Back to Journals » International Journal of Nanomedicine » Volume 15
Polydopamine Nanoparticles Camouflaged by Stem Cell Membranes for Synergistic Chemo-Photothermal Therapy of Malignant Bone Tumors
Authors Zhang M, Zhang F, Liu T, Shao P, Duan L, Yan J, Mu X ![]() , Jiang J
, Jiang J
Received 19 September 2020
Accepted for publication 20 November 2020
Published 14 December 2020 Volume 2020:15 Pages 10183—10197
DOI https://doi.org/10.2147/IJN.S282931
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Meng Zhang, Fuqiang Zhang, Te Liu, Pu Shao, Lian Duan, Jun Yan, Xupeng Mu,* Jinlan Jiang*
Scientific Research Center, China-Japan Union Hospital of Jilin University, Changchun, Jilin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jinlan Jiang; Xupeng Mu
Scientific Research Center, China-Japan Union Hospital of Jilin University, No. 126 Xiantai Street, Changchun 130033, Jilin, People’s Republic of China
Email [email protected]; [email protected]
Purpose: Nanoparticle (NP)-based chemo-photothermal therapy (CPT) has been shown to be a promising non-invasive approach for antitumor treatment. However, NPs must overcome the limitations of opsonization, clearance of the reticuloendothelial system, and ineffective targeting of tumor tissue sites. To solve these problems, stem cell membrane (SCM)-camouflaged polydopamine nanoparticles (PDA@SCM NPs) carrying the hydrophobic anticancer drug 7-ethyl-10-hydroxycamptothecin (SN38) were constructed for CPT of malignant bone tumors.
Methods: We developed umbilical-cord mesenchymal stem cell membrane-coated polydopamine nanoparticles encapsulating SN38 (PDA-SN38@SCM NPs) as an efficient tumor-targeting drug-delivery platform for CPT of malignant bone tumors. We characterized PDA@SCM NPs and evaluated the biocompatibility and anti-phagocytosis properties of PDA@SCM NPs. The antitumor activity of PDA-SN38@SCM NPs was evaluated in MG63 lines and an MG63 xenograft model in mice.
Results: Synthesized PDA-SN38@SCM NPs retained an excellent photothermal effect after SN38 loading. The drug release of PDA-SN38@SCM NPs could be triggered by near-infrared irradiation and an acidic stimulus. PDA@SCM NPs exhibited lower nonspecific macrophage uptake, longer retention in blood, and more effective accumulation at tumor sites than that shown by PDA NPs. Confocal laser scanning microscopy (CLSM) and flow cytometry showed that MG63 cells took up more PDA-SN38@SCM NPs than PDA-SN38 NPs. In vitro and in vivo antitumor studies demonstrated the outstanding performance of PDA-SN38@SCM NPs in synergistic CPT for bone tumors.
Conclusion: PDA-SN38@SCM NPs demonstrated an extraordinary synergistic CPT effect and could be a promising strategy for the treatment of malignant bone tumors.
Keywords: stem cell membrane, polydopamine nanoparticles, chemo-photothermal therapy
Introduction
In recent years, nanotechnology has become increasingly prevalent in tumor research owing to its efficacy and safety compared with current therapies.1–3 Extensive efforts have been made in the development of NP-based drug-delivery systems (DDSs) for minimizing side effects and improvement of therapeutic efficacy for targeting tumors.4,5 NPs loaded with various antitumor drugs can alter the biodistribution and pharmacokinetics of drugs. Such NPs can accumulate at tumor sites via the enhanced permeability and retention (EPR) effect.6,7 Due to various biological barriers at systemic, tissue, cell, and subcellular levels resulting after an intravenous injection, the dysfunction of tumor vessels and high tissue pressure of the tumor itself, the EPR effect is insufficient to complete effective delivery of drugs in vivo.8,9 With increased understanding of the interactions between NPs and the tumor microenvironment, development of functionalized NPs for active targeting of cancer is necessary. Functionalized NPs can increase the efficiency of delivery of antitumor drugs.10–12 Typically, the size and surface morphology of synthetic NPs is substantially similar to that of pathogens such as bacteria and viruses, so they can be subjected to similar clearance mechanisms mediated by the innate immune system.13 Traditionally, polyethylene glycol (PEG) is reacted with the NP surface, which reduces the interaction of the NP with its surrounding environment and inhibits uptake in the reticuloendothelial system.14 Recent observations have indicated that PEG can decelerate opsonization by reducing nonspecific interactions between NPs and the biosystem. However, PEG can trigger another type of immune response resulting in anti-PEG antibodies, which may adversely affect multiple administrations of NPs as well as their therapeutic potential.15,16 Therefore, rational design of advanced NPs to overcome physical and biological barriers as well as to bridge the gap between synthetic NPs and biological entities to achieve the desired biodistribution and therapeutic effects remains a challenge.
