Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 12
Ponesimod in the Treatment of Relapsing Forms of Multiple Sclerosis: An Update on the Emerging Clinical Data
Authors Ruggieri S ![]() , Quartuccio ME, Prosperini L
, Quartuccio ME, Prosperini L ![]()
Received 22 January 2022
Accepted for publication 10 March 2022
Published 22 March 2022 Volume 2022:12 Pages 61—73
DOI https://doi.org/10.2147/DNND.S313825
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Müller
Serena Ruggieri,1,2 Maria Esmeralda Quartuccio,3 Luca Prosperini3
1Department of Human Neurosciences, Sapienza University, Rome, 00185, Italy; 2Neuroimmunology Unit, Santa Lucia Foundation, Rome, 00143, Italy; 3Department of Neurosciences, S. Camillo-Forlanini Hospital, Rome, 00152, Italy
Correspondence: Luca Prosperini, Department of Neurosciences, S. Camillo-Forlanini Hospital, C.ne Gianicolense 87, Rome, 00152, Italy, Tel +39-6-58704349, Fax +39-6-58704206, Email [email protected]
Abstract: Sphingosine 1-phosphate (S1P) receptors are bioactive lipid metabolites that bind five different types of receptors expressed ubiquitously in human body and mediate a broad range of biological functions. Targeting S1P receptors is nowadays a well-established pharmacological strategy to treat multiple sclerosis (MS). However, the adverse events associated with the ancestor (fingolimod), especially in terms of heart conduction and slow reversibility of its pharmacodynamics effect on lymphocytes, have stimulated a search for a S1P modulator with greater selectivity for S1P1 (the most important immune mechanism to prevent MS-related neuroinflammation). Ponesimod is a second-generation, orally active, directly bioavailable, highly selective, and rapidly reversible modulator of the S1P1 receptor. Gradual 14-day up-titration of ponesimod mitigates its first-dose effects on heart rate and facilitates its use over fingolimod, as it does not require first-dose cardiac monitoring. Ponesimod is rapidly eliminated within 1 week of discontinuation, thereby representing a more manageable approach in case of vaccination, pregnancy, or adverse events. However, the fast reversibility of ponesimod may also raise concerns about the possibility of a rapid reactivation of disease activity following its discontinuation. Ponesimod was recently approved for the treatment of relapsing MS forms on the basis of a Phase III, double-blind, double-dummy, randomized clinical trial (OPTIMUM) that demonstrated the superiority of ponesimod over teriflunomide on disease activity markers, without unexpected safety concerns. This review summarizes the pharmacodynamic and pharmacokinetic characteristics of ponesimod, and the main Phase II and III studies that led to its approval. Comparisons of ponesimod with other S1P receptor modulators currently available for MS (fingolimod, ozanimod, siponimod) are also provided.
Keywords: multiple sclerosis, disease-modifying treatments, ponesimod, sphingosine-1-phosphate, sphingosine-1-phosphate receptor modulators
Introduction
Multiple sclerosis (MS) is a chronic inflammatory and neurodegenerative immune-mediated disease of the central nervous system (CNS) that represents the lead cause of irreversible disability among young adults, thus placing a high burden both on individual patients and on society.1 MS affects about 2.8 million people worldwide, and its incidence and prevalence are extremely variable depending on genetics and several environmental factors.2 In spite of such a wide incidence variability, MS is still the commonest non-traumatic cause of neurological disability in young adults in Western Europe and North America and the global distribution of MS generally growths with increasing distance from the equator.3
The onset of MS usually arises in young subjects, with a female-to-male ratio of approximately 2–3:1.4 In most patients (70–80%), MS begins with a relapsing-remitting course (RR), which is characterized by recurring attacks of acute focal neurological deficits, or exacerbations of existing deficits (relapses) followed gradually by partial or full recovery (remission).5 According to its natural history, after 20 years approximately 80% of patients convert to the secondary progressive (SP) course, which is described by an accumulation of irreversible neurologic deficits regardless of clinical relapses.5 The remaining 10–20% of patients have primary progressive (PP) disease course, characterized by progressive clinical deterioration since the disease onset.5 In contrast with RRMS, people with PPMS are older and a higher proportion of them are men.6
The diagnosis of MS requires objective evidence of an inflammatory event occurring into the CNS, and additional demonstration of dissemination of the disease process “in space and in time”, ie involving more than one CNS location with evolution over time.7 The main paraclinical test to corroborate diagnoses are magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analysis.7
From an etiological viewpoint, MS is considered to be an autoimmune, inflammatory disease of the CNS caused by the interaction of environmental, genetic, and epigenetic factors.1 However, there is mounting evidence that MS may be better represented by an “immunological convolution” between an underlying primary degenerative disorder and the host’s aberrant immune response.8 The wide between-individual heterogeneity of the disease in its phenotypical and clinical manifestations implies indeed a different extent of the individual immune response and neurodegenerative process.8
Although the exact cause of MS is not well known and the disease results from an interplay of several individual and external factors, two main assumptions named as “inside-out” model and “outside-in” model try to capture the immunological cascade possibly triggering the disorder.8 The inside-out theory postulates that events occurring into the CNS may activate disease accrual, with the infiltration of autoreactive lymphocytes happening as a following phenomenon.9 Although not yet identified, the hypothesized triggering mechanisms include inflammatory responses to an unrevealed CNS viral infection or to processes leading to primary cytodegeneration, as observed in other primitive neurodegenerative diseases (eg: Alzheimer disease, Parkinson's disease, etc.).10 In the “outside-in” model, autoreactive T cells are initiated at peripheral sites, likely through molecular mimicry between myelin and viral antigens (the Epstein-Barr virus has been identified as the main putative causal agent11), thus determining an activation and/or the co-expression of T cell receptors (TCRs) with different specificities and transfer to the CNS along with activated B cells and monocytes.12 This latter assumption is coherent, and probably derived, with data from the experimental model known as experimental autoimmune encephalomyelitis (EAE), where combined CNS antigen is administered along with immune stimulants, resulting in the generation of pathogenic CD4+ T-helper 1 (TH1) cells and TH17 cells in the draining lymph nodes.13 These cells then enter the systemic circulation and eventually express their effector functions within the CNS, having crossed the Blood Brain Barrier (BBB) or the blood-CSF barrier at the choroid plexus.14
During MS, from a pathological perspective, active tissue injury is associated with inflammation, but the inflammatory response in the progressive phase occurs at least partly trapped by the BBB, which makes it more difficult to be targeted by treatments mainly affecting neuroinflammation.15 Other mechanisms that occur during the disease are thought to include oxidative stress resulting in mitochondrial injury that might participate in the induction of demyelination and neurodegeneration in both the RR and progressive phases of MS.16 Thus, even though no pathological or mechanistic features are unique hallmarks in either the RR or progressive stages of MS, differences might accrual in term of quantitative rather than qualitative between these stages leading to each predominant phenotype.15,16
Treatment of MS typically consists of direct symptom management, brief corticosteroid administration for acute exacerbations, and the regular use of disease-modifying treatments (DMTs). According to more recent studies, the natural history of the disease has significantly changed in patients treated with immunological agents, showing more favourable outcomes during the newest treatment era.17 Continued treatment with MS immunotherapies reduces disability accrual by 19–44% and the risk of requiring a walking aid by 67%, regardless of the type of DMT considered.18
The evolving landscape of effective therapies for RRMS, and partially successful treatment for PPMS and SPMS, represents an outstanding achievement that has radically changed the prognosis of people living with MS.19 However, the high level of efficacy of DMTs targeting neuroinflammation (clinical exacerbation and radiological activity) in RRMS has revealed a relapse-independent “silent” progression that was previously under covered by the natural course of the disease.20 This perception has also shifted the clinician approach to an increasing use of highly effective therapies in the early phase of MS to minimize the occurrence of both clinical attacks and progression.21 The counterpart of this successful attitude is the emerging of potential safety issues, often focused on long-term safeness of treatment that need to be taken chronically.22 Consequently, there is still room for the development of new treatments that can combine efficacy with long-term safety and meet patients’ satisfaction. In this review, we summarize recent advances in MS management and focus on the sphingosine 1-phosphate (S1P) modulator ponesimod, an upcoming new treatment for MS.
