Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Post-stroke dyskinesias
Authors Nakawah MO, Lai EC
Received 28 July 2016
Accepted for publication 23 September 2016
Published 7 November 2016 Volume 2016:12 Pages 2885—2893
DOI https://doi.org/10.2147/NDT.S118347
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Mohammad Obadah Nakawah, Eugene C Lai
Stanely H. Appel Department of Neurology, Houston Methodist Neurological Institute, Houston, TX, USA
Abstract: Strokes, whether ischemic or hemorrhagic, are among the most common causes of secondary movement disorders in elderly patients. Stroke-related (vascular) movement disorders, however, are uncommon complications of this relatively common disease. The spectrum of post-stroke movement disorders is broad and includes both hypo- and hyperkinetic syndromes. Post-stroke dyskinesias are involuntary hyperkinetic movements arising from cerebrovascular insults and often present with mixed phenotypes of hyperkinesia which can sometimes be difficult to classify. Nevertheless, identification of the most relevant motor phenotype, whenever possible, allows for a more specific phenomenological categorization of the dyskinesia and thus helps guide its treatment. Fortunately, post-stroke dyskinesias are usually self-limiting and resolve within 6 to 12 months of onset, but a short-term pharmacotherapy might sometimes be required for symptom control. Functional neurosurgical interventions targeting the motor thalamus or globus pallidus interna might be considered for patients with severe, disabling, and persistent dyskinesias (arbitrarily defined as duration longer than 12 months).
Keywords: vascular dyskinesia, stroke, movement disorders
Introduction
Strokes and movement disorders are quite common diseases and come to the attention of all physicians, but most often neurologists, emergency physicians, and primary care providers. Stroke-related (vascular) movement disorders, however, are relatively uncommon and represent a recognized complication of either ischemic or hemorrhagic strokes. Although the prevalence of post-stroke movement disorders is unclear because of the lack of prospective studies, data from retrospective studies have estimated the overall prevalence to range from 1% to 4% of all strokes with both sexes affected equally.1–3
The spectrum of post-stroke movement disorders is broad and includes both hypo- and hyperkinetic syndromes. The latter often present as variable combinations of hyperkinetic movements (dyskinesias) and can be broadly classified into three main phenotypes: choreiform dyskinesias (ballism, chorea, and athetosis), dystonia, and non-choreo-dystonic dyskinesias (eg, tremor, asterixis, and myoclonus) (Figure 1). The prevalence of vascular dyskinesias remains uncertain. In the Lausanne Stroke Registry from Switzerland, 29 (1%) of 2,500 patients admitted to the registry over 14 years developed hyperkinetic movement disorders with an estimated incidence of 0.08% per year. However, the large loss to follow-up has likely underestimated the prevalence, particularly for delayed movement disorders.1 In another stroke registry from Ecuador, 56 (3.7%) of 1,500 patients admitted to the registry over 9 years developed post-stroke movement disorders within the first year after stroke: 50 patients (3.3%) had hyperkinetic movements and 6 patients (0.4%) parkinsonism.2 Although data from these two studies suggest that vascular hyperkinetic movement disorders are likely to regress spontaneously, it remains unknown whether that decreases their prevalence over time as the incidence of delayed vascular dyskinesias is largely unidentified.
 | Figure 1 Major types of post-stroke movement disorders. Hyperkinetic disorders are the most frequent, at least in the first year following the stroke. Choreiform dyskinesias are the most common phenotype, followed by dystonia, but non-choreo-dystonic dyskinesias (eg, tremor and asterixis) are generally far less common. |
This review summarizes the current knowledge about the complex pathophysiology, overlapping phenomenology, and treatment options for common post-stroke hyperkinetic movement disorders. We searched for related published literature via PubMed through June 2016 by using the search terms “stroke”, “vascular”, “dyskinesia”, “movement disorders”, “basal ganglia circuit”, “cerebellar circuit”, and “motor control”.
