")
Back to Journals » Journal of Pain Research » Volume 15
Potent Analgesic Action of 2-acetoxy-5-(2-4 (trifluoromethyl)-phenethylamino)-benzoic Acid (Flusalazine) in Experimental Mice
Authors Kim SS , Won S, Lee HE, Ryu SH, Choi DJ, Cho SI, Gwag BJ, Youn HY, Lee JH
Received 16 August 2022
Accepted for publication 25 November 2022
Published 9 December 2022 Volume 2022:15 Pages 3869—3879
DOI https://doi.org/10.2147/JPR.S385617
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert B. Raffa
Sung-Soo Kim,1,* Sojung Won,2,* Ha Eun Lee,2 Seung Hyun Ryu,2 Dong Joon Choi,2 Sung Ig Cho,2 Byoung Joo Gwag,2 Hwa-Young Youn,3 Jin Hwan Lee2
1VIP Animal Medical Center KR, Seoul, 02830, Republic of Korea; 2GNT Pharma, Yongin, Gyeonggi, 17096, Republic of Korea; 3Laboratory of Veterinary Internal Medicine, College of Veterinary Medicine, Seoul National University, Seoul, 08826, Republic of Korea
*These authors contributed equally to this work
Correspondence: Jin Hwan Lee, GNT Animal Health Business Unit, Yongin, Gyeonggi, 17096, Republic of Korea, Tel +82 10 8878 0456, Fax +82 31 8005 9931, Email [email protected]
Purpose: Nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase (COX)-2 selective inhibitors are the most widely used drugs to treat pain. Conventional NSAIDs and COX-2 selective inhibitors, however, cause several side effects such as gastric damage, kidney damage, and cardiovascular problems. Our previous study showed that 2-acetoxy-5-(2-4-(trifluoromethyl)-phenethylamino)-benzoic acid ie, flusalazine (also known as ND-07), which exerts dual actions by serving both as an anti-inflammatory agent and a free radical scavenger, is an effective and safe treatment for severe inflammatory diseases in mice. The goal of the present study was to examine the potential analgesic action and safety of flusalazine in mice models of pain.
Methods and Results: Flusalazine showed a significant analgesic effect in an acetic acid-induced abdominal constriction model. Likewise, total paw licking was reduced significantly in neurogenic (early stage) and inflammatory (late stage) pain induced by formalin in flusalazine-treated mice. In the tail immersion test, flusalazine significantly increased tail withdrawal time at 2 h after its administration. Also, the formation of paw edema in the flusalazine-treated group was significantly inhibited in a carrageenan-induced inflammatory pain model. Gastric damage was not induced by flusalazine even up to 1000 mg/kg, while aspirin and indomethacin caused critical gastric bleeding.
Conclusion: These findings suggest that flusalazine’s safety profile and analgesic effects have high translational potential for the clinical treatment of patients experiencing pain.
Keywords: analgesic effect, aspirin, flusalazine, indomethacin, NSAIDs
Introduction
Pain is defined as an unpleasant sensation and emotional experience associated with actual or potential damage to a specific part of the body. It is one of the most common symptoms in many medical conditions and can significantly diminish quality of life. Over the decades, nonsteroidal anti-inflammatory drugs (NSAIDs) have been commonly prescribed for the management of acute and chronic pain, bringing about their effects by inhibiting the cyclooxygenase (COX) enzyme.1 COX is responsible for converting arachidonic acid into prostaglandins, which plays a key role in the inflammatory response and mediation of pain.
The two currently identified isoforms of COX, COX-1 and COX-2, are known to perform unique functions. COX-1 exists as a constitutive form and plays an important role in regulating normal physiological functions such as platelet activation and gastrointestinal protection. On the other hand, COX-2 exists as an inducible isoform that mediates pro-inflammatory activity by creating prostaglandin E2 (PGE2), resulting in the sensation of pain when activated by inflammatory stimuli such as proinflammatory cytokines and bacterial toxins.1,2 Thus, the prolonged use of non-specific COX inhibitors for attenuating inflammatory responses leads to numerous side effects due to the cytotoxic effects caused by inhibiting the COX enzymes. To overcome the side effects of NSAIDs observed frequently in association with gastro-intestinal lesions, the COX-2 selective NSAIDs were developed. Unfortunately, selective COX-2 inhibitors have been linked to an increased risk of myocardial infarction and renal toxicity due to the inhibition of prostacyclin (PGI2) and thromboxane A2 (TXA2) as with pathogenic PGE2 resulting in gastro-intestinal, renal, and cardiovascular complications.3–7 There are three different PGE synthases (PGESs): cytosolic PGES (cPGES) and two microsomal PGES (mPGES-1 and mPGES-2). cPGES and mPGES-2 are constitutive enzymes, whereas mPGES-1 is induced by inflammatory stimuli.8–11 Therefore, a selective mPGES-1 inhibitor to only inhibit the terminal PGE2 biosynthesis might be a potential alternative with much safer gastro-intestinal, renal, and cardiovascular complication profiles than traditional NSAIDs and COX-2 inhibitors in treating patients with acute or chronic pain.1,8–15
On the other hand, evidence supports the central role of oxidative stress in the pathogenesis of pain. Elevated oxidative stress generated by superoxide anions, hydroxyl radicals, and hydrogen peroxide plays a central role in multiple pathological pain states, such as neuropathic pain, migraine-related pain, and inflammatory pain, suggesting that antioxidant therapy might be beneficial in patients experiencing pain.16–19
We previously reported that 2-acetoxy-5-(2-4-(trifluoromethyl)-phenethylamino)-benzoic acid ie, flusalazine (also known as ND-07), reduces the severity of acute pancreatitis through targeting both free radicals and PGE2-mediated inflammation.20 Furthermore, flusalazine has anti-inflammatory action through specific mPEGS-1 inhibition, not COX-1 or COX-2 inhibition.20 In the present study, we evaluated the anti-analgesic action and safety of flusalazine in animal models to confirm its potential as a novel analgesic agent for treating pain.
Materials and Methods
Experimental Animals
Male ICR mice with a body weight of 20–25 g were supplied from Orient Bio Co., Ltd. (Seoul, South Korea) and acclimated for 7 days. Animals were maintained in separate cages with laboratory chow and water ad libitum. During the experiment, animals were kept at 23±3°C with a relative humidity of 50±10% in a 12-h light-dark cycle. Animals were handled in accordance with a protocol approved by the GNT Pharma Institutional Animal Care Committee.
