Back to Journals » International Journal of Nanomedicine » Volume 15
Polymeric Nanoparticles-Based Brain Delivery with Improved Therapeutic Efficacy of Ginkgolide B in Parkinson’s Disease
Authors Zhao Y, Xiong S, Liu P, Liu W, Wang Q, Liu Y, Tan H ![]() , Chen X
, Chen X ![]() , Shi X, Wang Q, Chen T
, Shi X, Wang Q, Chen T ![]()
Received 1 August 2020
Accepted for publication 20 November 2020
Published 24 December 2020 Volume 2020:15 Pages 10453—10467
DOI https://doi.org/10.2147/IJN.S272831
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Yuying Zhao,1 Sha Xiong,1 Piaoxue Liu,1 Wei Liu,1 Qun Wang,1 Yao Liu,1 Hanxu Tan,2 Xiaojia Chen,3 Xuguang Shi,4 Qi Wang,1 Tongkai Chen1
1Science and Technology Innovation Center, Guangzhou University of Chinese Medicine, Guangzhou 510405, People’s Republic of China; 2Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing 100700, People’s Republic of China; 3State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau 999078, People’s Republic of China; 4School of Pharmaceutical Sciences, Guangzhou University of Chinese Medicine, Guangzhou 510006, People’s Republic of China
Correspondence: Tongkai Chen; Qi Wang
Science and Technology Innovation Center, Guangzhou University of Chinese Medicine, 12 Jichang Road, Guangzhou 510405, People’s Republic of China
Email [email protected]; [email protected]
Purpose: Ginkgolide B (GB) is a terpene lactone derivative of Ginkgo biloba that is believed to function in a neuroprotective manner ideal for treating Parkinson’s disease (PD). Despite its promising therapeutic properties, GB has poor bioavailability following oral administration and cannot readily achieve sufficient exposure in treated patients, limiting its clinical application for the treatment of PD. In an effort to improve its efficacy, we utilized poly(ethylene glycol)-co-poly(ϵ-caprolactone) (PEG-PCL) nanoparticles as a means of encapsulating GB (GB-NPs). These NPs facilitated the sustained release of GB into the blood, thereby improving its ability to accumulate in the brain and to treat PD.
Methods and Results: Using Madin-Darby canine kidney (MDCK) cells, we were able to confirm that these NPs could be taken into cells via multiple nonspecific mechanisms including micropinocytosis, clathrin-dependent endocytosis, and lipid raft/caveolae-mediated endocytosis. Once internalized, these NPs tended to accumulate in the endoplasmic reticulum and lysosomes. In zebrafish, we determined that these NPs were readily able to undergo transport across the chorion, gastrointestinal, blood–brain, and blood-retinal barriers. In a 1-methyl-4-phenylpyridinium ion (MPP+)-induced neuronal damage model system, we confirmed the neuroprotective potential of these NPs. Following oral administration to rats, GB-NPs exhibited more desirable pharmacokinetics than did free GB, achieving higher GB concentrations in both the brain and the blood. Using a murine PD model, we demonstrated that these GB-NPs achieved superior therapeutic efficacy and reduced toxicity relative to free GB.
Conclusion: In conclusion, these results indicate that NPs encapsulation of GB can significantly improve its oral bioavailability, cerebral accumulation, and bioactivity via mediating its sustained release in vivo.
Keywords: drug delivery system, blood–brain barrier, endocytosis, zebrafish, PD treatment
Introduction
Parkinson’s disease (PD) is a very common neurodegenerative disease that impacts roughly 2% of adults over the age of 65.1 At a molecular level, PD is associated with progressive dopaminergic (DA) neuron degeneration, Lewy body development in these neurons, and accumulation of α-synuclein within the substantia nigra pars compacta.2 At the clinical level, PD patients suffer from significant motor abnormalities postural instability, tremors, altered gait, bradykinesia, and rigidity. Many pharmacological approaches have sought to treat PD in recent years, but most compounds used for such approaches have poor oral bioavailability and brain penetration and as such are of limited therapeutic value. Owing to these limitations, increasing numbers of studies have sought to utilize compounds derived from herbal sources in order to treat PD.3,4
Ginkgolide B (GB) is a diterpene derivative of Ginkgo biloba leaves, and it has frequently been explored as a neuroprotective agent for PD treatment.5 GB is known to disrupt the degradation of the rate-limiting enzyme responsible for dopamine synthesis, tyrosine hydroxylase (TH), thereby protecting against DA neuron damage or loss induced by agents such as 6-hydroxydopamine6 or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).7 Despite this promising efficacy, however, GB exhibits very poor water solubility. As a result, oral administration of GB achieves very poor brain and blood exposure, restricting its clinical utility. A number of approaches have been examined as a means of improving GB bioavailability including soybean phospholipid-stabilized nanosuspension,8 nanoemulsion,9 and polymeric nanocapsules.10 These approaches have been hampered by their poor drug-loading capacity and by a lack of data pertaining to their pharmacokinetic profiles in the blood and the brain of treated animals. Therefore, in the present study, we sought to develop a new formulation capable of enhancing oral bioavailability, plasma pharmacokinetics and brain accumulation of GB.
Polymeric nanoparticles (NPs) are optimal candidates for drug delivery strategies owing to their limited toxicity, high biocompatibility, desirable biodegradability, and long circulation time.11,12 Poly(ethylene glycol) (PEG) and poly(ε-caprolactone) (PCL) polymers have been approved by the FDA for pharmaceutical use.13 Amphiphilic copolymers composed of PEG and PCL can adopt a number of different structures based upon the molecular weights of the hydrophilic (PEG) and hydrophobic (PCL) components thereof.14 For the treatment of PD, NPs <100 nm in size are ideal as they have been shown to be well suited to delivering cargos through the brain capillary endothelial cell layer.15 Therapeutically viable NPs also need to be capable of achieving sustained drug release in vivo in order to effectively treat chronic conditions such as PD. Therefore, in this study, we endeavored to develop small NPs capable of releasing GB in a sustained fashion in order to achieve anti-Parkinsonian efficacy. To that end, we encapsulated GB in PEG-PCL via an antisolvent precipitation approach, yielding GB-NPs. In addition, NPs coated with Poloxamer 188 have been shown to be effective for transporting drugs across the blood–brain barrier (BBB).16 We have successfully developed six-armed star-shaped NPs to deliver drugs across the BBB.15 However, the endocytic mechanisms responsible for the uptake and transport of such NPs remain uncertain. To that end, we explored the endocytic mechanisms of NPs in Madin–Darby canine kidney (MDCK) cells, as these cells represent an ideal in vitro epithelial model owing to their polarity, tight junction formation, and relatively thin mucus layer.17 Moreover, the neuroprotective effects of NPs were evaluated using human neuroblastoma SH-SY5Y cells, a neuron-like cell model that has been widely used to evaluate intracellular events linked to neurotoxic insults. To extend this work, we then employed a zebrafish model system in order to explore the ability of coumarin 6 (C6)-labeled NPs (C6-NPs) to cross specific biological barriers in vivo.