Terminology
Stroke is broadly defined as an episode of neurological dysfunction caused either by a focal infarction of the central nervous system (CNS) or by a nontraumatic intracerebral or subarachnoid hemorrhage.4 Although acute focal neurological deficits (eg, hemiparesis) are the clinical hallmark of stroke presentation, nonfocal deficits (eg, impaired consciousness) or positive neurological phenomena (eg, hemidyskinesia) can occasionally be the initial manifestation of stroke resulting in diagnostic uncertainty and delayed treatment.4–6
Movement disorders are a group of basal ganglia and/or cerebellar circuit disorders characterized by impaired ability to control or coordinate movements. They are usually classified first based on an accurate description of the abnormal movement (phenomenology), and then subdivided according to the underlying cause (etiology). The “phenomenological classification” divides movement disorders into the following types: 1) Hypokinetic (parkinsonian) disorders, dominated by poverty (hypokinesia) or slowness (bradykinesia) of movement, 2) Hyperkinetic (dyskinetic) disorders, characterized by excessive, abnormal involuntary movements, and 3) Other movement disorder syndromes, cannot easily be grouped under the previous two categories, such as ataxia and akathisia. Furthermore, hyperkinesias (dyskinesias) are classified as “focal” if only one body region is involved, “segmental” if ≥2 adjacent body regions are affected, “multifocal” if ≥2 noncontiguous body regions are affected, “hemibody” if the ipsilateral arm and leg are involved, and “generalized” if the trunk and ≥2 other body regions are involved. The “etiological classification” of movement disorders, on the other hand, subdivides them into the following types: 1) Primary (genetic or idiopathic) disorders, without an identifiable secondary cause; 2) Secondary (symptomatic) disorders, due to a known acquired etiology such as vascular, toxic, or metabolic abnormalities; and 3) Psychogenic (functional) disorders, commonly due to conversion, somatic symptom, or factitious disorders.7
Although “dyskinesia” is a broad term that can indicate any hyperkinesia, it is often employed in clinical practice to indicate mixed, unusual, or complex hyperkinetic movements, especially when these movements are challenging to describe; nevertheless, a more specific phenomenological categorization should be used whenever possible. For example, variable mixtures of ballism, chorea, and athetosis are often seen simultaneously in the same patient and are commonly referred to as “choreiform dyskinesias”. Not infrequently, dystonia coexists with choreiform movements and the term “choreo-dystonic dyskinesia” is often used in this clinical setting. Interestingly, the choreiform dyskinesias are considered variants within the same phenotypic spectrum, with ballism and athetosis representing its fastest and slowest motor phenotypes, respectively (Table 1).7–11 Finally, the term “dyskinesia” is also used more specifically to indicate certain movement disorders such as levodopa-induced dyskinesia (due to chronic exposure to levodopa in Parkinson’s disease patients) and tardive dyskinesia (due to prolonged exposure to dopamine-blocking agents).12,13
 | Table 1 Major phenomenological types (phenotypes) of hyperkinetic movement disorders |
Pathophysiology
Motor control is a complex process that is governed by a sophisticated motor circuitry involving both pyramidal (cortical) and extrapyramidal (basal ganglionic and cerebellar) circuits. Motor commands are generated in the motor cortex, but basal ganglia and cerebellum closely refine these signals by acting as feedback loops to allow for smooth, accurate, coordinated movements. While the cortico-basal loop (basal ganglia circuitry) provides a tonic inhibitory output to the thalamus and thus the motor cortex, the output of the cortico-cerebellar loop (cerebellar circuitry) is tonically excitatory. Glutamate is the major excitatory neurotransmitter and gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in this motor network (Figure 2).14–18 Despite the significant overlap in their functions, the basal ganglia are particularly important for gross motor skills, whereas the cerebellum is crucial for fine or complex motor skills (eg, speech and writing) in which accurate performance is necessary.14–18
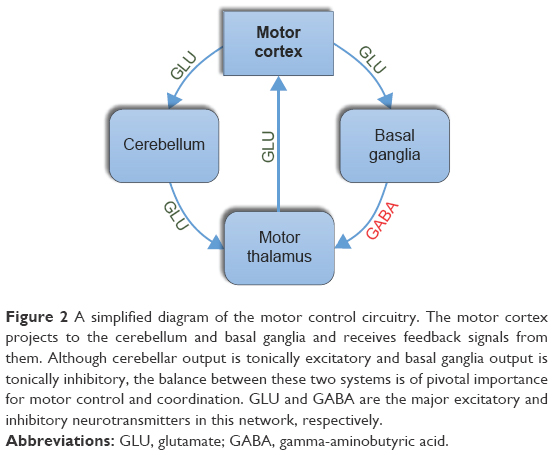 | Figure 2 A simplified diagram of the motor control circuitry. The motor cortex projects to the cerebellum and basal ganglia and receives feedback signals from them. Although cerebellar output is tonically excitatory and basal ganglia output is tonically inhibitory, the balance between these two systems is of pivotal importance for motor control and coordination. GLU and GABA are the major excitatory and inhibitory neurotransmitters in this network, respectively. |