In recent years, due to the challenges faced by synthetic functionalized NPs, surface modification using a top-down approach by coating with naturally derived bio-membranes represents an emerging strategy for developing bioinspired nanotechnology, which has garnered interest in nanomedicine.17,18 Zhang and colleagues were the first to report that coating poly(lactic-co-glycolic acid) (PLGA) nanoparticles with red blood cell (RBC) membranes can retain membrane proteins, thereby resulting in improved stability of NPs and prolonged circulation in blood.19 These NPs “disguised” by a cell membrane consist of two components: a synthetic NP core and a layer of natural cell membrane. Coating NPs with membranes derived from different types of cells or biological vesicles results in a molecule that is considered “self” and, therefore, has higher biocompatibility and lower toxicity. This membrane-based DDS can be produced by a simple method, and the loss of membrane protein can be minimized. To date, pioneering research in this field has involved several types of membranes, from cells such as RBCs, platelets, white blood cells, cancer cells, and stem cells to construct “bionic” core-shell NPs for cancer treatment.20–27
Among the various types of cells, mesenchymal stem cells (MSC) have demonstrated hypo-immunogenicity and migration towards tumor microenvironments. Although the specific mechanism of the tumor tropism of MSCs is not clear, studies have demonstrated use of stem cells as an anti-cancer DDS to improve the therapeutic effect on tumors.28–30 However, certain studies have observed that MSCs may promote the progression and metastasis of tumor cells, which limits their application in DDSs. Similar to MSC-based DDSs, coating SCM on NPs is regarded as a safer DDS akin to stem cells with low immunogenicity and tumor targeting. Most importantly, such a DDS does not require complex production of targeting molecules and their incorporation into passive carriers.31,32 It is a simpler and clinically more relevant method for avoiding clearance by the system of mononuclear phagocytes, overcoming vascular barriers, and actively targeting tumor tissue compared with that using existing nano-drug delivery vehicles.
Chemotherapy is one of the most commonly employed cancer treatments. However, administration of chemotherapy alone most likely will not eliminate the tumor owing to drug resistance and tumor heterogeneity. It is often necessary to combine multiple therapies with different modes of action. Photothermal therapy (PTT) is a novel non-invasive type of cancer treatment. PTT employs the property of photothermal agents accumulating in tumors and reacting to near-infrared (NIR) light by converting light energy into heat energy for killing tumor cells directly at >45°C.33 Recently, several NIR-response DDSs have been used in a combination of CPT to treat cancer cells through thermal stimuli releasing chemotherapeutic drugs and photothermal conversion.34,35 CPT has been considered to be a promising cancer treatment strategy that can minimize use of photothermal agents and chemical drugs to reduce dose-related side effects, as well as the tissue damage and inflammation caused by hyperthermia.36,37
We developed umbilical-cord mesenchymal stem cell (UCMSC) membrane-coated polydopamine NPs encapsulating SN38 as an efficient tumor-targeting DDS for CPT of malignant bone tumors (Figure 1). Polydopamine nanoparticles (PDA NPs) exhibit excellent biocompatibility and high photothermal performance, biodegrade within the body, and can be loaded with various anti-cancer drugs for controlled release and DDSs.38 SN38 is a hydrophobic drug that can be loaded efficiently into PDA mediated by π-π stacking interactions.37 The SCM not only camouflaged PDA-SN38 NPs but also enhanced their active targeting abilities. SN38 release of PDA-SN38@SCM NPs exhibited pH-dependence/photothermal dependence, in which drug release was accelerated in the acidic tumor microenvironment and irradiated by a laser at 808 nm. Compared with uncoated PDA-SN38 NPs, PDA-SN38@SCM NPs mimicked the cancer-targeting ability of MSCs better in vitro and showed increased accumulation at the tumor site in vivo. PDA-SN38@SCM NPs demonstrated an outstanding synergistic effect in CPT for malignant bone tumors.