The overall goal of this study was to determine whether GB-NPs were capable of releasing GB in a sustained manner in vivo, thereby improving the bioavailability of this compound and enhancing its therapeutic efficacy for the treatment of PD. As such, after conducting the studies discussed above, we used a rat model system to analyze the pharmacokinetics of these NPs, and we used an MPTP-induced mouse model of PD to explore their anti-Parkinsonian activity. Together, our results indicated that GB-NPs were successfully able to enhance the oral bioavailability, cerebral delivery, and therapeutic utility of GB.
Materials and Methods
Materials
PEG-PCL (PEG, MW, 2000 Da; PCL, MW 6000Da, mol/mol of lactide/glycotide, 75/25) were purchased from the Glaco Ltd. (Beijing, China). GB, Poloxamer 188 (F68), 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), rabbit polyclonal anti-TH antibody, MPTP, 1-methyl-4-phenylpyridinium ion (MPP+), and Selegiline were all obtained from Sigma-Aldrich (St. Louis, MO, USA). MDCK and SH-SY5Y cells were provided by Guangzhou Jenniobio Biotechnology Co., Ltd. (Guangzhou, China). Methyl-β-cyclodextrin (MβCD), Chlorpromazine (CPZ), hypertonic sucrose and 5-(N-ethyl-N-isopropyl)-amiloride (EIPA) were purchased from Sigma Aldrich (St. Louis, MO, USA). Coumarin-6 (C6), LysoTracker, ER Tracker and Mito Tracker were purchased from Molecular Probes Inc. (OR, USA).
Cell Culture and Animal Care
MDCK and SH-SY5Y cells were cultured in high glucose DMEM containing 10% FBS and antibiotics (1% penicillin and streptomycin mixture) at 37 °C in a humidified 5% CO2 incubator.
Wild-type zebrafish (Danio rerio) were obtained from the China Zebrafish Resource Center (Wuhan, China), and were housed and bred under standard conditions.18 Briefly, these zebrafish were exposed to a 14 h/10 h light/dark cycle. When zebrafish were mature, males and females were separated from one another at a 1:2 ratio in a 1 L tank overnight. Following the light cycle, the screen was separated, and embryos that had been fertilized were collected. Pigment formation was blocked using 1-phenyl-2- thiourea (PTU).
Male Sprague-Dawley (SD) rats (6–8 weeks old) and C57BL/6 mice (8 weeks old) were provided by the Experimental Animal Center of Guangzhou University of Chinese Medicine (Guangzhou, China), and were housed with free food and water access in a 25 ± 2 °C facility with 55 ± 5% relative humidity and a 12 h light/dark cycle. The Animal Ethics Committee of Guangzhou University of Chinese Medicine approved all studies described herein, which were consistent with regulations for the care and use of experimental animals defined by the Chinese government.
GB-NPs Preparation and Characterization
1H-nuclear magnetic resonance1H-NMR and Fourier-transform infrared (FTIR) spectroscopy were used to evaluate the properties of the PEG-PCL used for NPs preparation. Detailed methods are available in Supporting Information (Section S1). Next, GB-NPs were prepared via an antisolvent precipitation and stabilizer-based approach.15 Briefly, we injected 0.2 mL volumes of GB and PEG-PCL (20 mg/mL in acetone) into a 10 mL volume of dH2O containing the stabilizer F68 (0.5 mg/mL). This solution was constantly stirred at 1000 rpm at room temperature. The resultant GB-NPs were then collected and subjected to dynamic light scattering (DLS) in order to quantify their size distribution, polydispersity index (PDI), and zeta potential. The morphology of these NPs was then further evaluated via transmission electron microscopy (TEM), while high-performance liquid chromatography (HPLC) was used to quantify drug loading (DL) and encapsulation efficiency (EE), and differential scanning calorimetry (DSC) was used to assess the thermal properties of these NPs. Lastly, we used dialysis to assess drug release of GB-NPs. Detailed methods are available in Supporting Information (Section S1). C6-NPs were prepared in the same manner as GB-NPs, substituting GB for C6 and conducting the procedure in the dark.
GB-NPs Cellular Uptake and Permeation Measurements
In order to assess the impact of GB-NPs or free GB on the viability of MDCK cells, an MTT assay was conducted. Briefly, MDCK cells were added to 96-well plates overnight (5000 cells/cm2), after which media was exchanged for 100 μL of fresh media containing GB-NPs or GB. After an additional 24 h, 20 μL of MTT solution (5 mg/mL in PBS) was added to each well for 4 h. Media was then removed, and replaced with 150 μL DMSO. A microplate reader (Thermo Multiskan FC, USA) was then used to quantify absorbance at 570 nm.
Cellular GB-NPs and GB uptake were measured by replacing MDCK cell culture media with fresh media supplemented with 20 μM of GB-NPs or free GB (GB concentration of 20 μM). Cells were then incubated for 2 h, after which they were washed thrice using cold PBS (4°C) prior to lysis. Methanol was then used to facilitate protein precipitation, and samples were spun for 20 minutes at 15,000 rpm at room temperature. GB levels were then measured via HPLC using an Agilent 1100 instrument (CA, USA) with a DAD detector, an auto-sampler, and an Ecosil C18 analytical column (4.6 mm × 150 mm, 5μm). For this analysis, a 20 µL volume of each sample was injected into the system, with a mobile phase composed of methanol/water (50:50), a 1.0 mL/min flow rate, and a 220 nm detection wavelength.
We additionally analyzed the ability of free GB, GB-NPs, and a mixture of GB, PEG-PCL, and F68 (GB-PM) to permeate across MDCK cell monolayers. For such permeability analyses, transepithelial electrical resistance (TEER) values were measured prior to experimentation in order to confirm the integrity of these MDCK cell monolayers. Apical-to-basolateral transepithelial transport was tested via the apical addition of 20 μM GB (or equivalent GB-NPs or GB-PM doses) in 500 μL HBSS (pH 7.4). 1500 μL HBSS was added to the basolateral side of the monolayer. After 15, 30, 60, 90, and 120 minutes, 200 μL samples of this basolateral solution were collected. GB concentrations in these samples were quantified via HPLC, with the apparent permeability coefficient, Papp (cm/s), being calculated.19
MDCK Endocytic Uptake of C6-NPs
We utilized C6-labeled NPs (C6-NPs) to monitor cell-mediated endocytosis of these NPs. For this analysis, MDCK cells were plated at 1 × 105 cells/well in 12-well plates overnight, after which they were washed thrice with PBS prior to incubation with serum-free media supplemented with C6-NPs at final C6 concentrations of 0.5 μg/mL, 1 μg/mL or 2 μg/mL. After appropriate periods of incubation at 37 °C, cells were collected, washed using cold PBS, and fixed using 4% paraformaldehyde (PFA). A confocal laser scanning microscope (CLSM; TCS SPE Ⅱ, Leica, Germany) was then used to image cells.