The basal ganglia are a large collection of subcortical nuclei, which play a central role in modulating motor cortical activity through the selection and execution of appropriate motor programs.16 Almost all inputs to the basal ganglia circuitry arrive via the striatum (caudate and putamen). The motor signals are then processed through the pallidum (globus pallidus interna “GPi” and externa “GPe”), motor thalamus (ventral anterior “VA” and ventral lateral “VL” nuclei), and then back to the motor cortex. The normal function of this motor loop is influenced by two additional nuclei: substantia nigra and subthalamic nucleus, which regulate the activity of the striatum and pallidum, respectively.16–20 While glutamate and GABA are the major excitatory and inhibitory neurotransmitters, respectively, dopamine and acetylcholine act as neuromodulators in this circuit that regulate striatal activity. Dopamine is the transmitter of the nigrostriatal pathway, whereas acetylcholine is a major transmitter of striatal interneurons (Figure 3).21,22
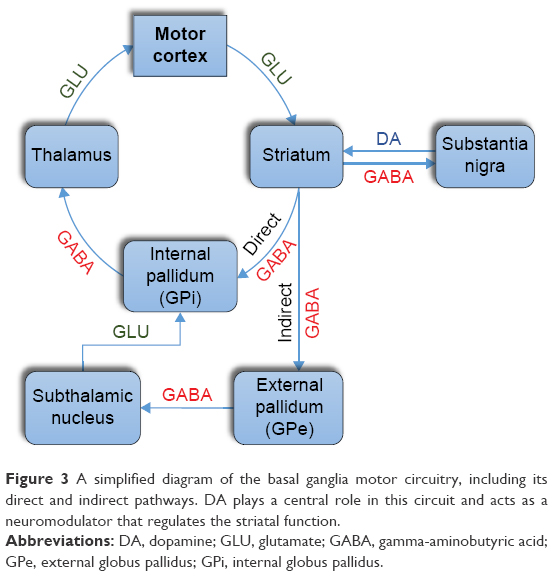 | Figure 3 A simplified diagram of the basal ganglia motor circuitry, including its direct and indirect pathways. DA plays a central role in this circuit and acts as a neuromodulator that regulates the striatal function. |
The striatum is the largest component of the basal ganglia motor circuit, and the vast majority of its neurons are GABAergic medium-sized spiny neurons whose axons comprise the striatal output pathways. These neurons are divided into two subsets of approximately equal numbers and provide two projection systems that have opposite effects on movement, the direct and indirect pathways. The direct pathway (GO pathway) originates from dopamine D1 receptor-expressing neurons and projects directly to the GPi. It is an excitatory pathway because its net effect is to disinhibit the thalamus and thus the motor cortex. The indirect pathway (NO-GO pathway) originates from D2 receptor-expressing neurons and projects to the GPe and subthalamic nucleus before terminating in the GPi. It is an inhibitory pathway because its net effect is to inhibit the thalamus and thus the motor cortex. Dopamine has differential effects on these two pathways: it activates D1 receptors and facilitates the direct pathway, but it inhibits D2 receptors and suppresses the indirect pathway. Therefore, the net effect of dopamine is to facilitate voluntary movements by promoting transient interruptions of the tonic inhibitory output of the basal ganglia via the simultaneous activation and suppression of the direct and indirect pathways, respectively.16–20
The cerebellum consists of a cortex, white matter, and deep cerebellar nuclei. It acts as a sensorimotor information processor by receiving information from all parts of the nervous system and comparing the motor commands of the cortex with the proprioceptive information coming from joints and muscles. This enables the cerebellum to detect errors in muscle contractions during active movements and thus to contribute to motor accuracy and coordination.15 Two cerebellar feedback loops are critical for this task, the dentato-rubro-olivary loop and the cortico-cerebellar loop. The dentato-rubro-olivary circuit (Guillain-Mollaret triangle) connects the dentate nucleus in the cerebellum with the contralateral red nucleus and inferior olivary nucleus in the brainstem via the superior cerebellar peduncle, the central tegmental tract, and the inferior cerebellar peduncle, respectively.23 This subcortical circuit is itself part of the larger cerebellar motor network, the cortico-cerebellar circuitry, in which the cerebral and cerebellar cortices are connected together indirectly. The motor cortex projects to the cerebellar cortex (via pontine nuclei) which, in turn, projects primarily to the VL nuclei of thalamus and then back to the cerebral cortex (Figure 4).14,15,23 As the inferior olivary nucleus receives collateral inputs from all afferent pathways projecting to the cerebellar cortex via mossy fibers, it compares intended with executed movements and conveys error signals to the cerebellar cortex via climbing fibers.14,15,24
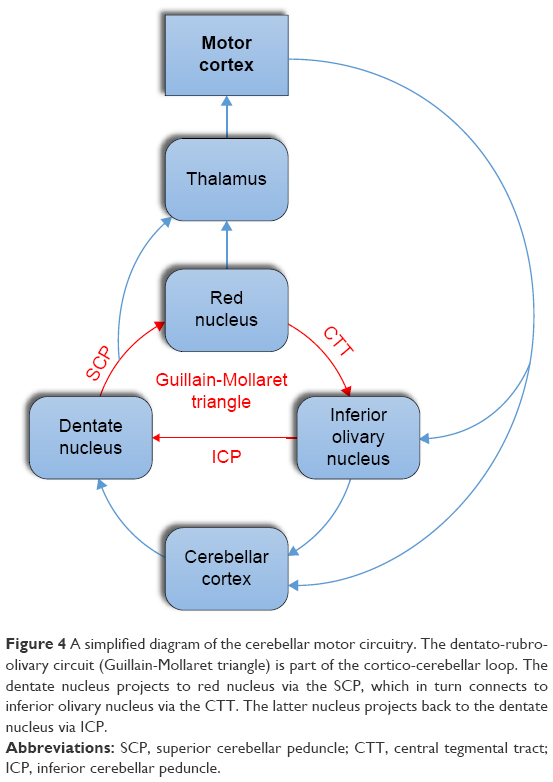 | Figure 4 A simplified diagram of the cerebellar motor circuitry. The dentato-rubro-olivary circuit (Guillain-Mollaret triangle) is part of the cortico-cerebellar loop. The dentate nucleus projects to red nucleus via the SCP, which in turn connects to inferior olivary nucleus via the CTT. The latter nucleus projects back to the dentate nucleus via ICP. |
The pathogenesis of stroke-related dyskinesias is still incompletely understood but suggested mechanisms include post-synaptic denervation hypersensitivity, trans-synaptic neuronal degeneration, as well as aberrant axonal and dendritic plasticity (remodeling) after the cerebrovascular injury.3 Post-stroke dyskinesias are relatively rare even with marked lesions, but they can arise after any stroke subtype at any level within the motor circuitry and after any interval period. Furthermore, no specific anatomical locations in the motor circuitry are reliably predictive of a particular dyskinesia, whereas the same dyskinesia can be caused by lesions in different locations in this circuitry. Due to this overlap, stroke-related movement disorders cannot be predicted from the location, size, or number of vascular insults.1,2,19 Interestingly, though the pathogenesis of vascular and primary movement disorders is different, they share similar underlying pathophysiology.