Currently Available Treatments for Multiple Sclerosis
During the last 30 years the treatment scenario of DMTs targeting the immunological cascade of MS has enriched dramatically thank to a better understanding of the pathophysiology of the disease.19 The main therapeutic goals of these treatments consist in relapse rate reduction, extension of time to next relapse, reduction of new lesions detectable by MRI, and decreasing the long-term accumulation of disability.23 This is achieved by acting against the inflammatory component of the disease, thus exerting also a partial and indirect effect on the underlying neurodegenerative process, measured in term of whole or regional brain volume loss.24 For these reasons, all but one of the currently available DMTs are approved to treat RRMS or active SPMS, which are supposed to be sustained by a more inflammatory component disease activity.23 Conversely, SPMS without evidence of disease activity and PPMS are mainly characterized by a greater amount of trapped inflammation and neurodegeneration, thus more difficult to halt; consequently, only specific treatments have been approved for these population, but with detailed disease characteristics.6
The Food and Drug Administration (FDA) approved the first DMT for RRMS in 1993, the subcutaneous interferon beta (IFNB)-1b (marketed as Betaseron® [Bayer HealthCare Pharmaceuticals, Montville, NJ, USA] in United States, and as Betaferon [Bayer Schering Pharma AG, Berlin-Wedding, Germany] in the rest of the world).25 Starting from this milestone start, a huge number of other drugs have reached the market upon the completion of randomized clinical trials (RCT). Several classes of DMTs, with varying mechanisms of action and routes of administration, are currently available for treating RRMS: self-administered subcutaneous or intramuscular IFNB −1a or −1b formulations; self-administered subcutaneous glatiramer acetate (GA); orally administered teriflunomide; orally administered S1P receptor modulators (fingolimod, ozanimod); orally administered fumarate derivatives (dimethyl fumarate and diroximel fumarate); orally administered cladribine; three types of intravenous monoclonal antibodies (alemtuzumab, natalizumab, ocrelizumab), and one, administered subcutaneously (ofatumumab). The S1P receptor modulator siponimod and the monoclonal antibody ocrelizumab have been approved to treat SPMS and PPMS, respectively.
According to their mechanisms of action to selectively suppress or modulate the immune system, especially lymphocytes, DMTs can be classified as immunomodulatory, sequestering, and depletive agents.26,27 Immunomodulatory agents include all IFNB formulations (−1a and −1b), pegylated IFNB −1a, GA, teriflunomide, dimethyl and diroximel fumatares. All these compounds have a pleiotropic effect on immune system without affecting immune surveillance, therefore being considered lower-efficacy but safer DMTs. Among the few safety concerns raised by immunomodulatory agents, there is the risk of persistent grade 2 or 3 lymphopenia (ie: <800 cells per µL) related with dimethyl fumarate that, in turn, has been associated with an increased risk of progressive multifocal leukoencephalopathy (PML) – an opportunistic infection of CNS caused by reactivation of the John Cunningham virus (JCV) – especially in older patients.28
Sequestering agents include S1P receptor modulators, such as fingolimod, ozanimod and siponimod, and natalizumab, a monoclonal antibody binding the very late antigen-4 (VLA-4), an integrin expressed on activated leukocytes that enables them to cross the BBB. Despite their different mechanisms of action, both S1P receptor modulators and natalizumab have an “anti-trafficking” mechanism that inhibits CNS entry of lymphocytes via compartmentalization in lymphatic tissue and peripheral blood, respectively. The efficacy of sequestering agents is higher as compared to immunomodulatory agents, but at the price of two main safety concerns. The first one is related to the reduced immune surveillance into CNS leading to an increased risk of PML (especially for natalizumab) and other opportunistic infections.29 The second one is somewhat consequence of the previous one: the discontinuation of sequestering agents, that is mainly driven by the need to prevent opportunistic infections in high-risk patients, may cause a quick disease reactivation due to the rapid re-entry of lymphocytes into the CNS.30,31
Depletive agents include the oral compound cladribine, and several monoclonal antibodies such as alemtuzumab, ocrelizumab, ofatumumab. Cladribine is a purine analogue drug that inhibited adenosine deaminase, an enzyme that is biologically active in B and T cells, where a preferential accumulation of cladribine phosphates occurs because of a high intracellular ratio of deoxycytidine kinase to 5’-nucleotidases. Therefore, cladribine induces a dose-dependent cytotoxic effects on B and T lymphocytes through impairment of DNA synthesis, resulting in depletion of lymphocytes by apoptosis or programmed cell death.
By targeting specific molecules expressed on lymphocyte surface, monoclonal antibodies induce long-lasting depletion of B and T cells (alemtuzumab binds CD52) or only B cells (ocrelizumab and ofatumumab bind CD20) via antibody-dependent cell-mediated and complement-dependent cytolysis.