Abdominal Writhing Induced by Acetic Acid in Mice
In the writhing test, male mice were divided into six groups. The procedure described by Gaertner et al21 was used with slight modifications. The mice received an intraperitoneal injection of 1% acetic acid solution in normal saline at a dose of 10 mL/kg. The number of writhes was counted starting 5 min after the injection and lasted for 15 min. The response consisted of abdominal wall contractions and pelvic rotation followed by hind limb stretches. Antinociceptive activity was expressed as a reduction in the number of abdominal constrictions between the control animals and the mice that were treated with the compounds. Flusalazine (10–250 mg/kg), aspirin (25–300 mg/kg), indomethacin (10, 100 mg/kg), and vehicle (10% Lutrol F127 in water) were administered orally 30 min prior to acetic acid injection.
Formalin Test in Mice
The procedure described by Santos et al22 was used with slight modifications. Pain was induced by injecting 0.05 mL of 2.5% formalin in normal saline in the subplantar of the right hind paw. The amount of time spent licking the injected paw was indicative of pain. The number of lickings from 0–5 min (first phase) and 25–35 min (second phase) were counted after the injection of formalin. Antinociception was calculated as a percentage of inhibition of writhing constrictions by using the formula [(control group mean – test group mean)/(control group)] 100%.23 Mice (six per group) were given flusalazine (25, 100 mg/kg), aspirin (300 mg/kg), indomethacin (10 mg/kg) and vehicle 30 min prior to injecting formalin.
Tail Immersion Test
Tail immersion was conducted as described by Asongalem et al.24 It involved immersing the extreme 3 cm of the mouse’s tail in a water bath containing water at a temperature of 55°C. Each animal served as its own control and two readings were obtained for the control at 0- and 10-min intervals. The average of the two values was the initial reaction time (pre-treatment value). Flusalazine (25, 100 mg/kg), aspirin (300 mg/kg), indomethacin (10 mg/kg), and vehicle were administered orally to mice. Thirty minutes later, tail reaction response was tested with one-third of the tail immersed in a water bath heated to 55°C. The response latency between the onset of immersion and the withdrawal of the tail was recorded.
Carrageenan-Induced Hind Paw Edema and Hyperalgesia
Acute hind paw edema and thermal hyperalgesia were produced by injecting 0.05 mL of carrageenan (4% suspension in normal saline) locally into the subplantar of the right hind paw of mice. Animals were divided into five groups with five mice per group. Flusalazine (25, 100 mg/kg), aspirin (300 mg/kg), and vehicle were administered (P.O.) 30 min prior to injection of carrageenan in the right hind paw subplantar of each mouse. The thermal hyperalgesia and paw edema were measured 2 h after the injection of carrageenan. The paw edema was measured by a plethysmometer (Ugo Basile S.R.L., Gemonio, Italy) just before and 2 h after the injection of carrageenan. Edema was expressed as the difference between the vehicle, flusalazine, aspirin, or vehicle and the basal level.
Thermal hyperalgesia was assessed using latency of paw withdrawal25 from a radiant heat source applied to the plantar surface of the hind paws. Animals were placed in transparent acrylic boxes for 20–30 min to adapt before application of radiant heat through the glass flooring. Latency from stimulus onset to paw withdrawal was measured across three trials with a cutoff of 30s. The formula used for calculating the percent change was (baseline latency-postdrug latency) x 100 (baseline latency)−1 or [(latency observed) – (latency control) x 100] / [(cut-off) – (latency control)].26
Gastrointestinal Safety Test
Mice were divided into four groups (n = 5 each) and deprived of food but not water for 24 h prior to treatment. Flusalazine (1000 mg/kg), aspirin (100, 300 mg/kg), indomethacin (10, 100 mg/kg), and vehicle (1% Lutrol F127 in water) were administered orally to mice. Animals were anesthetized with 2% isoflurane 5 h after drug administration. The stomachs were removed and then inflated with 3.7% formalin and placed in fresh formalin before assessment. The following day, the stomachs were opened, and gastric mucosal lesions were examined microscopically. The stomach was removed, opened along the greater curvature, washed with normal saline, and flattened on a piece of cardboard. The gross damage of the gastric mucosa was assessed by an experienced gastroenterologist, who was blind to the treatments, using a gross ulcer index.27 The gastric mucosal membrane was exposed on a microscope with a video camera (Kaiser, Seda Media).
PGES Activity Assay
PGES activity in cell lysates was analyzed by measuring the conversion of PGH2 to PGE2 as previously reported.28 PGES activities in cell lysates were measured by assessment of the conversion of PGH2 to PGE2. The cells were scraped from the dishes and disrupted by sonication (10 s three times at 1 min intervals) in 250 μL 0.2 M Tris-HCl, pH 8.0. After centrifugation of the sonicates at 15,000 rpm for 10 min at 4℃, the supernatant fluids were used as the enzyme source. An aliquot of each lysate (90 μg protein equivalents) was incubated with 2 μg PGH2 for 30s at 24℃ in 100 μM 0.1 M Tris-HCl, pH 8.0, containing 2 mM glutathione and 14 μM indomethacin. After terminating the reaction by the addition of 100 mM FeCl2, PGE2 contents in the supernatant fluids were quantified using an enzyme immunoassay (EIA) kit (Cayman Chemical Inc., Ann Arbor, MI, USA; Cat. No. 514010 for PGE2).
Prostaglandin Production in the Peritoneal Cavity of Mice
Acetic acid-induced prostaglandin formation in the peritoneal cavity of mice was based on the method described by Tong et al,29 which was used with slight modification. At 20 min after injection of 1% acetic acid, the mice were killed by cervical dislocation. The peritoneal cavity was exposed through a 15 mm incision, and the lavage fluid was pipetted off and transferred to a polypropylene tube. The samples were centrifuged at 2500 rpm for 10 min at 4℃ and the supernatant (0.5 mL) was used for PGE2 measurement. The level of PGE2 in the peritoneal cavity was determined by an EIA kit (Cayman Chemical Inc., Ann Arbor, MI, USA) in accordance with the manufacturer’s instructions.
Statistical Analysis
Data are expressed as mean ± SEM. Data were analyzed with the SPSS software package (Statistical Program for Social Science, version 12.0, SPSS Inc., Chicago, IL, USA). Different groups were compared using a one-way analysis of variance (ANOVA) and Tukey analysis. Statistical significance was set at p<0.05 and p<0.01.