If symptomatic, vascular lesions involving the basal ganglia circuitry usually present with contralateral abnormal involuntary movements, but there have been rare case reports of ipsilateral dyskinesias.25 The most commonly involved areas in this regard are the striatum followed by the thalamus, but other reported locations include the fronto-parietal cortex, caudate, subthalamic nucleus, corona radiata, internal capsule, and pons.1–3,19 These vascular dyskinesias are believed to arise from underactivity of the indirect pathway (and/or overactivity of the direct pathway) leading to a decreased pallidal inhibitory output to the thalamus. The resultant thalamic disinhibition releases the motor cortex and allows movements that are normally suppressed.2,19,26,27 Vascular parkinsonism, on the other hand, is usually caused by diffuse or multiple insults in the basal ganglia loops and presents with non-tremulous, lower-body parkinsonism (ie, predominantly affecting the legs).2,19,28 It is believed to arise from overactivity of the indirect pathway (and/or underactivity of the direct pathway) leading to an increased pallidal inhibitory output to the thalamus. The resultant thalamic inhibition suppresses the motor cortex and leads to hypokinetic movements.3,23,29
The cortico-cerebellar and dentato-rubro-olivary circuits are critically involved in tremor genesis.30 Insults to the various structures of this network can give rise to cerebellar tremor, palatal tremor (previously known as palatal myoclonus), or Holmes tremor (also known as thalamic, rubral, midbrain, or cerebral outflow tremor). The latter occurs when the lesion involves both the nigrostriatal and cerebellar pathways resulting in resting and action tremor, respectively.31–33 Dystonic and parkinsonian tremors are special forms of tremor that occur in association with vascular dystonia and parkinsonism, respectively.
Differential Diagnosis
Acute focal, segmental, or hemibody dyskinesia should always raise a suspicion of structural lesions involving the motor circuitry.11,33 Although stroke is the most common cause after the age of 50 years, other structural pathologies have also been reported, including tumors, arteriovenous malformation, cerebral abscess, encephalitis, and multiple sclerosis.11,33–36 Non-structural causes, especially hypoglycemia and nonketotic hyperglycemia, can occasionally present with dyskinesia.37–41 Interestingly, hemichorea-hemiballism is the most commonly reported dyskinesia arising secondary to either structural (eg, vascular) or non-structural (eg, dysglycemic) etiologies.34,36,38,41
Diagnosis
A careful history (including medical, drug, and family history) and identifying the dominant phenomenology remain the first step in the clinical assessment, as that steers the differential diagnosis and the subsequent diagnostic trajectory. Neuroimaging studies, such as computed tomography (CT) or magnetic resonance imaging (MRI), are important to confirm the diagnosis of stroke or to evaluate for structural stroke mimics.
Post-stroke dyskinesias often present as variable combinations of hyperkinetic movements and can sometimes be challenging to classify.1,19 Nevertheless, identification of the dominant abnormal movement, whenever possible, allows for a more specific phenomenological categorization. Furthermore, the relative predominance of one type of phenomenology might be state dependent (eg, a resting athetosis might evolve into ballism with voluntary movements or when the patient is stimulated or excited). Hemichorea-hemiballism is the most common vascular dyskinesia, but other reported mixed movements include chorea-ballism-dystonia, chorea-athetosis-dystonia, and dystonia-athetosis with or without action tremor or myoclonic jerks.1,11,33,42–44 Tremor, asterixis, and myoclonus are generally far less common than choreo-dystonic dyskinesias.1
Post-stroke chorea usually presents as a hemichoreiform dyskinesia (hemichorea, hemiballism, and/or hemiathetosis) on the contralateral side of the vascular insult. Focal or segmental presentations are uncommon, and generalized forms are rare. The concomitant hemiparesis might initially prevent the expression of hemichorea, but improvement of the weakness unmasks this dyskinesia.1,2 Hemichorea can occasionally be the presenting sign of acute stroke, though careful examination often reveals an underlying mild hemiparesis with or without other subtle deficits.45,46
Post-stroke dystonia usually affects the contralateral limb, and it is often associated with hypertonia due to underlying spasticity (spastic dystonia).2,3,33 Interestingly, dystonia in combination with spasticity is usually intensified by muscle activation in the affected limb or elsewhere (overflow phenomenon). Cervical dystonia (eg, torticollis) and cranial dystonias (eg, blepherospasm or oromandibular dystonia) are rarely observed in association with cerebrovascular insults.47–49
Post-stroke tremor is a heterogeneous group of movement disorders characterized by various tremor phenomenologies, but isolated tremor unaccompanied by other abnormal movements is rare. Action tremor is by far the most common form, but rest tremor can occasionally be seen (eg, in vascular parkinsonism). Cerebellar tremor, the prototype vascular tremor, is primarily a slow intention tremor (<5 Hz) with frequent postural component.50 Holmes tremor is usually a low-frequency tremor (<5 Hz) with rest, postural, and intention components in the affected upper extremity.32 Palatal tremor is a slow tremor of the soft palate (<5 Hz) in which patients may complain of a disturbing clicking sound generated by palatal muscle contractions causing opening and closing of the eustachian tube.31 Dystonic tremor is usually a focal postural and/or kinetic tremor that occurs in association with dystonia, which might be subtle or overshadowed by the tremor. Despite being arrhythmic with variable frequencies, dystonic oscillations are traditionally referred to as “tremor”.50