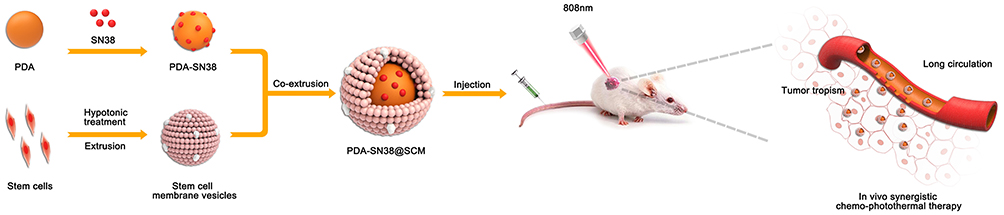 |
Figure 1 Preparation of PDA-SN38@SCM NPs and the synergistic chemo-photothermal treatment of malignant bone tumor. |
Materials and Methods
Materials
Dopamine hydrochloride (purity = 99%), SN38 (98%), dimethyl sulfoxide (DMSO), ammonia (28–30%), 3H-Indolium,5-[[[4-(chloromethyl)benzoyl]amino]methyl]-2-[3-(1,3-dihydro-3,3-dimethyl-1-octadecyl-2H-indol-2-ylidene)-1-propenyl]-3,3-dimethyl-1-octadecyl-,chloride (CM-Dil), fluorescein isothiocyanate (FITC), phenylmethylsulfonyl fluoride (PMSF), deionized water (pH 7.0) and absolute ethanol (99.8%) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM) with high glucose, Minimum Essential Medium α (MEM α), and fetal bovine serum (FBS) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). RIPA lysis buffer, a bicinchoninic acid (BCA) protein kit, enzyme-linked immunosorbent assay (ELISA) kit for tumor necrosis factor (TNF)-α, 4% paraformaldehyde solution, 4′,6-diamidino-2-phenylindole (DAPI), Triton X-100, and Coomassie Brilliant Blue were purchased from Beyotime (Shanghai, China). Other reagents were of analytical grade and purchased from Beijing Chemical Reagents Company (Beijing, China) unless stated otherwise.
Cell Culture
MG63, L929, and RAW246.7 cell lines and UCMSCs were purchased from American Type Culture Collection (Manassas, VA, USA). MG63, L929, and RAW246.7 cells were cultured in DMEM supplemented with 10% FBS in a humidified atmosphere of 5% CO2 at 37°C. UCMSCs were cultured in DMEM α supplemented with 10% FBS in a humidified atmosphere of 5% CO2 at 37°C.
Ethical Approval of the Study Protocol
Animals were treated according to the ethical guidelines set by Jilin University (Changchun, China) after obtaining approval from the Animal Welfare and Research Ethics Committee of Jilin University. Animal experiments were carried out following internationally accepted guidelines for animal care (EEC Directive of 1986; 86/609/EEC).
Synthesis and Characterization of PDA NPs
PDA NPs were prepared in accordance with methods described previously.33 Briefly, 16 mL of ethanol and 36 mL of deionized water were mixed with medium-speed magnetic stirring at room temperature, followed by the addition of 0.8 mL of an aqueous solution of ammonia with stirring for 30 min. Dopamine hydrochloride (50 mg/mL, 4 mL) in deionized water was injected into the reaction solution. The color of the solution changed gradually from pale-brown to dark-brown, and the reaction was allowed to proceed for 12 h. Products were obtained by centrifugation at 22,136 × g for 30 min at room temperature and washed with deionized water 3 times. The concentration of PDA NPs was determined by weight after lyophilization. FITC-labeled PDA NPs were prepared by mixing 5 mL of PDA NPs (5 mg/mL) with 10 mL of FITC (0.5 mg/mL) by magnetic stirring in the dark for 24 h. Then, the mixture was centrifuged (22,136 × g, 15 min, room temperature) and the pellet was washed with phosphate-buffered saline (PBS) 3 times to remove free FITC. The structure of PDA NPs was observed using a transmission electron microscope (TEM, Jeol, Tokyo, Japan). The hydrodynamic diameter and surface charge were measured by dynamic light scattering (DLS) using a Zetasizer Nano-ZS instrument (Malvern Instruments, Malvern, UK).
Preparation of SCM-Derived Vesicles
SCM-derived vesicles were prepared according to the previously published protocol with a few modifications.39 UCMSCs were maintained in DMEM α supplemented with 1% penicillin–streptomycin and 10% FBS. Cells were harvested, washed with PBS, and resuspended in cold hypotonic lysis buffer (1 mM NaHCO3, 0.2 mM EDTA, and 1 mM PMSF) overnight at 4°C. Cell lysates were centrifuged at 885 × g for 5 min at 4°C. The supernatant was centrifuged at 22,136 × g for 30 min at 4°C to obtain a cell-membrane precipitate, which was resuspended in PBS. SCM-derived vesicles were prepared by physically extruding cell-membrane through a 400-nm polycarbonate membrane (14 times) using a mini-extruder (Avanti Polar Lipid, Alabaster, AL, USA).