Results
Flusalazine Rescues Acetic Acid-Induced Abdominal Pain in Mice
The analgesic activity of flusalazine was evaluated using the abdominal writhing test. The acetic acid-induced abdominal constriction method is widely used for the evaluation of peripheral antinociceptive activity.21 As shown in Figure 1, 25, 50, 100, and 250 mg/kg of flusalazine showed significant inhibitory effects of 64%, 63%, 68.8%, and 70.2%, respectively. Aspirin (25–300 mg/kg) and indomethacin (10, 100 mg/kg) were used as positive controls, and they produced writhing inhibition in a dose-responsive manner. The inhibition of writhes in percentage ranged from 52.7–70.3% and 62.7–73.5% after the administration of aspirin and indomethacin, respectively.
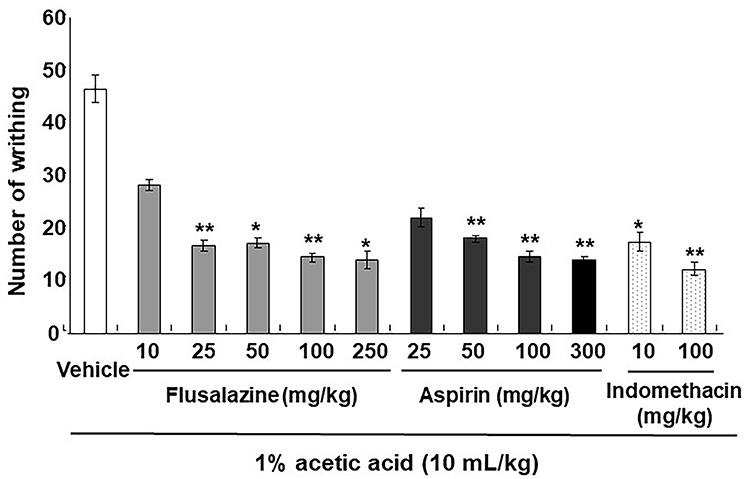 |
Figure 1 Analgesic effect of flusalazine on acetic acid-induced writhing in mice. The mice received 1% acetic acid solution in normal saline injected intraperitoneally at a dose of 10 mL/kg. The number of writhes was counted starting 5 min after injection and lasting for 15 min. Flusalazine (10, 25, 50, 100, and 250 mg/kg), aspirin (25, 50, 100, and 300 mg/kg), indomethacin (10, 100 mg/kg), and vehicle (1% Lutrol F-127 in water) were administered orally 30 min prior to acetic acid injection. Data are expressed as mean ± S.E.M. *p<0.05, **p<0.01 versus vehicle group compared by one-way ANOVA followed by Tukey. |
Flusalazine Reduces Formalin-Induced Neurogenic and Inflammatory Pain in Mice
The formalin test was performed to examine the antinociceptive effect of flusalazine in neurogenic and inflammatory pain. The intraplantar injection of formalin into the right hind paw generated a biphasic (early and late) nociceptive response. The early (neurogenic) phase was probably a direct result of stimulation of nociceptors in the paw and reflects centrally mediated pain while the late (inflammatory) phase appeared to be dependent on the combination of an inflammatory reaction in the peripheral tissue.30 As seen in Figure 2, flusalazine significantly reduced nociception in both the early (neurogenic) and late (inflammatory) phases in a dose-dependent manner (69.5% and 73.6% at 25 mg/kg; 50.0% and 66.0% at 100 mg/kg, respectively). The licking time in both the early and late phases were also significantly reduced in the aspirin (64.0% and 78.7% at 300 mg/kg, respectively)- and indomethacin (55.4% and 52.3% at 10 mg/kg, respectively)-treated groups.
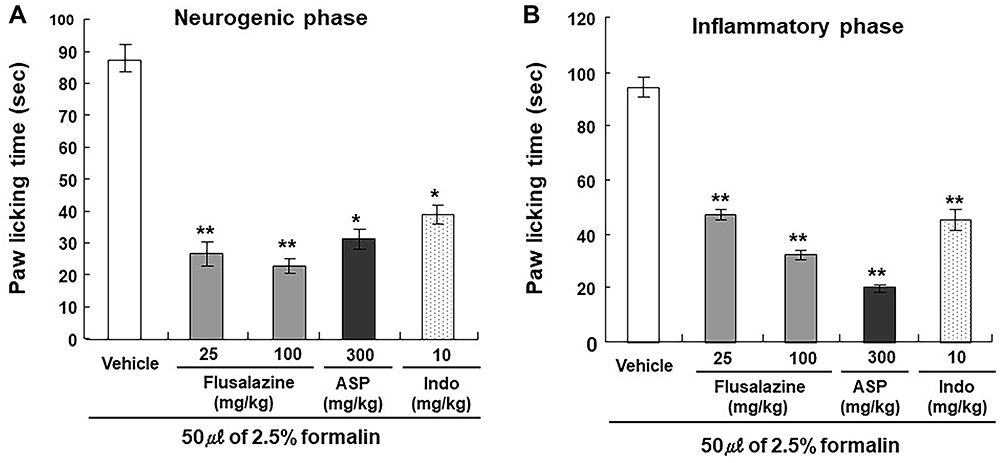 |
Figure 2 Antinociceptive effect of flusalazine on formalin-induced licking (early phase (A) and late phase (B)) in mice. Pain was induced by injecting 0.05 mL of 2.5% formalin in normal saline in the subplantar region of the right hind paw. The amount of time spent licking the injected paw was indicative of pain. The number of lickings from 0–5 min (early phase) and 25–35 min (late phase) was counted after the injection of formalin. Flusalazine (25, 100 mg/kg), aspirin (300 mg/kg), indomethacin (10 mg/kg) and vehicle (1% Lutrol F127 in water) were administered orally 30 min prior to formalin injection. Data are expressed as mean ± S.E.M. *p<0.05, **p<0.01 versus vehicle group compared by one-way ANOVA followed by Tukey. |
Flusalazine Prevents Thermal Nociceptive Pain in Mice
In an acute thermal nociception mice model, the effect of flusalazine was investigated. As shown in Table 1, flusalazine had anti-nociceptive effects in a dose-dependent manner. The most prominent analgesic effects for the 25 and 100 mg/kg of flusalazine were observed 90 and 120 min after oral administration, respectively. Aspirin (300 mg/kg) and indomethacin (10 mg/kg) also exhibited analgesic effects at 90 min after the treatment.