Post-stroke myoclonus and asterixis are usually focal or segmental with corresponding lesions reported in numerous, mostly contralateral, brain regions. Myoclonus is not an uncommon component of mixed vascular movement disorders, but isolated myoclonus is rare. Dystonic myoclonus (myoclonic dystonia) has been described in patients with vascular dystonia secondary to thalamic infarcts.2,3,19
Prognosis
The natural history of post-stroke dyskinesias is variable. Their onset can be either “early/acute”, occurring shortly after stroke, or “delayed/chronic”, emerging months to years later. On the other hand, their course can be transient, recurrent, persistent, or progressive.1,43,44 Although the latency interval seems to depend partially on the dyskinesia type, it still varies widely within each vascular hyperkinetic movement disorder.2,3 While dystonia is frequently delayed in onset, hemichorea-hemiballism usually occurs shortly after stroke and occasionally represents the initial manifestation of acute cerebral ischemia or intracerebral hemorrhage.2,33,45,51 Post-stroke dyskinesias are usually self-limited and resolve within 6 to 12 months of onset, but the overall long-term prognosis of the affected patients is similar to that of other stroke patients.1,2,46,52
Management
Although post-stroke dyskinesias tend to resolve spontaneously, a short-term treatment might sometimes be required for symptom control.1,2 As in all cases of secondary movement disorders, treatment of the underlying etiology is of paramount importance. Control of vascular risk factors is crucial in reducing the incidence of vascular dyskinesias. The discussion of stroke prevention and management, however, is beyond the scope of this article. Symptomatic pharmacotherapy might be necessary for severe dyskinesias, but periodic trials of therapeutic withdrawal (ie, for patients with controlled symptoms) are required due to the high likelihood of spontaneous regression. Medications should be started at low doses and gradually titrated up until an effective and tolerable dosage is reached. Although there are no established treatment guidelines, most of the treatment options are similar to those for primary movement disorders based on similar underlying pathophysiology.
The symptomatic pharmacotherapy for post-stroke choreiform dyskinesias consists mainly of anti-dopaminergic therapy with typical or atypical antipsychotics (neuroleptics). Blockade of striatal dopamine D2 receptors is believed to be responsible for their anti-dyskinetic activity, as D2 receptor antagonists disinhibit the indirect pathway and, therefore, suppress abnormal involuntary movements. Unfortunately, dopaminergic blockade carries the risk of acute dystonic reactions, tardive dyskinesia, and drug-induced parkinsonism. Atypical antipsychotics (eg, risperidone), however, are less likely to cause these side effects compared to typical antipsychotics (eg, haloperidol) and, therefore, are generally preferred for this use.53,54 Tetrabenazine, a presynaptic dopamine depletor with weak postsynaptic D2 receptor blocking activity, is a reasonable alternative for patients who are intolerant or unresponsive to dopamine receptor antagonists.55 Tetrabenazine acts primarily as a reversible inhibitor of the vesicular monoamine transporter 2 (VMAT2) in the presynaptic nerve terminals, thus exposing dopamine and other monoamines to monoamine oxidase and leading to their depletion.56 Unlike dopamine receptor blockers, tetrabenazine rarely causes tardive dyskinesia because of its dominant presynaptic anti-dopaminergic properties.3,57 Non-dopaminergic drugs have been tried in the management of vascular choreiform dyskinesias with varying success. Case reports and small case series have suggested a potential beneficial effect of antiepileptic drugs including levetiracetam, topiramate, gabapentin, clonazepam, and valproate.58–62
Chemodenervation with botulinum toxin (BTX) injections is the cornerstone of the symptomatic treatment of focal or segmental post-stroke dystonia.3 BTX is a neurotoxic protein with several serotypes that cleaves the synaptic proteins (SNARE) in the presynaptic nerve terminals, thereby blocking the release of acetylcholine at the neuromuscular junction. SNARE proteins are required for the fusion of presynaptic storage vesicles containing acetylcholine with the presynaptic membrane.63 BTX injections are given intramuscularly, often under electromyography (EMG) guidance, and need to be repeated every 3 to 6 months.64 Vascular dystonia usually has a poor response to oral pharmacotherapy, including dopamine blocking and depleting agents, anticholinergic drugs, baclofen, and benzodiazepines. Oral medications, however, are widely used in generalized dystonia, dystonia mixed with other movement disorders, or as an adjuvant therapy in focal or segmental dystonia when there is an unsatisfactory response to BTX.64–67
Post-stroke tremor is particularly refractory to pharmacotherapy. Trials of medications with GABA-agonistic activity (eg, clonazepam, valproate, topiramate, or primidone), alone or in combination, may be effective in individual cases. Because of the dopaminergic (nigrostriatal) system involvement in Holmes tremor, treatment with levodopa or dopamine agonists seems to be useful.31 Propranolol, one of the first-line treatments in essential tremor, is usually of limited benefit in vascular tremor. Dystonic and parkinsonian tremors are treated as vascular dystonia and parkinsonism, respectively.3,19
Post-stroke myoclonus and asterixis usually improve spontaneously and do not require pharmacotherapy. When interfering with the patient’s functional abilities, like eating or writing, myoclonus is most frequently treated with GABAergic medications (eg, clonazepam and valproate), but levetiracetam or piracetam can be very useful. Monotherapy should be attempted first, although eventually several drug combinations might be required.3,19,53 Dystonic myoclonus is treated as vascular dystonia.