The growing availability of treatments, along with revisions in the diagnostic criteria, have changed the characteristics of clinical trial MS population, preventing the implementation of placebo-controlled trials that are now considered unethical in RRMS.32 Furthermore, given the paucity of head-to-head RCTs, there is only little evidence on the relative efficacy and safety of these DMTs.33 Therefore, neurologists must rely on meta-analyses and post-marketing real world studies providing indirect and direct data, respectively, useful to suggest the best place-in-therapy for each DMT and to improve the decision-making process.34
Sphingosine 1-Phosphate Receptors
Sphingosines are structural components of the cell membrane from which many bioactive lipids originate, including S1P, the active terminal derivative of sphingosine metabolism generated by the action of sphingosine kinase.35,36 Structurally, S1P is a lysophospholipid with a polar head-group and lipophilic tail. Various hormones, immunoglobulins, trophic factors and cytokines can activate sphingosine kinase to produce S1P, a multifunctional bioactive lipid metabolite acting as soluble signalling molecule. S1P circulates in the blood and the lymphatic system, thus having autocrine and paracrine effects.37 S1P is involved in a wide range of physiological and pathophysiological events through its interaction with the S1P receptors that are high affinity G protein-coupled cell surface receptors expressed throughout the body.38
S1P has binding affinity for five different S1P receptor subtypes by which cellular signals are transduced. S1P1, S1P2 and S1P3 receptors are expressed ubiquitously, with each subtype exhibiting a different cell specificity, whereas the expression of S1P4 and S1P5 receptors is less widespread, being S1P4 receptors confined to lymphoid and hematopoietic tissues and S1P5 receptors located in the spleen and in white matter of the CNS, mainly in oligodendrocytes39 (Table 1).
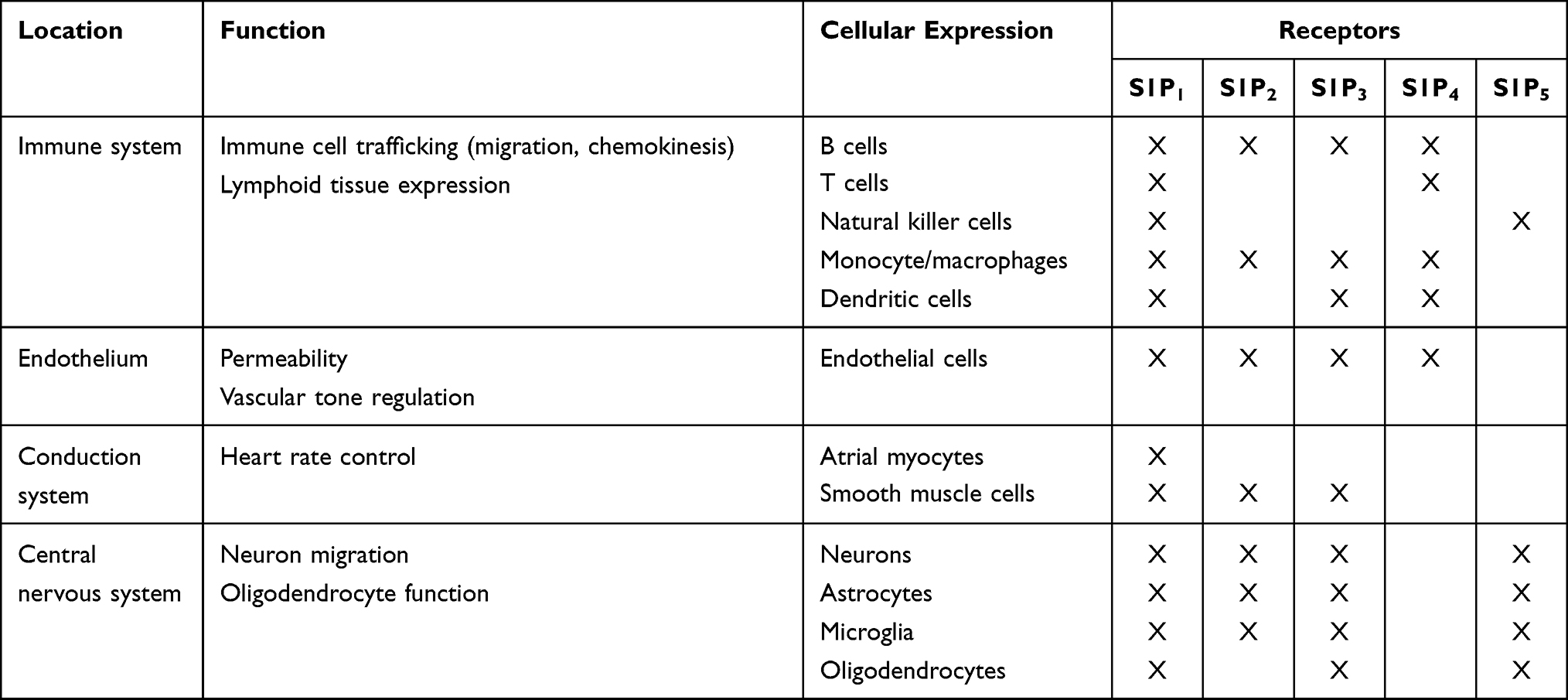 |
Table 1 Location, Function and Cellular Expression of Sphingosine 1-Phosphate Receptor Subtypes |
Consequently, S1P receptors mediate a broad range of biological functions, including cell proliferation, angiogenesis, cytoskeleton organization, endothelial cell chemotaxis, immune cell trafficking and mitogenesis. They are indeed involved in brain and cardiac development, proliferation of skin cells, vascular permeability, and regulation of vascular and bronchial tone.40
Overview on Sphingosine 1-Phosphate Receptor Modulators in Multiple Sclerosis
The widespread distribution and functional diversity of S1P receptors make their modulation an important therapeutic target in several diseases.40 S1P receptor modulators exert complex actions, as they can act as both functional antagonists and traditional agonists (depending on the targeted S1P receptor subtype or subtypes) and have different specificity.41 Structurally, S1P receptor modulators are small molecule drugs for oral administration that are S1P analogues.42 Some of these S1P receptor modulators are prodrugs (fingolimod, amiselimod) requiring phosphorylation or other metabolic steps to be converted into the pharmacologically active compound, whereas other ones are directly active drugs (siponimod, ozanimod, ponesimod) thereby resulting in different pharmacokinetic properties43 (Table 2).
 |
Table 2 Comparison of Sphingosine 1-Phosphate Receptor Modulators Currently Available for Treating Multiple Sclerosis |
The most important immunological mechanism of S1P receptor modulators relies on the regulation of lymphocyte trafficking by interacting with S1P1 receptor. This mechanism is based on a concentration gradient of S1P that, physiologically, is higher in the blood than in lymphatic tissues. When the ligand binds the receptor on cells expressing C-C Motif Chemokine Receptor 7 (CCR7), S1P1 receptor is internalised and degraded, resulting in loss of responsiveness to S1P gradient.44 Consequently, lymphocyte are retained in thymus and secondary lymphoid organs, leading to decreased peripheral blood lymphocyte count, limiting the inflammatory cells migration.
