V, Harland J, et al. HSV1716 injection into the brain adjacent to tumor following surgical resection of high-grade glioma: safety data and long term survival. Gene Ther. 2004;11:1648–1658. | |
MacKie RM, Stewart B, Brown SM. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet. 2001;357:525–526. | |
Mace AT, Ganly I, Soutar DS, Brown SM. Potential for efficacy of the oncolytic herpes simplex virus 1716 in patients with oral squamous cell carcinoma. Head Neck. 2008;30:1045–1051. | |
Argnani R, Marconi P, Volpi I, et al. Characterization of herpes simplex virus 1 strains as platforms for the development of oncolytic viruses against liver cancer. Liver Int. 2011;31:1542–1553. | |
Foka P, Pourchet A, Hernandez-Alcoceba R, et al. Novel tumour-specific promoters for transcriptional targeting of hepatocellular carcinoma by herpes simplex virus vectors. J Gene Med. 2010;12:956–967. | |
Santamaría E, Mora MI, Carro-Roldán E, et al. Identification of replication-competent HSV-1 Cgal+ strain targets in a mouse model of human hepatocarcinoma xenograft. J Proteomics. 2009;73:153–160. | |
Song TJ, Eisenberg DP, Adusumilli PS, Hezel M, Fong Y. Oncolytic herpes viral therapy is effective in the treatment of hepatocellular carcinoma cell lines. J Gastrointest Surg. 2006;10:532–542. | |
Miyatake S. Gene therapy using tissue-specific replication competent HSV. Hum Cell. 2002;15:130–137. | |
Xue F, Dong CY, Su Y, et al. Tumor-targeted therapy with a conditionally replicating mutant of HSV-1 induces regression of xenografted human hepatomas. Cancer Biol Ther. 2005;4:1234–1239. | |
Zager JS, Delman KA, Malhotra S, et al. Combination vascular delivery of herpes simplex oncolytic viruses and amplicon mediated cytokine gene transfer is effective therapy for experimental liver cancer. Mol Med. 2001;7:561–568. | |
Pawlik TM, Nakamura H, Yoon SS, et al. Oncolysis of diffuse hepatocellular carcinoma by intravascular administration of a replication-competent, genetically engineered herpesvirus. Cancer Res. 2000;60:2790–2795. | |
Miyatake SI, Tani S, Feigenbaum F, et al. Hepatoma-specific antitumor activity of an albumin enhancer/promoter regulated herpes simplex virus in vivo. Gene Ther. 1999;6:564–572. | |
Conner J, Braidwood L, Brown SM. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther. 2008;15:1579–1592. | |
Conner J, Braidwood L. Expression of inhibitor of growth 4 by HSV1716 improves oncolytic potency and enhances efficacy. Cancer Gene Ther. 2012;19:499–507. | |
Toyoda H, Kumada T, Kiriyama S, et al. Comparison of the usefulness of three staging systems for hepatocellular carcinoma (CLIP, BCLC, and JIS) in Japan. Am J Gastroenterol. 2005;100:1764–1771. | |
Yeo W, Mok TS, Zee B, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:1532–1538. | |
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. | |
Heo J, Breitbach CJ, Moon A, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19:1170–1179. | |
Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. | |
Kim JH, Oh JY, Park BH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther. 2006;14:361–370. | |
Parato KA, Breitbach CJ, Le Boeuf F, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20:749–758. | |
Kemeny N, Brown K, Covey A, et al. Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther. 2006;17:1214–1224. | |
Fong Y, Kim T, Bhargava A, et al. A herpes oncolytic virus can be delivered via the vasculature to produce biologic changes in human colorectal cancer. Mol Ther. 2009;17:389–394. | |
Geevarghese SK, Geller DA, de Haan HA, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther. 2010;21:1119–1128. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.