Preparation and Characterization of Polydopamine Nanoparticles Camouflaged by Stem Cell Membrane (PDA@SCM NPs)
Previously prepared PDA NPs (100 µg/mL, 50 µL) and 500 µL of SCM-derived vesicles (1 mg/mL) were mixed and sonicated for 5 min in a bath sonicator (53 kHz, 100 W) at 4°C. The mixture solution was co-extruded through a 200-nm polycarbonate membrane 14 times. Excess SCM vesicles were removed by centrifugation at 22,136 × g for 30 min at room temperature and washed with deionized water 3 times. PDA@SCM NPs were re-dispersed in PBS for subsequent experiments. The structure of PDA@SCM NPs was examined using a TEM. PDA@SCM NPs (100 µg/mL) were placed on a grid to dry. NPs were negatively stained with one drop of 3% phosphotungstic acid on the grid. Then, the grid was dried and observed using a TEM. The hydrodynamic diameter and surface charge were measured with the Zetasizer Nano-ZS instrument.
Characterization of SCM Proteins
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to analyze membrane proteins. Proteins present in SCM vesicles and PDA@SCM NPs were lysed with RIPA lysis buffer. The BCA protein kit was used to quantify the concentration of SCM proteins. After mixing with the sample loading buffer for SDS-PAGE and boiling for 10 min, samples (40 µg/well) were loaded on 10% SDS-PAGE gels and run at 120 V for 2 h. The SDS-PAGE gel was stained with Coomassie Brilliant Blue and de-stained in acetic acid overnight before imaging.
Colocalization of PDA@SCM NPs
CM-Dil-labeled SCM vesicles and FITC-labeled PDA NPs were added to MG63 cells. After incubation of PDA@SCM NPs with MG63 cells for 0.5 h, cells were washed 3 times with PBS, followed by staining with DAPI. Cells were observed by CLSM using a FluoView 500 (Olympus, Tokyo, Japan) system. Digital images of green, blue, and red fluorescence were acquired under FITC, DAPI, and CM-Dil channels, respectively.
Drug Loading
First, 1.5 mL of PDA (5.4 mg/mL) and 2.5 mg SN38 was dissolved in a solution of 10 mL of deionized water and 1 mL of DMSO and stirred magnetically for 24 h at room temperature. Undissolved SN38 crystals were removed by low-speed centrifugation (394 × g, 5 min, room temperature). The supernatant was collected and centrifuged at 22,136 × g for 10 min at room temperature to precipitate PDA-SN38. The resulting PDA-SN38 precipitate was washed with deionized water 3 times. PDA-SN38 NPs and SCM vesicles were sonicated for 5 min before co-extrusion with SCM vesicles using a polycarbonate membrane (14 times). The amount of loaded SN-38 was quantified with a UV–visible spectrophotometer by measuring absorbance at 365 nm (Synergy 2; BioTek Instruments, Winooski, VT, USA)
Photothermal Performance
To detect the photothermal-conversion capacity of PDA NPs and PDA@SCM NPs, a series of aqueous solutions of different concentrations comprising PDA NPs and PDA@SCM NPs (0, 50, and 100 μg/mL) were irradiated with a NIR laser (LEO Photonics, Beijing, China) at 808 nm at an intensity of 0.6 W/cm2 for 5 min and the temperature was measured (with a thermometer) every 25 s. The thermal stability of PDA NPs and PDA@SCM NPs was evaluated by irradiation with the NIR laser at 0.6 W/cm2 for 5 min and then cooling down to room temperature. Five cycles of irradiation were undertaken.
Stability of PDA@SCM NPs
To determine the stability of PDA@SCM NPs in vitro, NPs were added to PBS and 50% FBS for 14 days at 37°C. The change in particle size and size distribution was measured by DLS.
Drug Release
PDA-SN38@SCM NPs of volume 0.5 mL (100 μg/mL; drug-loading percentage = 6.36%) were suspended in PBS of different pH, sealed in a dialysis bag (molecular-weight cutoff = 4000 Da), and immersed in 5 mL of PBS with pH 5.0 or 7.4 at 37°C. PBS (1 mL) was collected at different time points and analyzed using the UV−visible spectrometer to evaluate drug release. After each measurement, 1 mL of fresh PBS was added into PBS to keep the volume constant. For photo-stimulated drug release, PDA-SN38@SCM NPs were suspended in deionized water and irradiated with the 808-nm NIR laser (0.6 W/cm2) at predetermined time intervals. The supernatant was collected by centrifugation (22,136 × g, 10 min, room temperature). The amount of released SN38 was measured at different intervals by the UV−visible spectrometer.