Endocytic Inhibitor Treatments
In order to better understand the role of specific endocytic pathways in the uptake of these NPs, we treated MDCK cells with the endocytic inhibitors shown in Table S2.20,21 Briefly, MDCK cells were plated at 1 × 105 cells/well in 12-well plates for 24 h, after which cells were washed and fresh media supplemented with methyl-β-cyclodextrin (MβCD, 10 mM), chlorpromazine (CPZ, 30 μM), hypertonic sucrose (HS, 0.4 M), or 5-(N-ethyl-N-isopropyl)-amiloride (EIPA, 40 μM) was added, followed by a 30-minute incubation at 37 °C. Fresh media containing C6-NPs at a final C6 concentration of 1 μg/mL was then added, and cells were incubated for 30 min at 37 °C. Cells were then rinsed with cold PBS, fixed in 4% PFA, stained with DAPI, and imaged via CLSM.
Intracellular C6-NPs Trafficking in MDCK Cells
C6-NPs trafficking to the lysosomal (Lyso), mitochondrial (Mito), and endoplasmic reticulum (ER) compartments was assessed in MDCK cells cultured as above. Cells were initially plated overnight, and were then washed with PBS and incubated with Lyso Tracker (10,000:1 dilution), ER Tracker (1000:1 dilution), and Mito Tracker (3000:1 dilution) for 120 min, 30 min, and 30 min, respectively. Cells were then washed three more times in PBS prior to the addition of C6-NPs at a final C6 concentration of 1 μg/mL for 1 h. Cells were then rinsed using cold PBS prior to fixation in 4% PFA and imaged via CLSM.
Analysis of GB-NPs-Mediated Cytoprotection
SH-SY5Y cells were plated at 5000 cells/cm2 in 96-well plates overnight, after which they were treated for 2 h with GB or GB-NPs (1, 5, 10, or 20 μM). Next, 2 mM MPP+ was added and cells were incubated for a further 36 h. An MTT assay was then used to assess the ability of GB or GB-NPs to modulate the survival of these SH-SY5Y cells.
Embryos and Larval Zebrafish Exposure to C6-NPs and Imaging
We exposed either embryonic (3 hours post-fertilization; hpf) or larval (7 days post-fertilization; dpf) zebrafish to C6 or C6-NPs (50, 100, or 200 ng/mL). After 10, 30, or 60 minutes, we then washed these zebrafish and collected them for microscopic imaging (Model DMi8, Leica, Germany).
Analysis of C6-NPs Endocytosis in Larval Zebrafish
We treated larval zebrafish (7 dpf) with MβCD, CPZ, HS, or EIPA for 30 minutes in order to inhibit endocytosis, after which fresh media supplemented with C6 or C6-NPs (100 ng/mL) was added followed by an additional 30-minute incubation. Zebrafish were then collected, washed, anesthetized using 0.04% MS-222 (tricaine methane sulfonate), oriented laterally in 1% low-melting agarose, and imaged via fluorescence microscopy.
In vivo GB-NPs Toxicity Evaluation
Zebrafish embryos (5 hpf) were hatched in the presence of a range of GB-NPs concentrations selected based upon the OECD Guidelines for the Testing of Chemicals (0–150 μg/mL; n=20 zebrafish/concentration). Media was refreshed once per day. Embryonic morphology was analyzed via microscopy, with survival rates being measured based upon movement, heart-beat, and circulation. At 96 hpf, we recorded the hatching rates, body length, and heart rates for all embryos.
Histological Analysis
Following the completion of the in vivo PD murine model experiments, mice were euthanized and their hearts, livers, spleens, lungs, and kidneys were collected and fixed using 4% PFA. Samples were them paraffin-embedded, cut into 4 μm sections, and stained using hematoxylin and eosin (H&E) to assess organ morphology.
GB-NPs Pharmacokinetic Analysis
SD rats were separated at random into two treatment groups that were orally administered either GB-NPs (4 mg/kg GB) or an equivalent GB dose. Over the 48 h following administration, serum samples were obtained via collecting 300 μL of blood from individual rats in each treatment group (n=6/time point). Blood samples were spun for 5 minutes at 5000 rpm, after which supernatants were used for downstream analyses. In addition, at 0, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h following dosing, 4 rats per treatment group were euthanized and their brains were perfused with physiological saline as in prior studies.19 The brains of these animals were then excised, weighed, and homogenized in cold saline. LC-MS/MS was then used to measure GB concentrations in these samples.22
The Drug and Statistics (DAS, v2.0) software was used to calculate pharmacokinetic parameters including elimination half-life (T1/2), time to maximal plasma or brain concentrations (Tmax), peak plasma or brain concentrations (Cmax), area under the concentration-time curve in the plasma or brain from time zero to time t (AUC0-t), and mean residence time (MRT0-t). A non-compartmental model was used to estimate these parameters. In addition, relative GB-NPs bioavailability (F) was calculated as follows:

Evaluation of the in vivo Neuroprotective Activity of GB-NPs
Mice were randomized into the following treatment groups: (1) control group (disease free); (2) MPTP; (3) Selegiline; (4) GB; (5) GB-NPs. PD model mice were generated via intraperitoneal injection of MPTP (18 mg/kg in 0.9% saline) four times with 2 h between injections.23,24 Mice in the GB and GB-NP treatment groups were gavaged with either GB (5 mg/kg) together with blank NPs or GB-NPs (5 mg/kg) for 14 days (once daily for 7 days prior to MPTP administration and twice daily for 7 days after MPTP administration).25 Control mice were administered only 0.9% saline, while animals in the Selegiline group were administered Selegiline (10 mg/kg). GB-NPs-mediated neuroprotection was quantified based on behavioral testing, TH+ neuron immunohistochemistry, and by measuring levels of DA and metabolites.26
Statistical Analysis
One- and two-way ANOVAs were used to compare data between groups. Data are given as means ± SD. The significance threshold was p < 0.05.