Finally, stereotactic functional neurosurgery, whether ablative or deep brain stimulation (DBS), should be considered for patients with severe and persistent dyskinesias (arbitrarily defined as duration longer than 1 year).3,19,53 While lesioning procedures (eg, pallidotomy or thalamotomy) are carried out using radiofrequency ablation or gamma knife radiosurgery, DBS (eg, pallidal or thalamic DBS) uses high-frequency electrical stimulation of the targeted nuclei. Although the therapeutic mechanisms of action are not completely understood, ablative surgeries are believed to destroy abnormally hyperactive circuits in deep brain nuclei, while DBS suppresses abnormally excessive activity in the motor circuitry without significant destruction of the brain tissue.68,69 Neurostimulation, therefore, has largely replaced neuroablation to treat refractory dyskinesias because it is less invasive, adjustable for maximal symptomatic benefit, and reversible in case of adverse effects. Stereotactic functional neurosurgery depends on the accurate identification of anatomically and functionally distinct deep brain structures to maximize therapeutic benefits and to minimize adverse neurological events of any surgical intervention. The contralateral motor thalamus or internal pallidum are the usual neurosurgical targets, but it is yet to be determined which target is more effective for specific dyskinesias.53,70 The ventrolateral “VL” nuclei, including ventralis intermedius and ventralis oralis posterior, are the most common nuclei targeted within the motor thalamus.71 Potential candidates with vascular dystonia should undergo surgery prior to the development of contractures and fixed deformities that may limit functional improvement as the dystonia improves.66
Conclusion
Stroke is the leading cause of focal or segmental limb dyskinesia as well as hemidyskinesia in elderly patients, but other structural and non-structural brain abnormalities should also be considered in the differential diagnosis. Post-stroke dyskinesias, which can arise from either ischemic or hemorrhagic cerebrovascular insults, are often mixed and variable with several components of hyperkinetic movements. A comprehensive clinical and neuroimaging assessment are essential to establish the correct diagnosis. Fortunately, post-stroke dyskinesias tend to resolve spontaneously within 6 to 12 months. A short-term symptomatic pharmacotherapy may be required in some patients, whereas surgical treatment is reserved for persistent, disabling, and medically intractable cases.
Acknowledgment
The authors would like to thank Yassine Rechoum, PhD, for his valuable comments and suggestions to improve the quality of this paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Ghika-Schmid F, Ghika J, Regli F, Bogousslavsky J. Hyperkinetic movement disorders during and after acute stroke: the Lausanne stroke registry. J Neurol Sci. 1997;146(2):109–116. | ||
Alarcón F, Zijlmans JC, Dueñas G, Cevallos N. Post-stroke movement disorders: report of 56 patients. J Neurol Neurosurg Psychiatry. 2004;75(11):1568–1574. | ||
Mehanna R, Jankovic J. Movement disorders in cerebrovascular disease. Lancet Neurol. 2013;12(6):597–608. | ||
Sacco RL, Kasner SE, Broderick JP, et al; American Heart Association Stroke Council, Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular and Stroke Nursing; Council on Epidemiology and Prevention; Council on Peripheral. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the AHA/ASA. Stroke. 2013;44(7):2064–2089. | ||
Schmahmann JD. Vascular syndromes of the thalamus. Stroke. 2003;34(9):2264–2278. | ||
Ghika J, Bogousslavsky J. Abnormal movements. In: Bogousslavsky J, Caplan L, editors. Stroke Syndromes. 2nd ed. Cambridge: Cambridge University Press; 2001:162–181. | ||
Fahn S, Jankovic J, Hallett M. Clinical overview and phenomenology of movement disorders. In: Fahn S, Jankovic J, Hallett M, editors. Principles and Practice of Movement Disorders. 2nd ed. Edinburgh, United Kingdom: Elsevier Saunders; 2011:1–35. | ||
Differential diagnosis of tardive dyskinesia. In: Tardive Dyskinesia: A Task Force Report of the American Psychiatric Association. Washington, DC: American Psychiatric Association Press; 1992. | ||
Albanese A, Jankovic J. Distinguishing clinical features of hyperkinetic disorders. In: Albanese A, Jankovic J, editors. Hyperkinetic Movement Disorders: Differential Diagnosis and Treatment. Oxford, United Kingdom: Wiley-Blackwell; 2012:3–14. | ||