Hemolysis
Fresh anticoagulated blood from mice (Vital River Company, Beijing, China) was centrifuged at 221 × g for 10 min at room temperature to collect the RBC pellet. RBCs in the pellet were washed repeatedly with PBS until a colorless supernatant was observed after centrifugation. Next, 0.5 mL of a 4% erythrocyte suspension was added to 0.5 mL of PDA@SCM NPs solutions of different concentrations so that the final concentrations obtained were 25, 50, 100, 200, and 400 μg/mL, respectively. Then, 0.5 mL of the RBC suspension was added to 0.5 mL of deionized water (positive control); 0.5 mL of PBS was added to 0.5 mL of the RBC suspension (negative control). After incubation for 4 h at room temperature, the supernatant was obtained by centrifugation, and absorbance was measured at 540 nm with a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA).
Cytotoxicity
The toxicity of PDA@SCM NPs against L929 cells was analyzed by the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. L929 cells were seeded in 96-well plates (1×104 cells per well) and incubated overnight under an atmosphere of 5% CO2 at 37°C. PDA NPs and PDA@SCM NPs of different concentrations (0, 25, 50, 100, 200, 400, 500, 600 μg/mL) were added to the cell-culture medium and incubated with cells for 24 h at 37°C. An MTT solution (0.5 mg/mL) was added to the medium in each well and incubation allowed for an additional 4 h. Then, the supernatant was removed and 150 µL of DMSO was added to each well. After shaking the 96-well plate evenly for 10 min, absorbance was measured at 490 nm with a microplate reader.
Uptake of PDA@SCM NPs by Macrophages
RAW246.7 macrophages (5×104) were seeded in 24-well plates and cultured in DMEM with 10% FBS and incubated in an atmosphere of 5% CO2 overnight at 37°C. FITC-labeled PDA NPs (100 μg/mL) and PDA@SCM NPs (100 μg/mL) were added to the culture medium, and cultured for an additional 12 h. Then, macrophages were washed with PBS three times for complete removal of the free NPs in the medium. Macrophages were digested and centrifuged, and flow cytometry using an FC500 system (Beckman Coulter, Fullerton, CA, USA) was used to detect NP phagocytosis by macrophages.
To determine the release of inflammatory factors from macrophages induced by NPs, PBS was selected as a negative control. After inoculation of macrophages in a 24-well plate for 24 h, LPS (1 μg/mL), PDA NPs (100 μg/mL), or PDA@SCM NPs (100 μg/mL) were added to the medium and the culture continued for an additional 12 h. Supernatants were collected carefully. The TNF-α content in supernatants was measured using the ELISA kit.
Targeted Delivery of SN38 in vitro
CLSM was used to observe the ability of SCMs to camouflage NPs to target cancer cells in vitro. MG63 cells (1×104) were seeded in 35-mm glass-bottom dishes and cultured for 24 h. Free SN38, PDA-SN38 NPs, or PDA-SN38@SCM NPs (equivalent concentrations of PDA and PDA@SCM = 39 μg/mL; equivalent SN38 concentration = 2.5 μg/mL) were added to the culture medium. After 4 h of co-cultivation, free SN38 was removed, washed with PBS 3 times, and fixed with 4% paraformaldehyde solution for 20 min at room temperature. Afterwards, cells were permeabilized with 0.1% (v/v) Triton X-100 in PBS for 5 min at room temperature, washed twice with PBS, and then blocked for 30 min in PBS containing 1% (w/v) bovine serum albumin. Propidium iodide (PI) was used to stain nuclei for 0.5 h followed by washing cells 3 times. SN38 and PI were excited at 405 nm and 603 nm, respectively, for observation by CLSM.
Flow cytometry was used to detect the phagocytosis of free SN38, PDA-SN38 NPs, and PDA-SN38@SCM NPs by MG63. MG63 cells were cultured in a 6-well plate (2×105 cells per well) for 12 h. Then, the medium was replaced with fresh medium containing PDA-SN38@SCM NPs or PDA-SN38 NPs. Cells were cultured further for 4 h, and then washed with PBS 3 times to remove excess NPs. Flow cytometry was employed to analyze the difference in the number of NPs on cancer cells.