Results and Discussion
GB-NPs Characterization
We began by characterizing the PEG-PCL polymer used for this study based upon its 1H-NMR spectrum, revealing that peaks at 3.34 ppm and 3.61 ppm were attributable to the protons of methylene and methyl in PEG (Figure S1). In addition, peaks at 1.65 ppm, 2.30 ppm and 3.99 ppm were assigned to the proton signals in PCL (Figure S1). We further employed FTIR spectroscopy to assess the structural conformation of the PEG-PCL polymer (Figure S2). This analysis revealed peaks at 2869 cm−1 and 2947 cm−1 consistent with C-H stretching, peaks at 957 cm−1 and 1245 cm−1 consistent with C-O stretching, peaks at 1728 cm−1 and 3443 cm−1 consistent with C=O and O-H, respectively, and peaks at 1090–1300 cm−1 consistent with C-C and C-O. We further detected peaks at 1085–1150 cm−1 that were attributable to the ether band of PEG. In addition, we measured the solubility of GB in different buffers (Table S1). We then characterized the optimized GB-NPs, which were found to have a particle size of 91.26 ± 1.34 nm, a PDI of 0.17 ± 0.01 (Figure 1A), a zeta potential of −12.09 ± 0.97 mV (Figure 1B), a DL capacity of 26.93%, and an EE of 87.52%. These GB-NPs were spherical in shape (Figure 1A, TEM image), and following a 4-week storage at room temperature, the size distribution of these NPs did not change (Figure S3). A single endothermic peak was detected at 130 °C when these NPs were analyzed via DSC, consistent with findings made upon the analysis of GB and GB-PM (Figure 1C). This suggested that the properties of GB within GB-NPs are not impacted by the antisolvent precipitation process. We also monitored in vitro GB release from GB-NPs over 48 h in PBS (pH 7.4), revealing a biphasic release pattern wherein ~30% of the total GB was released during the first 2 h, followed by more gradual sustained 94% release over the remainder of the 48 h period (Figure 1D). The rapid initial release of GB may be attributable to the release of free GB or surface GB into the PBS solution, with the following sustained release instead being attributable to more gradual diffusion and dissolution of these GB-NPs.27 Similar observations (rapid initial release and subsequent sustained release) are also found in the previous report.28
 |
Figure 1 GB-NPs characterization. (A) Size distribution. Insert: TEM image. Scale bar: 100 nm. (B) Zeta potential. (C) DSC profiles. (D) In vitro release profiles. |
Assessment of GB-NPs Cellular Uptake and Permeability with MDCK Cells
We examined the cytotoxic impact of GB and GB-NPs treatment on MDCK cells, revealing no significant cytotoxicity across a wide range of concentrations (1–100 μM) (Figure S4). However, GB-NPs were associated with significantly enhanced cellular GB uptake (3.18 ± 0.30 μg/mg protein) relative to the free GB (0.98 ± 0.07 μg/mg protein) and GB-PM (1.01 ± 0.11 μg/mg protein) groups, with no significant difference in uptake between the latter two groups. In addition, GB-NPs were associated with a higher Papp value (3.76 ± 0.39×10−5 cm/s) relative to free GB (1.08 ± 0.13×10−5 cm/s) and GB-PM (1.15 ± 0.16×10−5 cm/s), consistent with these GB-NPs being better-suited to transporting GB across MDCK cell monolayers. Importantly, TEER values were unchanged following treatment with these different GB formulations (Figure S5), suggesting that these NPs did not adversely impact MDCK monolayer integrity.
Endocytic Uptake of C6-NPs
We next sought to explore the mechanisms whereby NPs were taken into cells using C6-labeled NPs. C6-NPs were found to have a particle size of 90.40 ± 1.27 nm, a PDI of 0.17 ± 0.01 (Figure S6), a zeta potential of −11.85 ± 0.91 mV (Figure S7), which were similar with those of GB-NPs. In pilot experiments, we found that <4.5% of C6 was released from these C6-NPs over a 6-h period, suggesting that they are stably loaded with C6 even in acidic intracellular settings. When we treated MDCK cells with C6-NPs, we observed the time- and dose-dependent uptake of these NPs into cells (Figure S8).
Assessment of the Endocytic Mechanisms Governing C6-NPs Uptake into MDCK Cells
In order to explore the mechanisms whereby C6-NPs were taken up into MDCK cells, we treated these cells with a range of different inhibitors of endocytosis. We found that all tested inhibitors suppressed C6-NPs internalization to at least some degree (Figure 2A). Of the tested inhibitors, CPZ most significantly inhibited C6-NPs endocytosis (70.48%), followed by MβCD (68.86%), HS (38.94%) and EIPA (20.92%) (Figure 2B). Previous studies have found clathrin-mediated endocytosis to be one of the main mechanisms of NPs internalization,29 and consistent with these found that CPZ, which inhibits this process, interfered with C6-NPs uptake.30 As another mechanism of uptake, cells can internalize compounds via lipid raft-associated caveolae formation. MβCD inhibits this caveolar uptake pathway and was found to inhibit C6-NPs uptake in the present study, thus highlighting this as another mechanism of their endocytosis in MDCK cells.31 Macropinocytosis is another mechanism whereby extracellular macromolecules are internalized into cells, and we found that the micropinocytosis inhibitor EIPA interfered with C6-NPs uptake.32 As such, our results suggest that these NPs can be non-specifically internalized through multiple different endocytic mechanisms (Figure S9).
 |
Figure 2 Analysis of C6-NPs uptake and intracellular trafficking in MDCK cells. (A) C6-NPs were visualized within cells following treatment with endocytosis inhibitors (MβCD: methyl-β-cyclodextrin, CPZ: chlorpromazine, HS: hypertonic sucrose, EIPA: 5-(N-ethyl-N-isopropyl)-amiloride). Scale bar: 25 μm. (B) C6-NPs cellular uptake was quantified (n=3). *p<0.05, ***p<0.001 vs the Control group. (C) The co-localization of C6-NPs with lysosomes (Lyso), endoplasmic reticulum (ER), and mitochondria (Mito) was visualized using appropriate tracking dyes. Scale bar: 25 μm. |
C6-NPs Intracellular Localization in MDCK Cells
We analyzed the intracellular localization of C6-NPs within MDCK cells via confocal microscopy, revealing these NPs to co-localize with both the ER and with lysosomes (Figure 2C). Both of these organelles can play important roles in regulating the bioavailability and activity of drugs such as GB.33,34 In contrast, mitochondrial NPs accumulation is thought to induce high rates of cellular cytotoxicity.35 However, we observed negligible NPs co-localization with mitochondria in treated MDCK cells, suggesting that this is not a major cytotoxic mechanism associated with these NPs.