Moreover, S1P receptors are also present on microglia, oligodendrocytes, astrocytes, and neurons, thus their modulation can contribute to the beneficial effects on MS.13
Most S1P receptor modulators that are currently approved or in clinical development for MS treatment show high affinity for the S1P1 receptor, and typically for one or more of the other S1P receptor subtypes (Table 2).
To date, three S1P receptor modulators are licensed to treat MS: fingolimod, ozanimod and siponimod.45 Fingolimod was not only the first S1P receptor modulator approved for the treatment of any disease, but also the first oral drug licensed for adults and, more recently, for paediatric patients with MS.46–48 Second-generation S1P receptor modulators, such as ozanimod and siponimod, have been recently approved for treating adults with RRMS (Europe, USA) and active SPMS (USA).49–51 Next-generation S1PR modulators that are currently being developed for MS include amiselimod, ceralifimod, etrasimod, GSK2018682.44,45
Ponesimod: Pharmacokinetic and Pharmacodynamics Data
Structurally, ponesimod is an iminothiazolidinone derivative (Figure 1) acting as a selective, and rapidly reversible S1P1 receptor modulator by a functional antagonism mechanism.52
 |
Figure 1 Chemical structure of ponesimod (C23H25ClN2O4S). |
As compared with other S1P receptor modulators, ponesimod has a shorter half-life, allowing a faster reversibility of its effects. Ponesimod is rapidly absorbed in fasting state, with a median time to maximal concentration (tmax) from 2 to 4 hours according to the different doses tested (from 1 mg to 75 mg). The apparent terminal elimination half-life (t1/2) ranges from approximately 22 and 33 hours.52 Ponesimod has dose-proportional pharmacokinetic properties, as its elimination half-life (t1/2) increases with the increased single dose administered. Multiple-dose administration does not substantially modify the half-time, but induces an approximately two-fold increase of its plasma concentration, reaching the steady-state concentration at day 5.53 Food has a negligible effect on ponesimod pharmacokinetics: in fed state, absorption of ponesimod is delayed (tmax 5 versus 2.5 hours in fasting state), and its plasma concentrations is slightly higher (Cmax 81 versus 71 ng/mL in fasting state). After oral administration, ponesimod shows a bioavailability of 35–74%, low clearance, and high level of plasma protein binding about 99%. There is a sex difference in ponesimod absorption, which is increased in females compared to males, but in absence of sex difference in its metabolism and clearance.54 The major route of elimination is fecal, whereas urinary is only minor. Ponesimod does not reach the breast milk in large amounts, as highly bound in maternal plasma; however, being closely related to fingolimod, it should be avoided during breastfeeding because of its potential toxicity to the infant, especially in the absence of “ad hoc” studies.55
The oral administration of a single dose of 40 mg ponesimod determines a transient activation of S1P1 receptor, followed by its internalization and degradation. As a result, lymphocytes are sequestered in lymph nodes, thereby driving a dose-dependent reduction of circulating peripheral lymphocytes (up to above 80%) that prevents the lymphocyte-mediated immune response and the infiltration of autoimmune cells into the CNS.
Data from the phase II trial showed, after a 24-week follow-up, mean lymphocyte count reductions of 50%, 65% and 69% in patients with RRMS randomized to ponesimod 10, 20, or 40 mg, respectively. After ponesimod discontinuation, the mean lymphocyte count returns to baseline values within one week, thus suggesting a better safety profile (in terms of immune surveillance), but implying a faster disease activity reactivation, as compared to other S1P receptor modulators.
Notably, ponesimod has a differential effect on lymphocyte subpopulations, with decreased counts of T cells (CD3+) and B cells (CD20+) in a dose-dependent pattern, whereas there is no effect on natural killer cells (CD16+).56
Ponesimod: Efficacy
The efficacy of ponesimod has been determined earlier in a dose-finding phase II study and later confirmed in larger Phase III RCTs.
A 24-week, multicenter, randomized, double-blind, parallel-group, placebo-controlled, phase IIb dose-finding trial enrolled 464 patients with RRMS to evaluate the efficacy and safety of three doses of ponesimod.57 Patients were randomized to take either 10, 20, or 40 mg ponesimod daily or placebo for 24 weeks. From week 12 to 24, there was a significant reduction in the cumulative number of new gadolinium-enhancing (GD+) lesions for all ponesimod doses compared with placebo (–43%, –83%, and –77% for 10, 20, and 40 mg doses, respectively). However, the relapse rate reduction and the time to first confirmed relapse was statistically significant only for the 40 mg dose (–52% and –58%, respectively). Only the 20 mg dose showed a significant reduction in new or enlarging T2 lesions (–56%) at weeks 12–24, whereas the cumulative number of combined unique active lesions (CUALs), defined as the sum of GD+ lesions plus new or enlarging T2 lesions without double counting of lesions, was reduced by 42%, 80%, and 73% with all ponesimod doses (10, 20 and 40 mg, respectively), as compared to placebo. All ponesimod doses showed promising results on brain atrophy estimation, as revealed by a small mean increase in brain volume at week 24 (0.02%, 0.05% and 0.23% in the 10, 20 and 40 mg groups, respectively), in contrast with a mean decrease (–0.26%) of placebo.
Patients who completed the core study (n = 393) were eligible for enrolment in a long-term extension phase, initially at the same ponesimod dose they received in the primary phase, whereas patients originally receiving placebo were re-assigned randomly to ponesimod 10, 20, or 40 mg in a 1:1:1 ratio.58 A total of 326 patients received at least 48 weeks of study treatment, with a cumulative exposure across all doses of approximately 2372 patients/years. At week 48, the annualised relapse rate in the continuous-ponesimod group was 0.22, 0.23, and 0.15 for the 10, 20, and 40 mg doses, respectively. In the switched placebo-ponesimod group, the relapse rate decreased from 0.52 at week 24 to 0.25 at week 48. Patients receiving ponesimod 10 and 40 mg were later switched to 20 mg based on a better safety profile and confirmed efficacy. After an approximately 8-year follow-up, 214 patients remained on ponesimod therapy; the annualised relapse rate was 0.154, with 64.1% of patients free from confirmed relapse; the mean number of GD+ lesions per patient per scan was 0.448; the mean number of new or enlarging T2-hyperintense lesions per year was 0.718.58
In view of the phase II trial results, a larger multicenter, double-blind, active-comparator, superiority phase III RCT was established. The Oral Ponesimod Versus Teriflunomide In Relapsing Multiple Sclerosis (OPTIMUM) RCT allocated patients with RRMS in a 1:1 ratio to 20 mg ponesimod or 14 mg teriflunomide once daily for 108 weeks.59 The main eligibility criteria were: age 18 to 55 years; RRMS or SPMS with superimposed relapses; Expanded Disability Status Scale (EDSS) score60 between 0 and 5.5; recent clinical (either ≥1 relapse within 12 months or ≥2 relapses within 24 months prior to enrolment) or MRI activity (≥1 brain GD+ lesions on a scan performed within 6 months prior to enrolment).