Killing of Cancer Cells in vitro
MG63 cells were seeded in 96-well plates (1×104 cells per well) for 24 h. To evaluate the synergistic effects of CPT, cells were incubated with free SN38, PDA-SN38 NPs or PDA-SN38@SCM NPs (equivalent concentration for PDA and PDA@SCM = 80 μg/mL; equivalent SN38 concentration = 5 μg/mL). After incubation for 4 h, cells were irradiated with the 808-nm laser (0.6 W/cm2) for 5 min. Twenty-four hours later, the cell culture medium was removed, and 100 μL of MTT solution (0.5 mg/mL) was added to the culture medium. After incubation for 4 h, the supernatant was removed carefully and formazan crystals dissolved in 150 µL of DMSO. Cell viability was analyzed by measuring absorbance at 490 nm per well with a microplate reader. The antitumor efficacy was further studied by live/dead staining. MG63 cells were seeded in 6-well plates (2×104 cells per well) for 24 h. After different treatments, the cell culture medium was discarded. Then, fresh medium containing calcein-AM (5 μg/mL) and PI (15 μg/mL) was added. After incubating for 20 min, the cells were washed with PBS and imaged on an inverted fluorescence microscope (OlympusIX83, Tokyo, Japan).
Pharmacokinetics and Biodistribution of PDA@SCM NPs in vivo
To study the circulation time of NPs in blood as well as distribution in major organs and tumor tissues in the body of animals, 100 μL FITC-labeled PDA NPs (2 mg/mL) or FITC-labeled PDA@SCM NPs (2 mg/mL) were injected into the body of BALB/c nude mice with MG63 tumor xenografts via the tail vein. At predetermined intervals (1, 2, 4, 8, 12, 16, 24 h), blood was collected from the orbit. The fluorescence intensity of blood was analyzed at each time point. Tumors and main organs (heart, liver, spleen, lungs, kidneys) were collected and photographed under the IVIS (In Vivo Imaging Instruments; PerkinElmer, Waltham, MA, USA) spectrum imaging system at an excitation wavelength of 650 nm and emission wavelength of 665 nm. Then, major organs and tumors were weighed and homogenized in lysis buffer. Afterwards, the lysate of each tissue sample was centrifuged at 142 × g for 5 min at room temperature, and the supernatant was subjected to measurement of fluorescence intensity.
Anticancer Efficacy in vivo
Female BALB/c nude mice (4–6 weeks; mean weight, 20 g) were purchased from Vital River Company (Beijing, China) and maintained under specific pathogen-free conditions. MG63 cells (100 μL; 1×107/mL) were transplanted into the dorsal area of mice. After 7 days, when the MG63 xenograft tumor reached ~100 mm3, mice were divided randomly into eight groups (n =3). Treatment was administered with the following substances in the eight groups: (1) PBS; (2) 808-nm laser irradiation; (3) SN38; (4) PDA NPs plus 808-nm laser irradiation; (5) PDA-SN38 NPs; (6) PDA-SN38 NPs plus 808-nm laser irradiation; (7) PDA-SN38@SCM NPs and (8) PDA-SN38@SCM NPs plus 808-nm laser irradiation.
PBS (100 µL) containing SN38 (2.5 mg/kg), PDA NPs (39 mg/kg), PDA-SN38 NPs (39 mg/kg), or PDA-SN38@SCM NPs were injected into mice via the tail vein. After 24 h, mice were anesthetized with 5% chloral hydrate and irradiated with the 808-nm laser (0.6 W/cm2) for 5 min. After PTT, the tumor volume and body weight were measured every other day. After 14 days, mice were euthanized, and the main organs were collected for hematoxylin and eosin (H&E) staining. The tumor volume (V) was calculated as (tumor length) × (tumor width)2/2. The relative tumor growth was calculated as V/V0 (V and V0 are the tumor volume on day-14 and day-0, respectively).
Statistical Analyses
Data are the mean ± SD. Statistical analyses were carried out using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. P < 0.05 was considered significant.