GB-NPs Protected Against in vitro MPP+-Induced Cell Death
We next assessed the impact of GB-NPs on SH-SY5Y cells in vitro in an MTT assay (Figure 3A). These NPs failed to induce any significant toxicity in these cells across the tested concentration range. We then utilized MPP+-treated SH-SY5Y cells as an in vitro model of PD in order to assess the cytoprotective properties of these NPs. We found that a 2 mM treatment with MPP+ was sufficient to induce 47.6% cell death, while 1, 5, 10, and 20 μM GB-NPs significantly increased the viability of these cells to 67%, 76%, 85%, and 91%, respectively, whereas free GB increased the survival of these cells to a lesser extent (60, 65, 71, 78%, respectively) (Figure 3B). As such, these GB-NPs significantly enhanced the cytoprotective activity of GB in vitro.
 |
Figure 3 (A) MTT assays in response to the indicated treatments (n=3). (B) GB-NPs protected against MPP+-mediated cytotoxicity (n=3). ***p<0.001 vs the MPTP group. ##p<0.01 vs the GB group. |
Analysis of C6-NPs Uptake in Embryonic and Larval Zebrafish
We next utilized zebrafish as an in vivo model to explore the ability of NPs to cross biological barriers. In zebrafish, the functional gastrointestinal (GI) barrier slowly matures until 120 hpf,36 with tight junction proteins such as claudins being expressed at 56 hpf. Multidrug resistance protein expression can also be detected in both embryonic and adult zebrafish tissues.37 Zebrafish embryos also possess an additional barrier known as the chorion that protects against external threats.38 By 3 dpf, zebrafish exhibit a developed BBB with permeability restrictions similar to those observed in humans.39 Tight junction and efflux transport proteins are also evident in cerebral microvessels in these zebrafish. In addition, zebrafish embryos and larvae are transparent and can be used to easily track NPs distribution in vivo in a non-invasive manner.40 GB-NPs uptake was monitored by treating embryonic zebrafish with C6-NPs beginning at 3 hpf and then monitoring fluorescence within these embryos over time. We observed dose- and time-dependent increases in fluorescence within these embryos from 10 to 60 minutes post-NPs addition (Figure S10), consistent with the ability of these NPs to cross the chorion into the yolk sac. Fluorescence in control embryos was minimal. As such, these results confirmed the ability of GB-NPs to enhance GB permeability.
We next exposed 7 dpf zebrafish larvae to C6-NPs in order to explore the ability of these NPs to enter the GI tract and to cross the BBB in vivo. We observed significant fluorescence in both the GI tract and brain of treated larvae, consistent with the successful oral uptake and cerebral delivery of these NPs (Figure 4). We also observed C6-NPs distribution to the eyes of treated zebrafish (Figure 4), indicating that these NPs can also cross the blood-retinal barrier. As the BBB is mature in zebrafish at this study time point, this model system can effectively be used to gauge the BBB permeability of particular compounds. As free C6 is unable to cross the BBB, all C6-related fluorescence in the brain of these zebrafish is attributable to NPs-mediated transport across this barrier. We also found that the fluorescence in the eyes and GI tract of these zebrafish larvae increased in a dose- and time-dependent manner, confirming the ability of these particles to be readily internalized and to cross the GI, blood-retinal, and blood–brain barriers in vivo.
 |
Figure 4 C6-NPs were used to treat 7 dpf zebrafish larvae. Blue, yellow and red arrows are used to mark the eyes, brain and digestive system, respectively. Scale bar: 500 μm. |
Analysis of C6-NPs Endocytosis in Zebrafish Larvae
We next sought to understand the in vivo mechanisms underlying NPs internalization by treating zebrafish larvae (7 dpf) with MβCD, HS, EIPA, or CPZ. This analysis revealed findings similar with the results of our in vitro analysis. We found that CPZ and MβCD had the most profound inhibitory effect on this activity (Figure S11 and S12), suggesting that clathrin- and caveolae-mediated NPs uptake are the primary mechanisms whereby these NPs are internalized in vivo.
Vivo GB-NPs Toxicity Evaluation
We next sought to explore the in vivo toxicity of GB-NPs by using them to treat zebrafish embryos at 6 hpf across a range of concentrations (25, 50, and 100 μg/mL). We then monitored these embryos for any abnormalities in development, blood flow, or other changes at 24, 48, 72, and 96 hpf. No changes in larval morphology were observed upon treatment with GB-NPs (Figure S13A), nor were the survival, hatching, or heart rates of the body length of these zebrafish altered significantly at 96 hpf (Figure S13B-E). Therefore, these findings suggested that GB-NPs do not cause significant toxicity in vivo or in vitro across the tested concentration range.
Histological Analysis
Lastly, we examined the potential impact of GB-NPs on organs following treatment. To that end, treated animals were euthanized and key organs were collected and H&E stained for histological analysis (Figure S14). No clear histological abnormalities were observed in these stained samples, suggesting that GB-NPs do not induce significant toxicity within healthy organs.