Dewey RB Jr, Jankovic J. Hemiballism-hemichorea. Clinical and pharmacologic findings in 21 patients. Arch Neurol. 1989;46(8):862–867. | ||
Vidakovic A, Dragasevic N, Kostic VS. Hemiballism: report of 25 cases. J Neurol Neurosurg Psychiatry. 1994;57(8):945–949. | ||
Walker RH. Thoughts on selected movement disorder terminology and a plea for clarity. Tremor Other Hyperkinet Mov (N Y). 2013;3:tre-03-203-4656-2. | ||
Mehta SH, Morgan JC, Sethi KD. Drug-induced movement disorders. Neurol Clin. 2015;33(1):153–174. | ||
Manto M, Bower JM, Conforto AB, et al. Consensus paper: roles of the cerebellum in motor control – the diversity of ideas on cerebellar involvement in movement. Cerebellum. 2012;11(2):457–487. | ||
Apps R, Garwicz M. Anatomical and physiological foundations of cerebellar information processing. Nat Rev Neurosci. 2005;6(4):297–311. | ||
Smith Y, Bevan MD, Shink E, Bolam JP. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience. 1998;86(2):353–387. | ||
DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64(1):20–24. | ||
DeLong M, Wichmann T. Changing views of basal ganglia circuits and circuit disorders. Clin EEG Neurosci. 2010;41(2):61–67. | ||
Handley A, Medcalf P, Hellier K, Dutta D. Movement disorders after stroke. Age Ageing. 2009;38(3):260–266. | ||
Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50(4):381–425. | ||
Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18(12):527–535. | ||
Lim SA, Kang UJ, McGehee DS. Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci. 2014;6:22. | ||
Murdoch S, Shah P, Jampana R. The Guillain-Mollaret triangle in action. Pract Neurol. 2016;16(3):243–246. | ||
De Zeeuw CI, Simpson JI, Hoogenraad CC, Galjart N, Koekkoek SK, Ruigrok TJ. Microcircuitry and function of the inferior olive. Trends Neurosci. 1998;21(9):391–400. | ||
Kannepalli NRVL, Yadav R, Vazhayil V, Somanna S, Pal PK. Ipsilateral Hemichorea-hemiballism in a Case of Postoperative Stroke. Louis ED, ed. Tremor Other Hyperkinet Mov (N Y). 2016;6:359. | ||
Crossman AR. Neural mechanisms in disorders of movement. Comp Biochem Physiol A Comp Physiol. 1989;93(1):141–149. | ||
Hashimoto T, Yanagisawa N. Pathophysiology of involuntary movements in adults. No To Hattatsu. 1997;29(3):193–198. | ||
Fitzgerald PM, Jankovic J. Lower body parkinsonism: evidence for a vascular etiology. Mov Disord. 1989;4(3):249–260. | ||
Thiele SL, Chen B, Lo C, et al. Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l-DOPA induced dyskinesia in mouse models. Neurobiol Dis. 2014;71:334–344. | ||
Lefranc M, Manto M, Merle P, et al. Targeting the red nucleus for cerebellar tremor. Cerebellum. 2014;13(3):372–377. | ||
Deuschl G, Toro C, Valls-Solé J, Zeffiro T, Zee DS, Hallett M. Symptomatic and essential palatal tremor. 1. Clinical, physiological and MRI analysis. Brain. 1994;117(Pt 4):775–788. | ||
Raina GB, Cersosimo MG, Folgar SS, et al. Holmes tremor: clinical description, lesion localization, and treatment in a series of 29 cases. Neurology. 2016;86(10):931–938. | ||
Pettigrew LC, Jankovic J. Hemidystonia: a report of 22 patients and a review of the literature. J Neurol Neurosurg Psychiatry. 1985;48(7):650–657. | ||
Bhatoe HS. Movement disorders caused by brain tumours. Neurol India. 1999;47(1):40–42. | ||
Rana AQ, Yousuf MS, Hashmi MZ, Kachhvi ZM. Hemichorea and dystonia due to frontal lobe meningioma. J Neurosci Rural Pract. 2014;5(3):290–292. | ||
Rabhi S, Amrani K, Maaroufi M, et al. Hemichorea-hemiballismus as an initial manifestation in a Moroccan patient with acquired immunodeficiency syndrome and toxoplasma infection: a case report and review of the literature. Pan Afr Med J. 2011;10:9. | ||
Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a meta-analysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1–2):57–62. | ||
Felicio AC, Chang CV, Godeiro-Junior C, Okoshi MP, Ferraz HB. Hemichorea-hemiballism as the first presentation of type 2 diabetes mellitus. Arq Neuropsiquiatr. 2008;66(2A):249–250. | ||
Lai SL, Tseng YL, Hsu MC, Chen SS. Magnetic resonance imaging and single-photon emission computed tomography changes in hypoglycemia-induced chorea. Mov Disord. 2004;19(4):475–478. | ||
Shaw C, Haas L, Miller D, Delahunt J. A case report of paroxysmal dystonic choreoathetosis due to hypoglycaemia induced by an insulinoma. J Neurol Neurosurg Psychiatry. 1996;61(2):194–195. | ||