Results and Discussion
Preparation and Characterization of PDA@SCM NPs
PDA NPs were synthesized using the solution-oxidation method.38 The TEM image in Figure 2A demonstrated that the mean size of PDA NPs was ~128 nm. SCM vesicles derived from natural stem cells were coated on the surface of PDA NPs by an extrusion method reported previously to prepare PDA@SCM NPs.39 Briefly, stem cells were subjected to hypotonic treatment and centrifugation to yield “MSC ghosts” (Figure S1). The latter were sonicated and extruded physically to obtain SCM vesicles of diameter ~140 nm (Figure 2A). Finally, the resulting SCM vesicles were mixed with synthetic PDA NPs and extruded repeatedly through a 200-nm polycarbonate porous membrane. This extrusion promoted the effective fusion of the SCM with the NP surface, thereby resulting in PDA@SCM NPs. TEM images demonstrated that negatively stained PDA@SCM NPs with potassium phosphotungstate possessed a core-shell structure. The diameter of the PDA core was ~128 nm, and the thickness of the outer-membrane shell was ~10 nm, which verified the successful combination of SCMs with PDA NPs (Figure 2A). Consistently, DLS measurements showed that the mean hydrodynamic diameter of PDA@SCM NPs increased slightly from ~181 nm to ~191 nm following coating with SCMs (Figure 2B). This increase in size corresponded to the size of cell membranes ranging from 5 nm to 10 nm.32,40 After coating with the SCM, the zeta potential of PDA NPs changed from −43.5 to −28.4 mV, which was comparable with that of natural stem-cell vesicles (−32.5 mV), thereby confirming successful coating with the SCM (Figure 2C). To verify that the prepared PDA@SCM NPs contained SCM proteins, the protein content of the SCM and PDA@SCM NPs was measured in parallel using SDS-PAGE (Figure 2D). Compared with the SCM alone, cell-membrane proteins were retained in PDA@SCM NPs after extrusion. These results indicated that most of the membrane proteins were retained during preparation and that the PDA NPs were coated by SCMs.41 Furthermore, CLSM demonstrated that the green fluorescent signals (FITC) from the cores and red fluorescence (CM-Dil) from the membrane