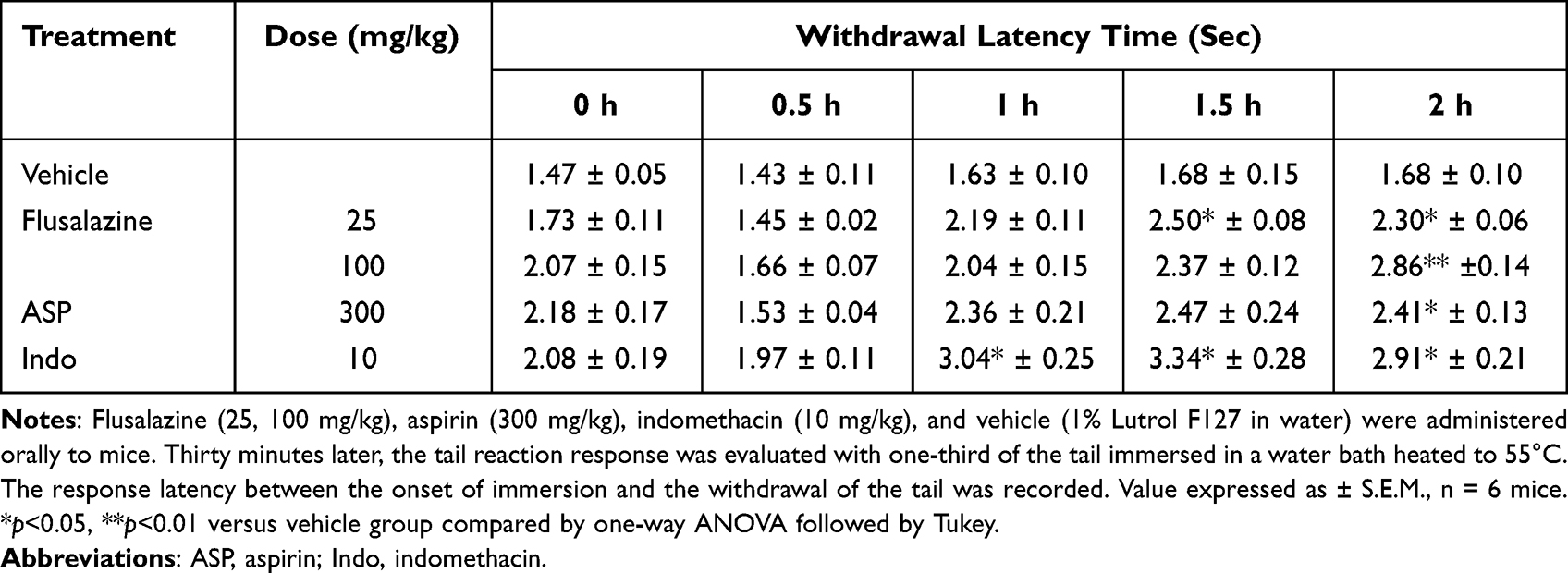 |
Table 1 Effect of Flusalazine on Pain Induced by the Tail Immersion Test in Mice |
Flusalazine Reduced Carrageenan-Induced Inflammatory Pain in Mice
Additionally, a carrageenan model was performed to examine the effects of flusalazine on inflammatory pain. The effects of orally administered flusalazine on paw edema and paw withdrawal induced by carrageenan are shown in Figure 3. Paw edema was significantly reduced by 46.5% at 25 mg/kg of flusalazine, and significantly decreased up to 52.9% in 100 mg/kg of flusalazine compared with the vehicle (Figure 3). Aspirin (300 mg/kg) also significantly inhibited (45.2%) the edema during the same period as compared with the vehicle. In paw withdrawal latency, vehicle decreased paw withdrawal latency compared with normal. However, 100 mg/kg of flusalazine significantly improved paw withdrawal latency by 45.7% compared with vehicle. Also, aspirin increased paw withdrawal latency by 24.0%.
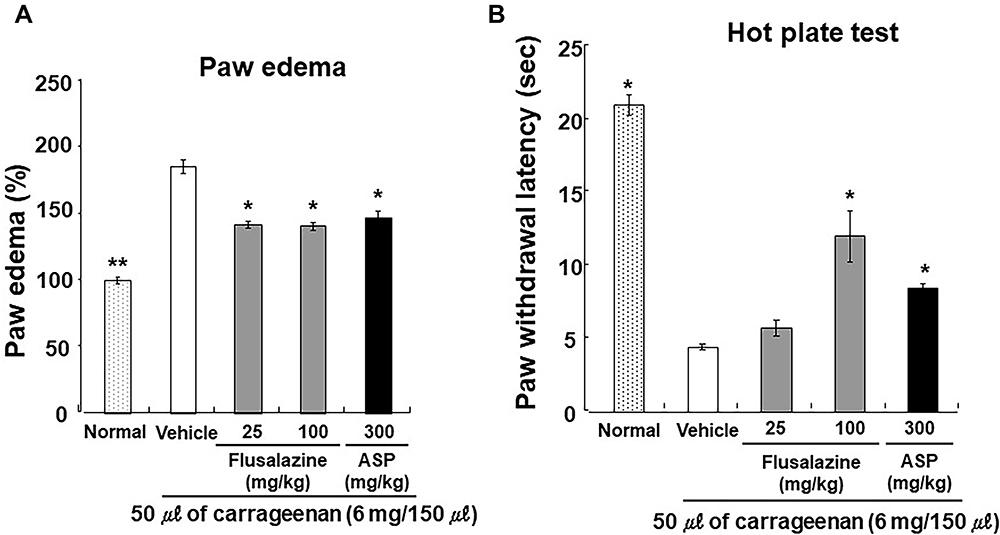 |
Figure 3 Effect of flusalazine on carrageenan-induced paw edema (A) and thermal hyperalgesia (B) in mice. The acute hind paw edema and thermal hyperalgesia were produced by injecting 0.05 mL of carrageenan (4% suspension in normal saline) locally into the subplantar region of the right hind paw of mice. Flusalazine (25, 100 mg/kg), aspirin (300 mg/kg), and vehicle (1% Lutrol F127 in water) were administered (P.O.) 30 min prior to injection of carrageenan in the right hind paw subplantar region of each mouse. The thermal hyperalgesia and paw edema were measured 3 h after the injection of carrageenan. Data are expressed as mean ± S.E.M. *p<0.05, **p<0.01 versus vehicle group compared by one-way ANOVA followed by Tukey. |
Flusalazine Causes No Gastric Damage
We evaluated the degree of gastric mucosal damage after the oral administration of flusalazine, aspirin, and indomethacin by macroscopic examination. While oral administration of 100 mg/kg aspirin and 10 mg/kg indomethacin caused severe gastric bleeding 24 h later (Figure 4), oral administration of flusalazine even up to 1000 mg/kg was not associated with any gastric damage.