During an initial two-week phase of the study (up-titration period), the study drugs were administered in a double-dummy fashion, ie: daily administration of one tablet (ponesimod or matching placebo) and one capsule (teriflunomide or matching placebo). The gradual up-titration of ponesimod from a 2 mg starting dose to a 20 mg maintenance was required to mitigate its first-dose effects.
The primary endpoint was the annualised relapse rate, whereas secondary endpoints included time to 12-week and 24-week confirmed disability accumulation, MRI endpoints (CUALs), and patient-reported outcomes, such as the Fatigue Symptom and Impact Questionnaire (FSIQ). Exploratory endpoints included the percentage change in brain volume derived from MRI and the percentage of patients with no evidence of disease activity (NEDA)-3, ie: no relapse, no 12-week confirmed disability accumulation, and no GD+ or new or enlarging T2 lesions; and NEDA-4, ie: a composite of NEDA-3 and no brain volume decrease of ≥0.4.61
A total of 1133 patients entered the study, of whom 567 receiving ponesimod and 566 receiving teriflunomide. At 108 weeks the annualized relapse rate was significantly lower for ponesimod (0.202) versus teriflunomide (0.290), with a –30.5% relative reduction (p < 0.001). The proportion of patients who experienced confirmed disability accumulation did not differ between the two groups: 10.8% versus 13.2% (for the 12-week confirmed outcome), and 8.1% and 9.9% (for the 24-week confirmed outcome) in the ponesimod arm and teriflunomide arm, respectively (p ≥ 0.29). Ponesimod was superior to teriflunomide in reducing the mean CUALs per year (1.405 versus 3.164, relative reduction –56%; p < 0.001). Mean FSIQ weekly symptoms score decreased by 0.01 point in ponesimod arm compared to a 3.56-point increase in teriflunomide arm (p = 0.002).
Among exploratory outcome, mean brain volume loss was –0.91% with ponesimod versus –1.25% with teriflunomide (p < 0.001). Ponesimod was superior to teriflunomide even in proportion of patients with NEDA-3 (25.0% versus 16.4%, p < 0.001) and NEDA-4 (11.4% versus 6.5%, p = 0.003). A summary of the main study findings is shown in Table 3. A long-term extension of the phase III OPTIMUM study (ClinicalTrials.gov registration: NCT03232073, OPTIMUM-LE) is ongoing.
 |
Table 3 Main Findings of the Oral Ponesimod versus Teriflunomide in Relapsing Multiple Sclerosis (OPTIMUM) |
The Clinical Study to Compare the Efficacy and Safety of Ponesimod to Placebo in Subjects With Active Relapsing Multiple Sclerosis Who Are Treated With Dimethyl Fumarate (POINT) was an add-on phase III RCT designed to evaluate the added benefit of ponesimod versus placebo in patients showing disease activity while taking dimethyl fumarate (ClinicalTrials.gov registration: NCT02907177). This RCT was designed on the basis of a preclinical study showing a synergistic effect of combining ponesimod and dimethyl fumarate in animal models of EAE.62 The study intended to recruit approximately 600 participants receiving dimethyl fumarate for at least 6 months and who had disease activity after at least 3 months of treatment with this compound. The primary endpoint was the annualised relapse rate, whereas the secondary endpoints were the proportions of patients experiencing 12-week confirmed disability accumulation, confirmed relapse, and adverse events (AEs). This RCT was ended in March 2020 because of low recruitment rates, as only 136 patients were randomized in a 1:1 ratio to ponesimod 20 mg or placebo in add-on to dimethyl fumarate. The primary endpoint was not met, as the annualised relapse rate was 0.237 in the ponesimod plus dimethyl fumarate arm versus 0.187 in the placebo plus dimethyl fumarate arm at 68 weeks (p = 0.52).63
Ponesimod: Safety
The majority of AEs observed during phase II and phase III RCTs occurring while taking ponesimod were mild or moderate in intensity, and were similar across all ponesimod groups and either placebo or teriflunomide.
In the dose-finding phase II RCT,57 the most frequently reported treatment-emergent AEs in the three ponesimod groups compared with placebo were anxiety, dizziness, dyspnoea, increased alanine aminotransferase, influenza, insomnia and peripheral oedema. Incidences of dyspnoea and peripheral oedema seemed to be dose-related, with substantially more cases reported in the highest dose ponesimod group compared with the other two dosages. Cardiac AEs associated with ponesimod treatment initiation involved first-degree (1.2%) and second-degree (0.9%; no cases of Mobitz type II) atrio-ventricular block and bradycardia (2%). All AEs relating to heart rhythm occurred on day 1 when all patients randomised to ponesimod were receiving a dose of 10 mg. The decrease in heart rate on day 1 reached a maximum at 2–3 hours post-dose and returned near to pre-dose values after 6 hours; on up-titration days (days 8 and 15), heart rate changes were small even with the higher ponesimod doses and consistent with those observed in the placebo group.
Ponesimod treatment results in a dose-dependent decrease in the Forced Expiratory Volume in the 1st second (FEV1), with a mean percentage change from baseline to week 24 of −5.2%, −6.0% and −10.3% in the ponesimod 10, 20 and 40 mg arms, respectively, and −0.6% in placebo arm. FEV1 returned to baseline values within 1 week of treatment discontinuation, as observed in patients who discontinued treatment prematurely or did not enter the extension study. There was also a decrease in forced vital capacity (FVC) with ponesimod treatment, but this was smaller than the decrease seen for FEV1 and also reversible, suggesting a functional obstructive change induced by ponesimod.