Analysis of GB-NPs Pharmacokinetics
We next sought to examine the pharmacokinetics of GB-NPs following their oral administration, with plasma concentration-time profile and pharmacokinetic parameters shown in Figure 5A and Table 1, respectively. We found that GB-NPs had a Cmax of 3.86 ± 0.33 μg/mL, which was 11.03-fold higher than associated with free GB (0.35 ± 0.04 μg/mL). This suggested that NPs loading of GB was able to significantly increase its systemic circulation owing to the advantageous particle sizes and surface properties of these NPs. In addition, we found these GB-NPs to have a T1/2 of 8.13 ± 0.84 h, which was 3.40-fold longer than that of free GB (2.39 ± 0.32 h), indicating that GB-NPs can reduce the rate of GB plasma elimination. Consistent with this, we found that the MRT0-t value for GB-NPs was increased by 1.58-fold relative to free GB. These increases in T1/2 and MRT0-t values were also in line with our in vitro findings, suggesting that these GB-NPs can prolong in vivo GB activity. It has been demonstrated that PEG-containing NPs could prolong drug residence in systemic circulation.41,42 We also determined that these GB-NPs had an AUC0-t of 47.81 ± 4.68 μg·h/mL, which was 13.94-fold higher than that of free GB (3.43 ± 0.37 μg·h/mL). This significant increase in oral GB bioavailability was attributable to the prolonged gradual release of GB from these NPs, shielding the drug from rapid excretion. Notably, following oral administration, GB-NPs has longer Tmax and MRT0-t and higher AUC0-t compared with the GB nanocrystals.25
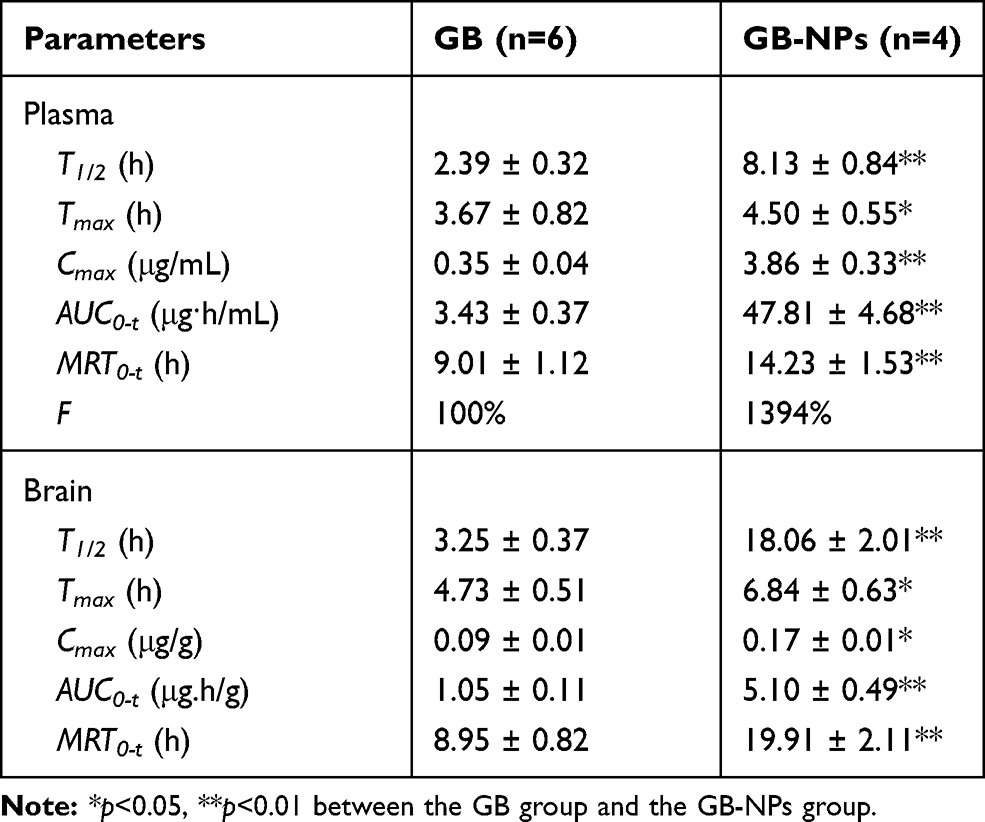 |
Table 1 GB-NPs Pharmacokinetic Parameters Following Oral Administration of GB-NPs |
 |
Figure 5 Plasma and brain pharmacokinetic profiles of GB and GB-NPs in rats. (A) Plasma concentration-time profile (n=6). (B) Brain concentration-time profile (n=4). |
We also quantified the cerebral GB concentration-time profile and associated pharmacokinetic parameters (Figure 5B and Table 1, respectively) after GB-NPs oral administration. We found that GB-NPs required a longer time (6.84 ± 0.63h) to reach Tmax relative to free GB (4.73 ± 0.51 h), but that GB-NPs were associated with a significantly higher Cmax than was free GB (0.17 ± 0.01 μg/g vs 0.09 ± 0.01 μg/g). In addition, the brain Tmax was significantly increased relative to the plasma value (6.84 ± 0.63 h vs 4.50 ± 0.55 h), consistent with the more gradual elimination of GB from the brain. In addition, the AUC0-t for GB-NPs in the brain was 4.86-fold higher than that for free GB (1.05 ± 0.11 μg·h/g vs 5.10 ± 0.49 μg·h/g). Together, these findings revealed that GB-NPs were able to reliably and rapidly transport GB into the brain. This is likely at least partially attributable to the higher plasma concentrations of GB in GB-NPs-treated animals (Figure 6), but the small size of these GB-NPs suggests that they can also effectively be transported across the BBB to combat PD.43,44
 |
Figure 6 Schematic overview of the mechanisms governing the absorption and accumulation of GB-NPs following oral uptake. |
Neuroprotective Effects of GB-NPs in a PD Mouse Model
In order to explore the therapeutic utility of GB-NPs, we utilized MPTP-treated mice as an acute model of PD (Figure S15), as these animals exhibit symptoms consistent with clinical PD including impaired motor functionality, TH+ neuron depletion, and low levels of dopamine within the brain.45 We utilized a pole test in order to assess how GB-NPs impacted bradykinesia in MPTP-lesioned mice, revealing that treatment with these NPs significantly reduced both turnaround time (t-turn) and total time to reach the bottom (t-total) after MPTP injection (Figure 7A and B). We also utilized a rotarod test to examine how these GB-NPs impacted the coordination and balance of these PD model mice (Figure 7C and D), revealing that they significantly increased the fall latency while simultaneously reducing the number of falls in treated animals. In an open field test aimed at examining spontaneous motor activity and exploratory behavior, we found that GB-NPs treatment was sufficient to reverse MPTP-induced reductions in animal movement, increasing both total distance traveled and average speed of travel (Figure 7E-G). As such, GB-NPs were able to improve the coordination, balance, and motor activity of MPTP PD model mice.
 |
Figure 7 The impact of GB-NPs on behavioral impairment in PD model mice. A pole test on days 3 and 5 post-MPTP injection (A, B) and a rotarod test on days 4 and 6 post-MPTP injection (C, D) were conducted (n=8). (E) Representative activity traces (green) derived from video tracking. Total distance traveled (F) and average speed (G) of treated mice (n=8). *p<0.05, **p<0.01, ***p<0.001 vs the Control group. #p<0.05, ##p<0.01, ###p<0.001 vs the GB group. |
The number of TH+ neurons in the substantia nigra pars compacta can serve as a gauge for DA neuron damage in this MPTP-induced PD model system. We were able to clearly detect evidence of GB-NPs-associated neuroprotection in vivo (Figure 8A), with treatment being associated with significant increases in numbers of TH+ neurons relative to free GB treatment (Figure 8B). MPTP treatment reduced levels of striatal dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA). However, treatment with GB-NPs was sufficient to reverse these MPTP-induced effects in these PD model mice (Figure 8C-E).
 |
Figure 8 The impact of GB-NPs in PD model mice. (A) Representative images of TH-stained sections of murine brain tissue. Scale bar: 100 μm. (B) Quantification of TH+ neurons (n=5). Striatal DA (C), DOPAC (D) and HVA (E) levels (n=4). *p<0.05 and **p<0.01 vs the MPTP group. #p<0.05 and ##p<0.01 vs the GB group. |
Conclusions
In this study, we developed NPs that were able to efficiently encapsulate substantial quantities of GB when formulated via an antisolvent precipitation method. We found that these GB-NPs were able to significantly enhance GB uptake when used to treat MDCK cells, with their internalization being mediated by nonspecific clathrin-mediated, lipid raft/caveolar, and micropinocytic uptake. Using zebrafish, we further demonstrated the ability of these NPs to transport across barriers including the chorion, the GI barrier, the BRB, and the BBB. In a pharmacokinetic study in rats, we were able to clearly demonstrate that these GB-NPs were associated with both enhanced brain uptakes of GB and higher levels of sustained GB in the plasma of treated animals. In a murine model of PD, we also found that administration of GB-NPs was sufficient to reduce behavioral deficits, attenuate DA depletion, and enhance DA, DOPAC, and HVA levels in treated animals. Our results thus offer evidence suggesting that GB-NPs can be utilized as a means of orally delivering GB in order to treat PD via improving its oral bioavailability and brain delivery.