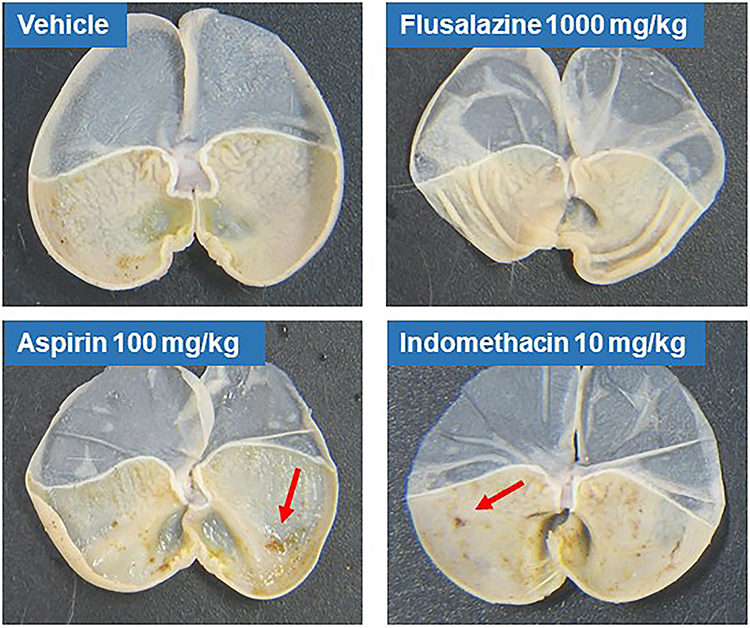 |
Figure 4 Gastrointestinal safety of flusalazine in mice. Mice were deprived of food but not water for 24 h prior to treatment. Flusalazine (1000 mg/kg), aspirin (100, 300 mg/kg), indomethacin (10, 100 mg/kg), and vehicle (1% Lutrol F127 in water) were administered orally to mice. Animals were anesthetized with pentobarbital sodium (45 mg/kg) by an intraperitoneal injection 5 h after drug administration. The stomach was removed, opened along the greater curvature, washed with normal saline, and flattened on a piece of cardboard. The gastric mucosal membrane was exposed on a microscope with a video camera. |
Prostaglandin Production in the Peritoneal Cavity of Mice
The effect of flusalazine on the inhibition of PGE2 levels in the peritoneal cavity of mice is shown in Figure 5. The levels of PGE2 in the peritoneal cavity of mice significantly increased at 20 min after acetic acid injection, however, flusalazine was associated with reduced levels of PGE2. The PGE2 level in the flusalazine group was similar to the control group and not significantly different compared to the NSAIDS group (aspirin, celecoxib, and indomethacin).
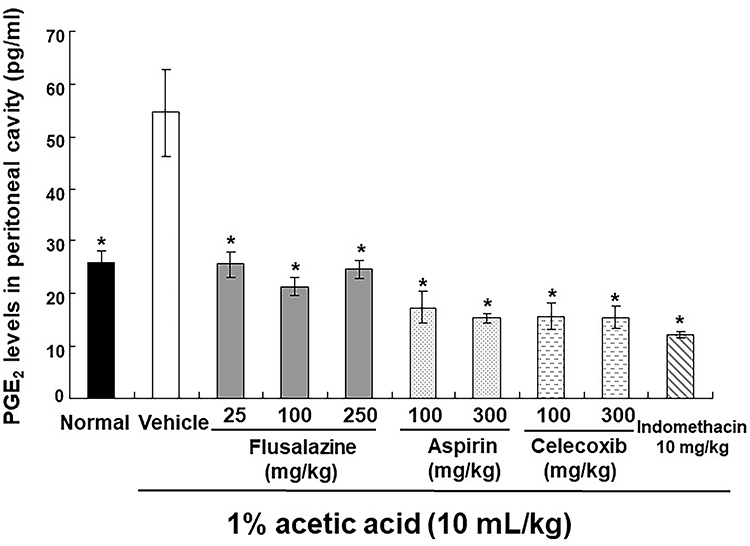 |
Figure 5 Inhibitory effect of flusalazine against PGE2 level on acetic acid-induced writhing response in mice. At 20 min after intraperitoneal injection of 1% acetic acid in normal saline, the mice were killed by cervical dislocation. Flusalazine (25, 100, 250 mg/kg), aspirin (100, 300 mg/kg), celecoxib (100, 300 mg/kg), indomethacin (10 mg/kg), and vehicle (1% Lutrol F-127 in water) were administered orally 30 min prior to acetic acid injection. The lavage fluid in the peritoneal cavity was collected and PGE2 was quantified by ELISA. Data are expressed as mean ± S.E.M. *p<0.05 versus vehicle group compared by one-way ANOVA followed by Tukey. |
Therapeutic Index Between the Effective Dose and Toxic Dose
Based on our current findings of gastric damage and acetic acid-induced pain models, the therapeutic index was considered. The therapeutic index was calculated from the following formula: therapeutic index = gastric toxic dose / maximum effective dose. The therapeutic index of flusalazine, aspirin, and indomethacin were represented above 40, below 1, and 1, respectively (Table 2).