The percentages of patients with infection-associated AEs were similar across the four groups (placebo: 45.5%; ponesimod 10 mg: 37.0%; 20 mg: 32.5%; 40 mg: 36.1%). As expected, lymphocyte counts rapidly decreased with ponesimod treatment in a dose-dependent manner; the mean reductions from baseline to week 24 were 50%, 65% and 69% for ponesimod 10, 20 and 40 mg, respectively, and 3% in the placebo group. However, there was neither treatment discontinuation due to lymphopenia, nor a correlation between infections and lymphocyte count decrease during the study. Specifically, infection-associated AEs was similar across the four groups (placebo: 45.5%; ponesimod 10 mg: 37.0%; 20 mg: 32.5%; 40 mg: 36.1%). Increased liver enzymes (defined as above three-fold increase with respect to the upper limit of the normal range) were observed more frequently in the four groups (ponesimod 10 mg: 2.8%; 20 mg: 4.5%; 40 mg: 4.2%) compared with no case in the placebo group.
Four macular oedema cases were described. Three cases occurred in the ponesimod 20 mg group, of whom two resolved after treatment discontinuation and the remaining one during the study treatment. The fourth case of macular oedema was reported in the placebo group and resolved during the study period.
The safety and tolerability profile of ponesimod has been furtherly confirmed in the OPTIMUM phase III RCT.59 Treatment discontinuation due to AEs was more frequent in the ponesimod arm than teriflunomide arm (8.7% versus 6.0%). Dyspnoea (1.1% versus 0%), increased alanine amino-transaminase level (0.9% versus 1.1%) or aspartate amino-transferase level (0.5% versus 0.9%), and macular edema (0.9% versus 0%) were the most reported reasons leading to treatment discontinuation.
With the optimization of treatment dose (20 mg) and 14-day up-titration regimen applied, the overall incidence of first-dose heart rate and rhythm changes on day 1 (at a 2-mg dose) of up-titration, or treatment re-initiation, was 2.1% in the ponesimod arm and 0.4% in the teriflunomide arm. None of the reported cardiac AEs was serious or required treatment discontinuation; no second-degree or higher-degree atrio-ventricular blocks were observed. The up-titration scheme of ponesimod that successfully mitigates its first-dose effects on heart rate is shown in Table 4.
 |
Table 4 Up-Titration Scheme of Ponesimod to Mitigate the First-Dose Effect on Heart Rate |
Most frequently occurring AEs of special interest with ponesimod versus teriflunomide were hepatobiliary disorders or liver enzyme increase (22.7% versus 12.2%), hypertension (10.1% versus 9.0%), pulmonary events (8.0% versus 2.7%), and herpetic infections (each 4.8%). Macular oedema were reported in 6 patients receiving ponesimod and none receiving teriflunomide; all cases resolved with treatment discontinuation. Five cases of skin malignancies were reported in the ponesimod group. Two patients had basal cell carcinomas, another two experienced excisions of pre-existing benign lesions, and one patient had malignant melanoma.
Ponesimod: Contraindications and Special Warnings
Ponesimod is contraindicated in the following cases: patients aged less than 18 years; patients with hypersensitivity to the active substance or to any of its excipients, immunocompromised patients; patients who experienced, in the previous 6 months, myocardial infarction, unstable angina, stroke, transient ischaemic attack, decompensated heart failure requiring hospitalisation, or New York Heart Association Class III or IV heart failure; patients with Mobitz type II second-degree, third-degree atrio-ventricular (AV) block, or sick sinus syndrome, unless a functioning pacemaker is present; patients with severe active infections, active chronic infections, or active malignancies; moderate or severe hepatic impairment (Child-Pugh class B and C, respectively); women of childbearing potential not using effective contraception; pregnancy and breast-feeding.
An electrocardiogram (ECG) should be obtained in all patients to exclude pre-existing conduction abnormalities. Cardiologist advice should be obtained before ponesimod start in patients with known heart diseases or conditions altering the heart conduction, and in those who are treated with drugs influencing the heart rate (class Ia and III anti-arrhythmic molecules, beta-blockers, non-dihydropyridine calcium channel blockers, digoxin). Special caution should be applied when ponesimod is initiated in patients receiving beta-blockers because of the additive effects on lowering heart rate: if resting heart rate is ≤55 bpm, temporary interruption of the beta-blocker treatment should be considered prior to ponesimod start. Switch to pharmacological treatments not affecting the heart rate should be considered before starting ponesimod.
First-dose 4-hour monitoring is recommended for patients with sinus bradycardia, defined as heart rate < 55 beats per minute (bpm); first- or second-degree (Mobitz type I) AV block, or a history of myocardial infarction or heart failure occurring more than 6 months prior to treatment initiation and in stable condition.
Additional monitoring after 4-hours is recommended in the following events: post-dose heart rate <45 bpm; post-dose heart rate is at the lowest value post-dose, thus suggesting that the maximum pharmacodynamic effect on the heart rate may not have occurred; post-dose ECG shows new onset second-degree or higher AV block.
Appropriate management, continuous ECG monitoring overnight, further 4-hour monitoring after the second dose, and pharmacological treatment (if necessary) are required in case of post-dose symptomatic bradycardia, bradyarrhythmia, conduction-related symptoms, or new onset second degree or higher AV block or QTc ≥500 msec (even if asymptomatic).
Conclusion
Ponesimod has been recently approved in both the United States and Europe for the treatment of patients with RRMS, on the basis of the OPTIMUM study that, for the first time, provided a comparison of two different oral DMTs.
Overall, the magnitude of treatment effects with ponesimod seems to be in line with other S1P receptor modulators, without unexpected safety concerns;46–51 nonetheless, comparisons across different RCTs suffer from several limitations and should be interpreted with caution in the absence of head-to-head comparison studies.33
The S1P1 selectivity of ponesimod may provide safety advantages over fingolimod, and to a lesser extent, over ozanimod and siponimod, but this hypothesis needs to be confirmed by long-term data.
Differently from fingolimod, but similarly to ozanimod and siponimod, the optimized up-titration regimen of ponesimod minimizes the first-dose heart rate effect and rhythm AEs, making prolonged cardiovascular monitoring at treatment start unnecessary.
Furthermore, the distinctive feature of a shorter half-life of ponesimod in comparison with fingolimod (resulting in a faster reversibility of its effects on the immune system) has a two-fold clinical implication. On one hand it may represent an advantage in case of vaccination or pregnancy and in managing AEs (opportunistic infections, macular oedema, pulmonary function changes, liver enzyme elevations), on the other hand it may raises concern about reactivation of disease activity following ponesimod discontinuation.
Given its favorable risk:benefit and convenience profile, and despite the availability of other S1P receptor modulators, ponesimod has the potential to become an important option among oral DMTs for treating RRMS.