Sweeney BJ, Edgecombe J, Churchill DR, Miller RF, Harrison MJ. Choreoathetosis/ballismus associated with pentamidine-induced hypoglycemia in a patient with the acquired immunodeficiency syndrome. Arch Neurol. 1994;51(7):723–725. | ||
Chung SJ, Im JH, Lee MC, Kim JS. Hemichorea after stroke: clinical-radiological correlation. J Neurol. 2004;251(6):725–729. | ||
Kim JS. Delayed onset mixed involuntary movements after thalamic stroke: clinical, radiological and pathophysiological findings. Brain. 2001;124(Pt 2):299–309. | ||
Scott BL, Jankovic J. Delayed-onset progressive movement disorders after static brain lesions. Neurology. 1996;46(1):68–74. | ||
McCollum D, Silvers S, Dawson SB, Barrett KM. Resolution of acute onset hemichorea–hemiballismus after treatment with intravenous tissue plasminogen activator. Neurohospitalist. 2013;3(3):131–134. | ||
Zijlmans JC. Vascular chorea in adults and children. Handb Clin Neurol. 2011;100:261–270. | ||
Dressler D. Nonprimary dystonias. Handb Clin Neurol. 2011;100:513–538. | ||
Lo SE, Rosengart AJ, Novakovic RL, et al. Identification and treatment of cervical and oromandibular dystonia in acutely brain-injured patients. Neurocrit Care. 2005;3(2):139–145. | ||
Khooshnoodi MA, Factor SA, Jinnah HA. Secondary blepharospasm associated with structural lesions of the brain. J Neurol Sci. 2013;331(1–2):98–101. | ||
Puschmann A, Wszolek ZK. Diagnosis and treatment of common forms of tremor. Semin Neurol. 2011;31(1):65–77. | ||
Das A, Baheti NN. Limb-shaking transient ischemic attack. J Neurosci Rural Pract. 2013;4(1):55–56. | ||
Ristic A, Marinkovic J, Dragasevic N, Stanisavljevic D, Kostic V. Long-term prognosis of vascular hemiballismus. Stroke. 2002;33(8):2109–2111. | ||
Bansil S, Prakash N, Kaye J, et al. Movement disorders after stroke in adults: a review. Tremor Other Hyperkinet Mov (N Y). 2012;2:tre-02-42-195-1. | ||
Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27–38. | ||
Calabrò RS, Polimeni G, Gervasi G, Bramanti P. Postthalamic stroke dystonic choreoathetosis responsive to tetrabenazine. Ann Pharmacother. 2011;45(12):e65. | ||
Kenney C, Jankovic J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev Neurother. 2006;6(1):7–17. | ||
LeWitt PA. Tardive dyskinesia caused by tetrabenazine. Clin Neuropharmacol. 2013;36(3):92–93. | ||
D’Amelio M, Callari G, Gammino M, et al. Levetiracetam in the treatment of vascular chorea, a case report. Eur J Clin Pharmacol. 2005;60(11):835–836. | ||
Gatto EM, Uribe Roca C, Raina G, Gorja M, Folgar S, Micheli FE. Vascular hemichorea/hemiballism and topiramate. Mov Disord. 2004;19(7):836–838. | ||
Driver-Dunckley E, Evidente VG. Hemichorea-hemiballismus may respond to topiramate. Clin Neuropharmacol. 2005;28(3):142–144. | ||
Kothare SV, Pollack P, Kulberg AG, Ravin PD. Gabapentin treatment in a child with delayed-onset hemichorea/hemiballismus. Pediatr Neurol. 2000;22(1):68–71. | ||
Shannon KM. Hemiballismus. Curr Treat Options Neurol. 2005;7(3):203–210. | ||
Brin MF. Botulinum toxin: chemistry, pharmacology, toxicity, and immunology. Muscle Nerve Suppl. 1997;6:S146–S168. | ||
Delnooz CC, van de Warrenburg BP. Current and future medical treatment in primary dystonia. Ther Adv Neurol Disord. 2012;5(4):221–240. | ||
Chuang C, Fahn S, Frucht SJ. The natural history and treatment of acquired hemidystonia: report of 33 cases and review of the literature. J Neurol Neurosurg Psychiatry. 2002;72(1):59–67. | ||
Cloud LJ, Jinnah HA. Treatment strategies for dystonia. Expert Opin Pharmacother. 2010;11(1):5–15. | ||
Umeh CC, Nichols P, Rosenthal LS, Mari Z. Dual treatment of hemichorea-hemiballismus syndrome with tetrabenazine and chemodenervation. Tremor Other Hyperkinet Mov (N Y). 2012;2. pii: tre-02-113-791-3. | ||
Montgomery EB Jr, Gale JT. Mechanisms of action of deep brain stimulation (DBS). Neurosci Biobehav Rev. 2008;32(3):388–407. | ||
Chiken S, Nambu A. Mechanism of deep brain stimulation: inhibition, excitation, or disruption? Neuroscientist. 2016;22(3):313–322. | ||
Katayama Y, Yamamoto T, Kobayashi K, Oshima H, Fukaya C. Deep brain and motor cortex stimulation for post-stroke movement disorders and post-stroke pain. Acta Neurochir Suppl. 2003;87:121–123. | ||
Hyam JA, Owen SL, Kringelbach ML, et al. Contrasting connectivity of the ventralis intermedius and ventralis oralis posterior nuclei of the motor thalamus demonstrated by probabilistic tractography. Neurosurgery. 2012;70(1):162–169. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.