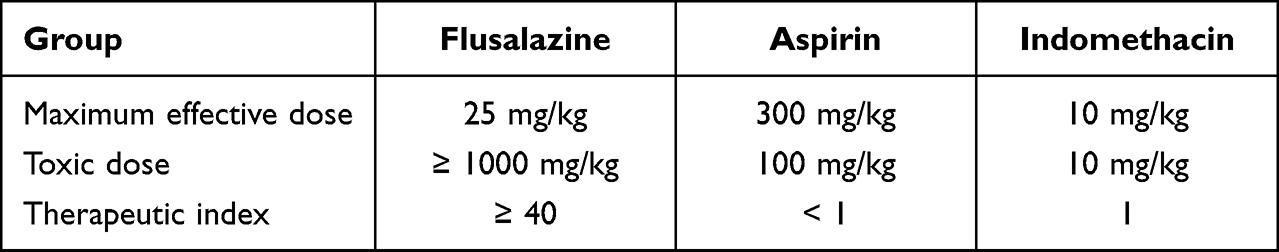 |
Table 2 Therapeutic Index of Flusalazine |
Discussion
The present investigation provides new evidence supporting the therapeutic benefits of flusalazine for treating pain. We previously reported that the potent spin trapping and anti-inflammatory action of flusalazine has significant preventive effects in severe inflammatory disease models.20 Because free radicals and the ensuing inflammation are principally involved in the pathogenesis of pain, we hypothesized that flusalazine would exhibit beneficial effects against pain through its potent ability to inhibit oxidative stress and inflammation. In the present study, flusalazine rescues the various pain responses induced by visceral, neurogenic, and inflammatory pain, successfully supporting the hypothesis. Although NSAIDs are the most commonly used analgesic, they have the limitation for treating pain patients due to undesirable side effects. Consistently, traditional NSAIDs such as aspirin and indomethacin cause severe gastric damage, whereas flusalazine even up to 1000 mg/kg does is not associated with any gastric damage, suggesting that flusalazine is a much safer compound. This report appears to be the first to demonstrate the therapeutic benefits of flusalazine in pain.
The acetic acid-induced writhing is a visceral pain model that is widely used to evaluate antinociceptive activity.21 Acetic acid produces pain via the activation of nociceptors or damage to the visceral surface, which leads to the release of histamine, bradykinin, prostaglandins, and serotonin.31 Among these mediators, increased levels of PGE2 in peritoneal fluid mainly contribute to the development of inflammatory pain.32 In addition, recent reports indicate that mPGES-1 plays a key proinflammatory role in fever and pain.33,34 This implies that, to prevent the development of pain, it is necessary to inhibit PGE2 production, which is generated by the activation of mPGES-1. The administration of 25 mg/kg of flusalazine inhibited acetic acid-induced abdominal constrictions as much as 50 mg/kg of aspirin and 10 mg/kg of indomethacin. We observed that the level of PGE2 in the peritoneal cavity of mice significantly increased in the acetic acid-induced writhing model but flusalazine inhibited the increased level of PGE2. Flusalazine reduced conversion of PGH2 to PGE2 with an IC50 of 0.33 μM in extracts of lipopolysaccharide (LPS)-treated BV2 cells, suggesting that flusalazine has a role as a mPGE-1 inhibitor at submicromolar concentrations in cells (data not shown). These findings indicated that flusalazine exerts a remarkable antinociceptive action through PGE2 reduction by selective mPGES-1 inhibition. This is supported by the efficacy of flusalazine in formalin or carrageenan-induced pain mice. In the formalin test, there is a distinctive biphasic nociceptive response termed the early phase and late phase. The early phase corresponds to neurogenic pain and is caused by the direct effect of formalin on the nociceptors of C-fibers (non-inflammatory pain).30,35 Drugs acting on the CNS such as morphine and codeine can inhibit this phase. The late phase is related with the inflammatory response development, releasing the nociceptive mediators in peripheral tissue and functional changes in the dorsal medulla body (inflammatory pain). This phase can be inhibited by NSAIDs and centrally acting drugs. Peripheral nociceptor in inflammatory process was sensitized by COX-2-derived prostanoids, which caused localized pain hypersensitivity.30,36,37
Interestingly, flusalazine and NSAIDs revealed the analgesic actions in both phases of the formalin test in the present study. As mentioned above, it has been believed that only neurogenic pain is involved in the early phase of formalin. However, there are some controversial data.38–40 Those reports showed the analgesic effects of NSAIDs in not only the late phase but also the early phase, indicating that prostaglandins production might be induced even in the early stage of the formalin test. Analgesic action during the early phase of flusalazine treatment may be caused by a reduction of prostaglandins production in the CNS. In the second phase, the antinociceptive effect of flusalazine may be ascribed to the inhibition of PGE2 production and other mediators involving in this test. In the carrageenan test, we also observed a significant decrease in paw edema and a significant increase in paw withdrawal latency with administration of flusalazine 2 h after administration of carrageenan. These results suggest that flusalazine has analgesic activities by decreasing PGE2-mediated inflammatory activity. Furthermore, accumulated reports showed that only opioid agonists (eg, morphine) could completely inhibit nociceptive pain.41 Interestingly, our results revealed that 25 and 100 mg/kg flusalazine significantly reduced the licking time during the early stage of formalin, and the reaction time was significantly increased at 2 h after administration of 100 mg/kg flusalazine in tail immersion test compared with vehicle. This result allows us to suggest that flusalazine can interfere with either the production or the release of nociception-related neuropeptides.
Taken together, flusalazine has analgesic activity not only against inflammatory pain but also against nociceptive pain, which suggests the possibility that at least some of the antinociceptive effects of flusalazine could be mediated via activation of the opioid receptor. Some evidence indicates that oxidative stress can play an essential role in the pathogenesis of pain. Superoxide, hydroxyl radical, and hydrogen peroxide-induced oxidative stress were involved in the inflammatory response of pathological pain states, such as neuropathic pain, migraine-related pain, and inflammatory pain.16–19 Reactive oxygen species (ROS) are one of the mediators of neuroinflammation, leading to an accelerated pain response.42 In addition, antioxidants such as 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxy (TEMPOL), phenyl-N-tert-butylnitrone (PBN), N-acetyl-L-cysteine (NAC), and 2-acetylphenothiazine (ML171; a specific inhibitor of NOX1) are known to attenuate both acute and late (tonic) formalin-induced nociceptive responses in mice.43,44 However, the intrathecal injection of TEMPOL effectively reduced nociception during the late (tonic) phase, implying that peripheral site of action of ROS may be more important in mediating acute phase of formalin-induced nociceptive responding.43 In the previous finding, flusalazine showed potent spin trapping of hydroxyl radicals. It was expected that flusalazine showed 120-fold better than vitamin E in a similar set of hydroxyl radical scavenging assays.20,45 Although further research might be needed to reveal the anti-oxidant roles of flusalazine in the pathogenesis of pain, these results suggest that the pharmacological property of flusalazine as a spin-trapping molecule helps to provide more analgesic effects.