Abbreviations
AE, adverse events; AV, atrio-ventricular; BBB, blood-brain barrier; bpm, beats per minute; CNS, central nervous system; CSF, cerebrospinal fluid; CUALs, combined unique active lesions; DMT, disease-modifying treatments; EAE, experimental autoimmune encephalomyelitis; EBV, Epstein-Barr virus; ECG, electrocardiogram; EDSS, Expanded Disability Status Scale; FSIQ, Fatigue Symptom and Impact Questionnaire; GD+, gadolinium-enhancing; IFNB, interferon beta; MRI, magnetic resonance imaging; MS, multiple sclerosis; PP, primary progressive; RCT, randomized clinical trial; RR, relapsing-remitting; S1P, sphingosine 1-phosphate; SP, secondary progressive; TCR, T cell receptors; TH, T-helper.
Disclosure
SR: personal fees and non-financial support from Biogen, Genzyme, Merck-Serono, Novartis, Teva, and Viatris. MEQ: personal fees and non-financial support from Roche. LP: consulting fees and/or speaker honoraria and/or travel grants from Biogen, Bristol-Meyers Squibb, Genzyme, Merck-Serono, Novartis, Roche, and Viatris; research grants from the Italian MS Society (Associazione Italiana Sclerosi Multipla) and Genzyme. The authors report no other conflicts of interest in this work.
References
1. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622–1636. doi:10.1016/S0140-6736(18)30481-1
2. Anon. Number of people with MS | Atlas of MS. Available from: https://www.atlasofms.org/map/global/epidemiology/number-of-people-with-ms.
3. Browne P, Chandraratna D, Angood C, et al. Atlas of multiple sclerosis 2013: a growing global problem with widespread inequity. Neurology. 2014;83(11):1022–1024. doi:10.1212/WNL.0000000000000768
4. McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: a review. JAMA. 2021;325(8):765–779. doi:10.1001/jama.2020.26858
5. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–286. doi:10.1212/WNL.0000000000000560
6. Ontaneda D. Progressive multiple sclerosis. Contin Minneap Minn. 2019;25(3):736–752.
7. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162–173. doi:10.1016/S1474-4422(17)30470-2
8. Stys PK, Zamponi GW, van Minnen J, Geurts JJG. Will the real multiple sclerosis please stand up? Nat Rev Neurosci. 2012;13(7):507–514. doi:10.1038/nrn3275
9. Almuslehi MSM, Sen MK, Shortland PJ, Mahns DA, Coorssen JR. CD8 T-cell recruitment into the central nervous system of cuprizone-fed mice: relevance to modeling the etiology of multiple sclerosis. Front Cell Neurosci. 2020;14:43. doi:10.3389/fncel.2020.00043
10. ’t Hart BA, Luchicchi A, Schenk GJ, Stys PK, Geurts JJG. Mechanistic underpinning of an inside-out concept for autoimmunity in multiple sclerosis. Ann Clin Transl Neurol. 2021;8(8):1709–1719. doi:10.1002/acn3.51401
11. Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296–301. doi:10.1126/science.abj8222
12. Croxford JL, Olson JK, Miller SD. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler’s virus-induced demyelinating disease model of multiple sclerosis. Autoimmun Rev. 2002;1(5):251–260. doi:10.1016/S1568-9972(02)00080-0
13. Titus HE, Chen Y, Podojil JR, et al. Pre-clinical and clinical implications of ‘inside-out’ vs. ‘outside-in’ paradigms in multiple sclerosis etiopathogenesis. Front Cell Neurosci. 2020;14:599717. doi:10.3389/fncel.2020.599717
14. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–558. doi:10.1038/nri3871
15. Tobin WO, Kalinowska-Lyszczarz A, Weigand SD, et al. Clinical correlation of multiple sclerosis immunopathologic subtypes. Neurology. 2021;97(19):e1906–e1913. doi:10.1212/WNL.0000000000012782
16. Lassmann H. Cortical lesions in multiple sclerosis: inflammation versus neurodegeneration. Brain. 2012;135(Pt 10):2904–2905. doi:10.1093/brain/aws260
17. Koch-Henriksen N, Magyari M. Apparent changes in the epidemiology and severity of multiple sclerosis. Nat Rev Neurol. 2021;17(11):676–688. doi:10.1038/s41582-021-00556-y
18. Kalincik T, Diouf I, Sharmin S, et al. Effect of disease-modifying therapy on disability in relapsing-remitting multiple sclerosis over 15 years. Neurology. 2021;96(5):e783–e797. doi:10.1212/WNL.0000000000011242
19. Ransohoff RM, Hafler DA, Lucchinetti CF. Multiple sclerosis—a quiet revolution. Nat Rev Neurol. 2015;11(3):134–142. doi:10.1038/nrneurol.2015.14
20. Cree BAC, Hollenbach JA, Bove R, et al.; University of California, San Francisco MS-EPIC Team. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653–666. doi:10.1002/ana.25463
21. Ontaneda D, Tallantyre E, Kalincik T, Planchon SM, Evangelou N. Early highly effective versus escalation treatment approaches in relapsing multiple sclerosis. Lancet Neurol. 2019;18(10):973–980. doi:10.1016/S1474-4422(19)30151-6
22. Jakimovski D, Vaughn CB, Eckert S, Zivadinov R, Weinstock-Guttman B. Long-term drug treatment in multiple sclerosis: safety success and concerns. Expert Opin Drug Saf. 2020;19(9):1121–1142. doi:10.1080/14740338.2020.1805430
23. Rotstein D, Montalban X. Reaching an evidence-based prognosis for personalized treatment of multiple sclerosis. Nat Rev Neurol. 2019;15(5):287–300. doi:10.1038/s41582-019-0170-8
24. Sormani MP, Arnold DL, De Stefano N. Treatment effect on brain atrophy correlates with treatment effect on disability in multiple sclerosis. Ann Neurol. 2014;75(1):43–49. doi:10.1002/ana.24018
25. IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43(4):655–661. doi:10.1212/WNL.43.4.655
26. Haghikia A, Hohlfeld R, Gold R, Fugger L. Therapies for multiple sclerosis: translational achievements and outstanding needs. Trends Mol Med. 2013;19(5):309–319. doi:10.1016/j.molmed.2013.03.004
27. Schweitzer F, Laurent S, Fink GR, et al. Effects of disease-modifying therapy on peripheral leukocytes in patients with multiple sclerosis. J Neurol. 2021;268:2379–2389.
28. Mills EA, Mao-Draayer Y. Aging and lymphocyte changes by immunomodulatory therapies impact PML risk in multiple sclerosis patients. Mult Scler. 2018 Jul;24(8):1014-1022.