Acknowledgments
We greatly acknowledge financial support from the Guangdong Basic and Applied Basic Research Foundation (2019B1515120043), the Guangdong Provincial Natural Science Foundation of China (2018A030310623), the Guangdong Provincial Medical Scientific Research Foundation of China (A2019027), the Research Fund of University of Macau (MYRG2019-00121-ICMS and MYRG2018-00207-ICMS), the National Natural Science Foundation of China (81774199 and 81973918), and the Guangzhou Science Technology and Innovation Commission Technology Research Projects (201805010005 and 201803010047).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas. All authors have drafted or written, or substantially revised or critically reviewed the article, and have agreed on the journal to which the article will be submitted. All authors have reviewed and agreed on all versions of the article before submission, during revision, the final version accepted for publication, and any significant changes introduced at the proofing stage. All authors agree to take responsibility and be accountable for the contents of the article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kalia LV, Lang AE. Parkinson’s disease. The Lancet. 2015;386(9996):896–912. doi:10.1016/S0140-6736(14)61393-3
2. Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016;15(12):1257–1272. doi:10.1016/S1474-4422(16)30230-7
3. Gaballah HH, Zakaria SS, Elbatsh MM, Tahoon NM. Modulatory effects of resveratrol on endoplasmic reticulum stress-associated apoptosis and oxido-inflammatory markers in a rat model of rotenone-induced Parkinson’s disease. Chem Biol Interact. 2016;251:10–16. doi:10.1016/j.cbi.2016.03.023
4. Zheng M, Liu C, Fan Y, Yan P, Shi D, Zhang Y. Neuroprotection by Paeoniflorin in the MPTP mouse model of Parkinson’s disease. Neuropharmacology. 2017;116:412–420. doi:10.1016/j.neuropharm.2017.01.009
5. Hua J, Yin N, Yang B, et al. Ginkgolide B and bilobalide ameliorate neural cell apoptosis in alpha-synuclein aggregates. Biomed Pharmacother. 2017;96:792–797. doi:10.1016/j.biopha.2017.10.050
6. Meng H, Li C, Feng L, et al. Effects of Ginkgolide B on 6-OHDA-induced apoptosis and calcium over load in cultured PC12. Int J Dev Neurosci. 2007;25(8):509–514. doi:10.1016/j.ijdevneu.2007.09.010
7. El-Ghazaly MA, Sadik NA, Rashed ER, Abd-El-Fattah AA. Neuroprotective effect of EGb761(R) and low-dose whole-body gamma-irradiation in a rat model of Parkinson’s disease. Toxicol Ind Health. 2015;31(12):1128–1143. doi:10.1177/0748233713487251
8. Wang P, Cao X, Chu Y, Wang P. Ginkgolides-loaded soybean phospholipid-stabilized nanosuspension with improved storage stability and in vivo bioavailability. Colloids Surf B Biointerfaces. 2019;181:910–917. doi:10.1016/j.colsurfb.2019.06.050
9. Yang P, Cai X, Zhou K, Lu C, Chen W. A novel oil-body nanoemulsion formulation of ginkgolide B: pharmacokinetics study and in vivo pharmacodynamics evaluations. J Pharm Sci. 2014;103(4):1075–1084. doi:10.1002/jps.23866
10. Ghosh S, Dungdung SR, Choudhury ST, Chakraborty S, Das N. Mitochondria protection with ginkgolide B-loaded polymeric nanocapsules prevents diethylnitrosamine-induced hepatocarcinoma in rats. Nanomedicine. 2014;9(3):441–456. doi:10.2217/nnm.13.56
11. Zhao J, Luo D, Zhang Z, et al. Celastrol-loaded PEG-PCL nanomicelles ameliorate inflammation, lipid accumulation, insulin resistance and gastrointestinal injury in diet-induced obese mice. J Control Release. 2019;310:188–197. doi:10.1016/j.jconrel.2019.08.026
12. Yang Z, Wang J, Liu S, et al. Defeating relapsed and refractory malignancies through a nano-enabled mitochondria-mediated respiratory inhibition and damage pathway. Biomaterials. 2020;229:119580. doi:10.1016/j.biomaterials.2019.119580
13. Grossen P, Witzigmann D, Sieber S, Huwyler J. PEG-PCL-based nanomedicines: a biodegradable drug delivery system and its application. J Control Release. 2017;260:46–60. doi:10.1016/j.jconrel.2017.05.028
14. Kubat P, Henke P, Raya RK, Stepanek M, Mosinger J. Polystyrene and Poly(ethylene glycol)-b-Poly(epsilon-caprolactone) nanoparticles with porphyrins: structure, size, and photooxidation properties. Langmuir. 2020;36(1):302–310. doi:10.1021/acs.langmuir.9b03468
15. Chen T, Liu W, Xiong S, et al. Nanoparticles mediating the sustained puerarin release facilitate improved brain delivery to treat Parkinson’s disease. ACS Appl Mater Interfaces. 2019;11(48):45276–45289. doi:10.1021/acsami.9b16047
16. Park J, Choi E, Shin S, et al. Nootropic nanocomplex with enhanced blood-brain barrier permeability for treatment of traumatic brain injury-associated neurodegeneration. J Control Release. 2018;284:152–159. doi:10.1016/j.jconrel.2018.06.021
17. Yang D, Liu D, Deng H, et al. Transferrin functionization elevates transcytosis of nanogranules across epithelium by triggering polarity-associated transport flow and positive cellular feedback loop. ACS Nano. 2019;13(5):5058–5076. doi:10.1021/acsnano.8b07231
18. Chen T, Li C, Li Y, et al. Small-Sized mPEG-PLGA nanoparticles of schisantherin a with sustained release for enhanced brain uptake and anti-parkinsonian activity. ACS Appl Mater Interfaces. 2017;9(11):9516–9527. doi:10.1021/acsami.7b01171
19. Chen T, Li C, Li Y, Yi X, Lee SM, Zheng Y. Oral delivery of a nanocrystal formulation of schisantherin a with improved bioavailability and brain delivery for the treatment of Parkinson’s disease. Mol Pharm. 2016;13(11):3864–3875. doi:10.1021/acs.molpharmaceut.6b00644