Due to adverse effects such as gastrointestinal damage, the use of traditional NSAIDs has been limited in terms of their use for pain relief, in spite of their excellent analgesic effects4,5 mainly caused by the inhibition of COX-1 in the gastrointestinal tract. In this study, aspirin and indomethacin caused evident gastric mucosal damage at doses of 100 mg/kg and 10 mg/kg, respectively. However, flusalazine showed no association with gastric mucosal damage up to a dose of 1000 mg/kg. In a mPGES-1 assay using microsomal fraction purified from an LPS-pretreated BV-2 murine microglial cell line, flusalazine directly inhibited mPGES-1 activity with an IC50 of 0.33 μM but with much weaker inhibition against ovine recombinant COX-1 (IC50=16.2 μM) and COX-2 enzymes (IC50=651.1 μM; data not shown). These results imply that flusalazine has a much higher safety margin compared to traditional NSAIDs.
It is important to note that anti-oxidants protect against NSAID-induced gastric injuries in rats, supporting that flusalazine could have an additional safety profile through its powerful spin trapping action. To examine the safety profile of flusalazine compared with NSAIDs such as indomethacin and aspirin, we also calculated the therapeutic index for the efficacy of each drug according to the pain model. This showed that the maximum effective dose of flusalazine, aspirin, and indomethacin was 25 mg/kg, 300 mg/kg, and 10 mg/kg, respectively. However, the therapeutic index of aspirin and indomethacin was either lower than or equal to its maximal efficacy doses. This is consistent with previous reports demonstrating that NSAIDs could cause gastric damage.46 Compared with NSAIDs, flusalazine showed a higher therapeutic index with an approximately 40-fold difference, suggesting that flusalazine could be substituted for NSAIDs without causing gastric damage during treatment. In addition, the no observable adverse effect levels (NOAELs) of flusalazine in a rat and dog during a 4-week repetitive toxicity study were 125 mg/kg (P.O. once daily) and >125 mg/kg, respectively, without significant adverse events to kidney, stomach, and heart which are the main toxic target organs of traditional NSAIDs including COX-2 inhibitors.
Conclusion
Our results demonstrate that flusalazine, (a compound with dual pharmacological antioxidant and anti-inflammatory actions), has analgesic effects with a much wider safety margin compared to traditional NSAIDs. Although further studies are required to verify the exact mechanism of the analgesic effects of flusalazine against pain, we suggest that flusalazine could be a potential drug candidate for pain management.
Ethics Approval
All procedures were conducted in compliance with the Animal Experiment Regulations of GNT Pharma Co. Ltd., the Animal Experiment Committee Regulations, and the Animal Experiment Approval Provisions (approval no. 20-0002).
Disclosure
The authors report no conflicts of interest in this work.
References
1. McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96(1):272–277. doi:10.1073/pnas.96.1.272
2. Garavito RM, DeWitt DL. The cyclooxygenase isoforms: structural insights into the conversion of arachidonic acid to prostaglandins. Biochim Biophys Acta. 1999;1441(2–3):278–287. doi:10.1016/S1388-1981(99)00147-X
3. Martinez-Gonzalez J, Badimon L. Mechanisms underlying the cardiovascular effects of COX-inhibition: benefits and risks. Curr Pharm Des. 2007;13(22):2215–2227. doi:10.2174/138161207781368774
4. Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med. 1992;327(11):749–754. doi:10.1056/NEJM199209103271101
5. Murray MD, Brater DC. Renal toxicity of the nonsteroidal anti-inflammatory drugs. Annu Rev Pharmacol Toxicol. 1993;33:435–465. doi:10.1146/annurev.pa.33.040193.002251
6. Hawkey CJ. COX-1 and COX-2 inhibitors. Best Pract Res Clin Gastroenterol. 2001;15(5):801–820. doi:10.1053/bega.2001.0236
7. Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol Rev. 2008;88(4):1547–1565. doi:10.1152/physrev.00004.2008
8. Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275(42):32775–32782. doi:10.1074/jbc.M003504200
9. Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96(13):7220–7225. doi:10.1073/pnas.96.13.7220
10. Murakami M, Naraba H, Tanioka T, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275(42):32783–32792. doi:10.1074/jbc.M003505200
11. Mancini JA, Blood K, Guay J, et al. Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001;276(6):4469–4475. doi:10.1074/jbc.M006865200
12. Burnier M. The safety of rofecoxib. Expert Opin Drug Saf. 2005;4(3):491–499. doi:10.1517/14740338.4.3.491
13. Wang D, Wang M, Cheng Y, Fitzgerald GA. Cardiovascular hazard and non-steroidal anti-inflammatory drugs. Curr Opin Pharmacol. 2005;5(2):204–210. doi:10.1016/j.coph.2005.02.001
14. Rudic RD, Brinster D, Cheng Y, et al. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96(12):1240–1247. doi:10.1161/01.RES.0000170888.11669.28
15. Xu XJ, Reichner JS, Mastrofrancesco B, Henry WL, Albina JE. Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol. 2008;180(4):2125–2131. doi:10.4049/jimmunol.180.4.2125
16. Yowtak J, Lee KY, Kim HY, et al. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain. 2011;152(4):844–852. doi:10.1016/j.pain.2010.12.034
17. De Logu F, Nassini R, Materazzi S, et al. Schwann cell TRPA1 mediates neuroinflammation that sustains macrophage-dependent neuropathic pain in mice. Nat Commun. 2017;8(1):1887. doi:10.1038/s41467-017-01739-2
18. Marone IM, De Logu F, Nassini R, et al. TRPA1/NOX in the soma of trigeminal ganglion neurons mediates migraine-related pain of glyceryl trinitrate in mice. Brain. 2018;141(8):2312–2328. doi:10.1093/brain/awy177
19. Ibi M, Matsuno K, Shiba D, et al. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci. 2008;28(38):9486–9494. doi:10.1523/JNEUROSCI.1857-08.2008