29. Winkelmann A, Loebermann M, Reisinger EC, Hartung H-P, Zettl UK. Disease-modifying therapies and infectious risks in multiple sclerosis. Nat Rev Neurol. 2016;12(4):217–233. doi:10.1038/nrneurol.2016.21
30. Barry B, Erwin AA, Stevens J, Tornatore C. Fingolimod rebound: a review of the clinical experience and management considerations. Neurol Ther. 2019;8(2):241–250. doi:10.1007/s40120-019-00160-9
31. Prosperini L, Kinkel RP, Miravalle AA, Iaffaldano P, Fantaccini S. Post-natalizumab disease reactivation in multiple sclerosis: systematic review and meta-analysis. Ther Adv Neurol Disord. 2019;12:1756286419837809. doi:10.1177/1756286419837809
32. Zhang Y, Salter A, Wallström E, Cutter G, Stüve O. Evolution of clinical trials in multiple sclerosis. Ther Adv Neurol Disord. 2019;12:1756286419826547. doi:10.1177/1756286419826547
33. Tur C, Kalincik T, Oh J, et al. Head-to-head drug comparisons in multiple sclerosis: urgent action needed. Neurology. 2019;93(18):793–809. doi:10.1212/WNL.0000000000008319
34. Trojano M, Tintore M, Montalban X, et al. Treatment decisions in multiple sclerosis - insights from real-world observational studies. Nat Rev Neurol. 2017;13(2):105–118. doi:10.1038/nrneurol.2016.188
35. Zhang H, Desai NN, Olivera A, et al. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol. 1991;114(1):155–167. doi:10.1083/jcb.114.1.155
36. Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171(15):3575–3594. doi:10.1111/bph.12678
37. Rosen H, Germana Sanna M, Gonzalez-Cabrera PJ, Roberts E. The organization of the sphingosine 1-phosphate signaling system. Curr Top Microbiol Immunol. 2014;378:1–21. doi:10.1007/978-3-319-05879-5_1
38. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12(9):688–702. doi:10.1038/nrd4099
39. Cartier A, Hla T. Sphingosine 1-phosphate: lipid signaling in pathology and therapy. Science. 2019;366(6463):eaar5551. doi:10.1126/science.aar5551
40. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22(1):50–60. doi:10.1016/j.tcb.2011.09.003
41. Subei AM, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs. 2015;29(7):565–575. doi:10.1007/s40263-015-0261-z
42. Edmonds Y, Milstien S, Spiegel S. Development of small-molecule inhibitors of sphingosine-1-phosphate signaling. Pharmacol Ther. 2011;132(3):352–360. doi:10.1016/j.pharmthera.2011.08.004
43. Pérez-Jeldres T, Alvarez-Lobos M, Rivera-Nieves J. Targeting sphingosine-1-phosphate signaling in immune-mediated diseases: beyond multiple sclerosis. Drugs. 2021;81(9):985–1002. doi:10.1007/s40265-021-01528-8
44. Chaudhry BZ, Cohen JA, Conway DS. Sphingosine 1-phosphate receptor modulators for the treatment of multiple sclerosis. Neurother J Am Soc Exp Neurother. 2017;14(4):859–873.
45. McGinley MP, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis and other conditions. Lancet. 2021;398(10306):1184–1194. doi:10.1016/S0140-6736(21)00244-0
46. Kappos L, Radue E-W, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. doi:10.1056/NEJMoa0909494
47. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. doi:10.1056/NEJMoa0907839
48. Chitnis T, Arnold DL, Banwell B, et al. Trial of fingolimod versus interferon beta-1a in pediatric multiple sclerosis. N Engl J Med. 2018;379(11):1017–1027. doi:10.1056/NEJMoa1800149
49. Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263–1273. doi:10.1016/S0140-6736(18)30475-6
50. Cohen JA, Comi G, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019;18(11):1021–1033. doi:10.1016/S1474-4422(19)30238-8
51. Comi G, Kappos L, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019;18(11):1009–1020. doi:10.1016/S1474-4422(19)30239-X
52. Brossard P, Derendorf H, Xu J, et al. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br J Clin Pharmacol. 2013;76(6):888–896. doi:10.1111/bcp.12129
53. Brossard P, Scherz M, Halabi A, et al. Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: favorable impact of dose up-titration. J Clin Pharmacol. 2014;54(2):179–188. doi:10.1002/jcph.244
54. Reyes M, Hoch M, Brossard P, Dingemanse J. Effects of ethnicity and sex on the pharmacokinetics and pharmacodynamics of the selective sphingosine-1-phosphate receptor 1 modulator ponesimod: a clinical study in Japanese and Caucasian subjects. Pharmacology. 2014;94(5–6):223–229. doi:10.1159/000368837
55. Anon. Ponesimod. In: Drugs and Lactation Database (Lactmed). Bethesda (MD): National Library of Medicine (US); 2006. Available from: http://www.ncbi.nlm.nih.gov/books/NBK575381/.
56. D’Ambrosio D, Steinmann J, Brossard P, Dingemanse J. Differential effects of ponesimod, a selective S1P1 receptor modulator, on blood-circulating human T cell subpopulations. Immunopharmacol Immunotoxicol. 2015;37(1):103–109. doi:10.3109/08923973.2014.993084
57. Olsson T, Boster A, Fernández Ó, et al. Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial. J. Neurol. Neurosurg. Psychiatry. 2014;85(11):1198–1208. doi:10.1136/jnnp-2013-307282
58. Freedman MS, Pozzilli C, Havrdova EK, et al. Long-term efficacy and safety of ponesimod, an oral S1P1 receptor modulator: results from randomized phase II core and extension studies in relapsing-remitting multiple sclerosis (1752). Neurology. 2020;94(15 Supplement).
59. Kappos L, Fox RJ, Burcklen M, et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active-comparator phase 3 OPTIMUM study: a randomized clinical trial. JAMA Neurol. 2021;78(5):558–567. doi:10.1001/jamaneurol.2021.0405
60. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–1452. doi:10.1212/WNL.33.11.1444
61. Giovannoni G, Tomic D, Bright JR, Havrdová E. ‘No evident disease activity’: the use of combined assessments in the management of patients with multiple sclerosis. Mult Scler. 2017;23(9):1179–1187. doi:10.1177/1352458517703193
62. Pouzol L, Piali L, Bernard CC, et al. Therapeutic potential of ponesimod alone and in combination with dimethyl fumarate in experimental models of multiple sclerosis. Innov Clin Neurosci. 2019;16(3–4):22–30.
63. Kappos L, Lindenstrom E, Freedman MS, et al. The POINT study: a randomized, double-blind, parallel-group, add-on, superiority phase 3 study to compare the efficacy and safety of ponesimod to placebo in subjects with active relapsing multiple sclerosis who are treated with dimethyl fumarate. Mult Scler. 2018;24(Suppl 2):271.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.