20. Roger E, Lagarce F, Garcion E, Benoit JP. Lipid nanocarriers improve paclitaxel transport throughout human intestinal epithelial cells by using vesicle-mediated transcytosis. J Control Release. 2009;140(2):174–181. doi:10.1016/j.jconrel.2009.08.010
21. Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123(5):1107–1117. doi:10.1083/jcb.123.5.1107
22. Wang S, Ouyang B, Aa J, et al. Pharmacokinetics and tissue distribution of ginkgolide A, ginkgolide B, and ginkgolide K after intravenous infusion of ginkgo diterpene lactones in a rat model. J Pharm Biomed Anal. 2016;126:109–116. doi:10.1016/j.jpba.2016.04.035
23. Huang D, Wang Z, Tong J, et al. Long-term changes in the nigrostriatal pathway in the MPTP mouse model of Parkinson’s disease. Neuroscience. 2018;369:303–313. doi:10.1016/j.neuroscience.2017.11.041
24. Liu J, Huang D, Xu J, et al. Tiagabine protects dopaminergic neurons against neurotoxins by inhibiting microglial activation. Sci Rep. 2015;5:15720. doi:10.1038/srep15720
25. Liu Y, Liu W, Xiong S, et al. Highly stabilized nanocrystals delivering Ginkgolide B in protecting against the Parkinson’s disease. Int J Pharm. 2020;577:119053. doi:10.1016/j.ijpharm.2020.119053
26. Xiong S, Liu W, Li D, et al. Oral delivery of puerarin nanocrystals to improve brain accumulation and anti-Parkinsonian efficacy. Mol Pharm. 2019;16(4):1444–1455. doi:10.1021/acs.molpharmaceut.8b01012
27. Xin H, Chen L, Gu J, et al. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(varepsilon-caprolactone) nanoparticles: in vitro and in vivo evaluation. Int J Pharm. 2010;402(1–2):238–247. doi:10.1016/j.ijpharm.2010.10.005
28. Xin H, Jiang X, Gu J, et al. Angiopep-conjugated poly(ethylene glycol)-co-poly(ε-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials. 2011;32(18):4293–4305. doi:10.1016/j.biomaterials.2011.02.044
29. Eom HJ, Choi J. Clathrin-mediated endocytosis is involved in uptake and toxicity of silica nanoparticles in Caenohabditis elegans. Chem Biol Interact. 2019;311:108774. doi:10.1016/j.cbi.2019.108774
30. Wang J, Gou W, Kim DS, Strange C, Wang H. Clathrin-mediated Endocytosis of Alpha-1 antitrypsin is essential for its protective function in islet cell survival. Theranostics. 2019;9(13):3940–3951. doi:10.7150/thno.31647
31. Zhao S, Dai W, He B, et al. Monitoring the transport of polymeric micelles across MDCK cell monolayer and exploring related mechanisms. J Control Release. 2012;158(3):413–423. doi:10.1016/j.jconrel.2011.12.018
32. Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15(1):176–188. doi:10.1091/mbc.e03-05-0319
33. He B, Lin P, Jia Z, et al. The transport mechanisms of polymer nanoparticles in Caco-2 epithelial cells. Biomaterials. 2013;34(25):6082–6098. doi:10.1016/j.biomaterials.2013.04.053
34. Muley H, Fado R, Rodriguez-Rodriguez R, Casals N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem Pharmacol. 2020;177:113959. doi:10.1016/j.bcp.2020.113959
35. Salnikov V, Lukyánenko YO, Frederick CA, Lederer WJ, Lukyánenko V. Probing the outer mitochondrial membrane in cardiac mitochondria with nanoparticles. Biophys J. 2007;92(3):1058–1071. doi:10.1529/biophysj.106.094318
36. Wallace KN, Akhter S, Smith EM, Lorent K, Pack M. Intestinal growth and differentiation in zebrafish. Mech Dev. 2005;122(2):157–173. doi:10.1016/j.mod.2004.10.009
37. Lu X, Long Y, Lin L, Sun R, Zhong S, Cui Z. Characterization of zebrafish Abcc4 as an efflux transporter of organochlorine pesticides. PLoS One. 2014;9(12):e111664. doi:10.1371/journal.pone.0111664
38. Jia HR, Zhu YX, Duan QY, Chen Z, Wu FG. Nanomaterials meet zebrafish: toxicity evaluation and drug delivery applications. J Control Release. 2019;311-312:301–318. doi:10.1016/j.jconrel.2019.08.022
39. Jeong JY, Kwon HB, Ahn JC, et al. Functional and developmental analysis of the blood-brain barrier in zebrafish. Brain Res Bull. 2008;75(5):619–628. doi:10.1016/j.brainresbull.2007.10.043
40. Li Y, Miao X, Chen T, et al. Zebrafish as a visual and dynamic model to study the transport of nanosized drug delivery systems across the biological barriers. Colloids Surf B Biointerfaces. 2017;156:227–235. doi:10.1016/j.colsurfb.2017.05.022
41. Cherukula K, Uthaman S, Park IK. “Navigate-dock-activate” anti-tumor strategy: tumor micromilieu charge-switchable, hierarchically activated nanoplatform with ultrarapid tumor-tropic accumulation for trackable photothermal/chemotherapy. Theranostics. 2019;9(9):2505–2525. doi:10.7150/thno.33280
42. Kuplennik N, Lang K, Steinfeld R, Sosnik A. Folate receptor alpha-modified nanoparticles for targeting of the central nervous system. ACS Appl Mater Interfaces. 2019;11(43):39633–39647. doi:10.1021/acsami.9b14659
43. Liu D, Lin B, Shao W, Zhu Z, Ji T, Yang C. In vitro and in vivo studies on the transport of PEGylated silica nanoparticles across the blood-brain barrier. ACS Appl Mater Interfaces. 2014;6(3):2131–2136. doi:10.1021/am405219u
44. Yan XQ, Shi YL, Jiang QF, Ping GF, ZJ D. Design of amphiphilic PCL-PEG-PCL block copolymers as vehicles of Ginkgolide B and their brain-targeting studies. J Biomater Sci Polym Ed. 2017;28(14):1497–1510. doi:10.1080/09205063.2017.1332470
45. Wang LY, Yu X, Li XX, et al. Catalpol exerts a neuroprotective effect in the MPTP mouse model of Parkinson’s disease. Front Aging Neurosci. 2019;11:316. doi:10.3389/fnagi.2019.00316
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.