20. Lee JH, An CS, Yun BS, et al. Prevention effects of ND-07, a novel drug candidate with a potent antioxidative action and anti-inflammatory action, in animal models of severe acute pancreatitis. Eur J Pharmacol. 2012;687(1–3):28–38. doi:10.1016/j.ejphar.2012.04.048
21. Gaertner M, Muller L, Roos JF, et al. Analgesic triterpenes from Sebastiania schottiana roots. Phytomedicine. 1999;6(1):41–44. doi:10.1016/S0944-7113(99)80033-6
22. Santos AR, Filho VC, Niero R, et al. Analgesic effects of callus culture extracts from selected species of Phyllanthus in mice. J Pharm Pharmacol. 1994;46(9):755–759. doi:10.1111/j.2042-7158.1994.tb03897.x
23. Ait Tastift M, Makbal R, Bourhim T, Omari Z, Isoda H, Gadhi C. Safety assessment and pain relief properties of saffron from Taliouine Region (Morocco). Molecules. 2022;27(10):3339. doi:10.3390/molecules27103339
24. Asongalem EA, Foyet HS, Ekobo S, Dimo T, Kamtchouing P. Antiinflammatory, lack of central analgesia and antipyretic properties of Acanthus montanus (Ness) T. Anderson. J Ethnopharmacol. 2004;95(1):63–68. doi:10.1016/j.jep.2004.06.014
25. Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32(1):77–88. doi:10.1016/0304-3959(88)90026-7
26. Svensson CI, Zattoni M, Serhan CN. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J Exp Med. 2007;204(2):245–252. doi:10.1084/jem.20061826
27. Chandranath SI, Bastaki SM, Singh J. A comparative study on the activity of lansoprazole, omeprazole and PD-136450 on acidified ethanol- and indomethacin-induced gastric lesions in the rat. Clin Exp Pharmacol Physiol. 2002;29(3):173–180. doi:10.1046/j.1440-1681.2002.03626.x
28. Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem. 2005;94(6):1546–1558. doi:10.1111/j.1471-4159.2005.03302.x
29. Tong Y, Zhou XM, Wang SJ, Yang Y, Cao YL. Analgesic activity of myricetin isolated from Myrica rubra Sieb. et Zucc. leaves. Arch Pharm Res. 2009;32(4):527–533. doi:10.1007/s12272-009-1408-6
30. Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51(1):5–17. doi:10.1016/0304-3959(92)90003-T
31. Garcia MD, Fernandez MA, Alvarez A, Saenz MT. Antinociceptive and anti-inflammatory effect of the aqueous extract from leaves of Pimenta racemosa var. ozua (Mirtaceae). J Ethnopharmacol. 2004;91(1):69–73. doi:10.1016/j.jep.2003.11.018
32. Verma PR, Joharapurkar AA, Chatpalliwar VA, Asnani AJ. Antinociceptive activity of alcoholic extract of Hemidesmus indicus R. Br. in mice. J Ethnopharmacol. 2005;102(2):298–301. doi:10.1016/j.jep.2005.05.039
33. Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59(3):207–224. doi:10.1124/pr.59.3.1
34. Zhou Z, Yuan Y, Zhou S, Ding K, Zheng F, Zhan CG. Selective inhibitors of human mPGES-1 from structure-based computational screening. Bioorg Med Chem Lett. 2017;27(16):3739–3743. doi:10.1016/j.bmcl.2017.06.075
35. Connor J, Makonnen E, Rostom A. Comparison of analgesic effects of khat (Catha edulis Forsk) extract, D-amphetamine and ibuprofen in mice. J Pharm Pharmacol. 2000;52(1):107–110. doi:10.1211/0022357001773580
36. Shields SD, Cavanaugh DJ, Lee H, Anderson DJ, Basbaum AI. Pain behavior in the formalin test persists after ablation of the great majority of C-fiber nociceptors. Pain. 2010;151(2):422–429. doi:10.1016/j.pain.2010.08.001
37. Puig S, Sorkin LS. Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain. 1996;64(2):345–355. doi:10.1016/0304-3959(95)00121-2
38. Kunnaja P, Wongpalee SP, Panthong A. Evaluation of anti-inflammatory, analgesic, and antipyretic activities of the ethanol extract from Murdannia loriformis (Hassk.) Rolla Rao et Kammathy. Bioimpacts. 2014;4(4):183–189. doi:10.15171/bi.2014.018
39. Moniruzzaman M, Imam MZ. Evaluation of antinociceptive effect of methanolic extract of leaves of Crataeva nurvala Buch.-Ham. BMC Complement Altern Med. 2014;14:354. doi:10.1186/1472-6882-14-354
40. Hunskaar S, Berge OG, Hole K. Dissociation between antinociceptive and anti-inflammatory effects of acetylsalicylic acid and indomethacin in the formalin test. Pain. 1986;25(1):125–132. doi:10.1016/0304-3959(86)90014-X
41. Figueiredo JG, da Silveira Bitencourt F, Beserra IG, et al. Antinociceptive activity and toxicology of the lectin from Canavalia boliviana seeds in mice. Naunyn Schmiedebergs Arch Pharmacol. 2009;380(5):407–414. doi:10.1007/s00210-009-0448-2
42. Xu J, Wu S, Wang J, et al. Oxidative stress induced by NOX2 contributes to neuropathic pain via plasma membrane translocation of PKCepsilon in rat dorsal root ganglion neurons. J Neuroinflammation. 2021;18(1):106. doi:10.1186/s12974-021-02155-6
43. Hacimuftuoglu A, Handy CR, Goettl VM, Lin CG, Dane S, Stephensjr RL. Antioxidants attenuate multiple phases of formalin-induced nociceptive response in mice. Behav Brain Res. 2006;173(2):211–216. doi:10.1016/j.bbr.2006.06.030
44. Kumar S, Vinayak M. NADPH oxidase1 inhibition leads to regression of central sensitization during formalin induced acute nociception via attenuation of ERK1/2-NFkappaB signaling and glial activation. Neurochem Int. 2020;134:104652. doi:10.1016/j.neuint.2019.104652
45. Kim JI, Lee JH, Choi DS, Won BM, Jung MY, Park J. Kinetic study of the quenching reaction of singlet oxygen by common synthetic antioxidants (tert-butylhydroxyanisol, tert-di-butylhydroxytoluene, and tert-butylhydroquinone) as compared with alpha-tocopherol. J Food Sci. 2009;74(5):C362–C369. doi:10.1111/j.1750-3841.2009.01160.x
46. Fesharaki M, Nasimi A, Mokhtari S, Mokhtari R, Moradian R, Amirpoor N. Reactive oxygen metabolites and anti-oxidative defenses in aspirin-induced gastric damage in rats: gastroprotection by Vitamin E. Pathophysiology. 2006;13(4):237–243. doi:10.1016/j.pathophys.2006.08.003
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.