showed a high degree of overlap (Figure 2E). Hence, the core-shell structure of PDA@SCM NPs was stable after internalization by tumor cells. To determine the stability of PDA@SCM NPs, they were suspended in PBS and FBS at a concentration of 1 mg/mL for 14 days and the particle size was monitored by DLS. The change in particle size of PDA@SCM NP was negligible (Figure 2F). Collectively, these results indicated that the PDA NPs were coated by SCMs and exhibited good physiological stability.
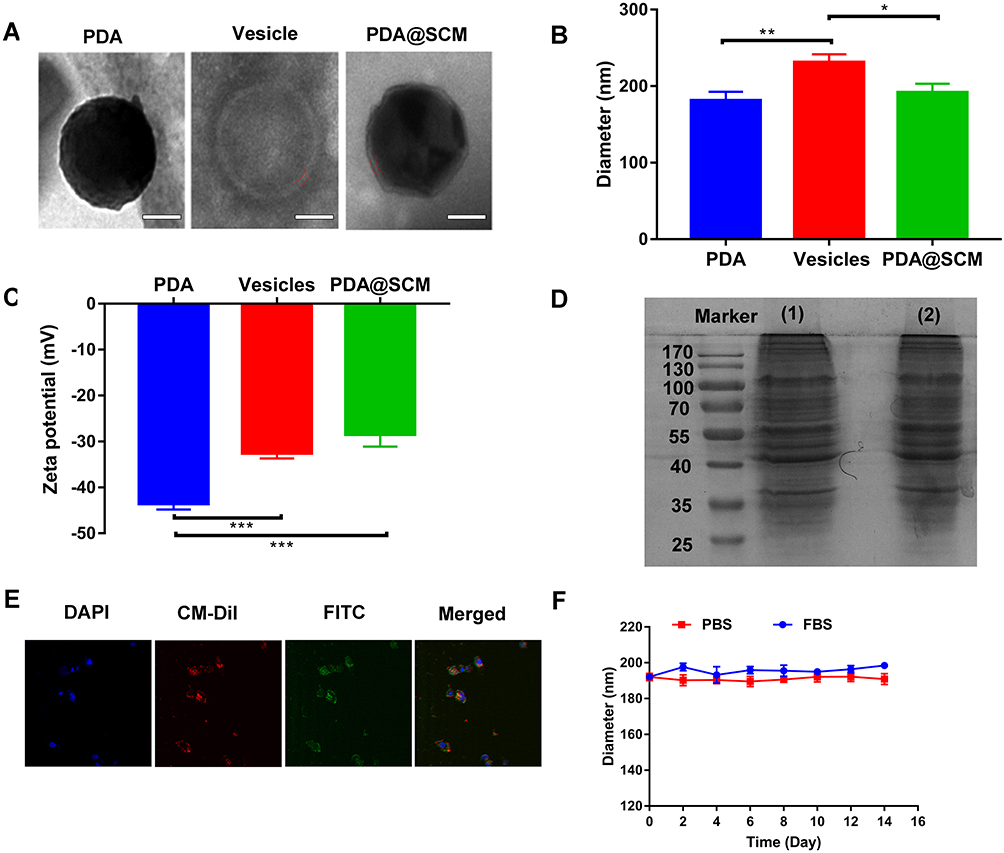 |
Figure 2 Characterization of PDA@SCM NPs. (A) TEM images of synthesized PDA NP (left), SCM vesicle (middle) and PDA@SCM NPs (right). Scale bar = 50 nm. (B) Hydrodynamic size of PDA NPs, SCM vesicles, and PDA@SCM NPs. (C) Surface charge of PDA NPs, SCM vesicles and PDA@SCM NPs. (D) SDS-PAGE-based protein analyses of (1) SCM and (2) PDA@SCM NPs. (E) CLSM images of PDA@SCM NPs illustrating colocalization of PDA NPs (FITC channel) and SCMs (CM-Dil channel). Scale bar = 50 μm. (F) Stability of PDA@SCM NPs in PBS and FBS by measurement of particle size. *p < 0.05, **p < 0.01, ***p < 0.001. |
Biocompatibility and Immune Evasion Ability of PDA@SCM NPs
The toxicity of PDA@SCM NPs against L929 cells was examined first using MTT. When the concentration increased from 25 μg/mL to 500 μg/mL, neither PDA NPs nor PDA@SCM NPs exerted toxicity against L929 cells (Figure 3A). For concentrations >500 μg/mL, PDA@SCM NPs were less toxic than PDA NPs, indicating that the camouflage property attributed by SCMs improved the biocompatibility of PDA NPs. The results of the hemolysis test demonstrated that PDA@SCM NPs did not rupture RBCs within 4 h of co-incubation with blood, indicating that PDA@SCM NPs exhibited good compatibility with blood (Figure 3B, Figure S2).
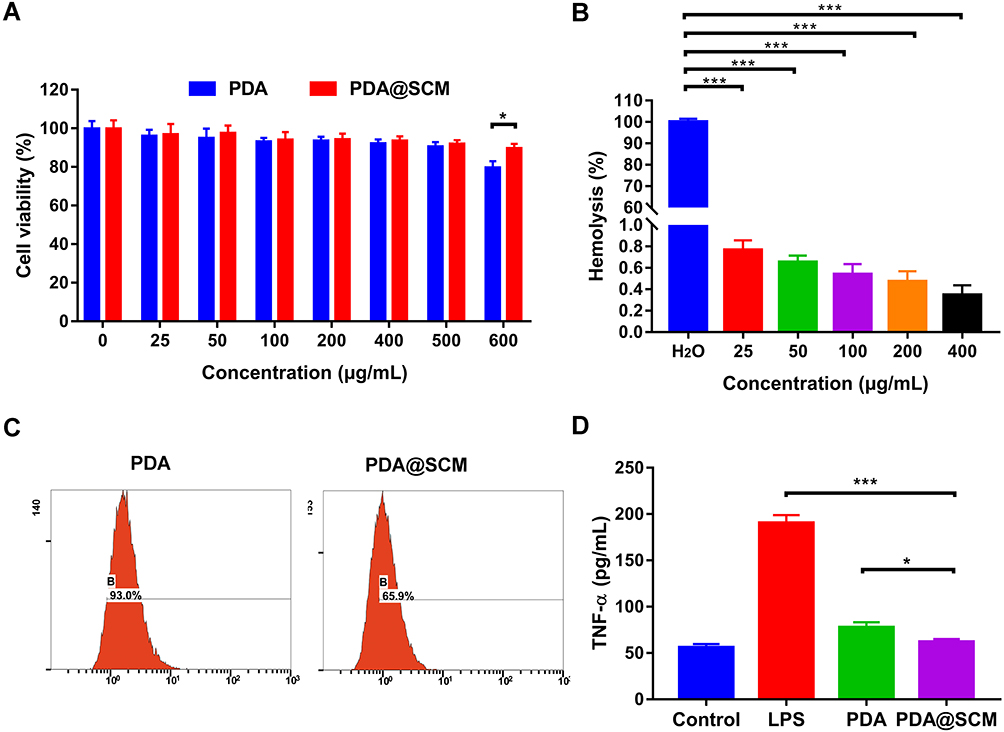 |
Figure 3 Biocompatibility and immune-evasion ability of PDA@SCM NPs. (A) Toxicity of PDA NPs and PDA@SCM NPs against L929 cells. (B) Hemolysis study of PDA@SCM NPs at various concentrations. (C) Uptake of PDA NPs and PDA@SCM NPs by macrophages evaluated by flow cytometry. (D) TNF-α concentrations in cell-culture supernatants of macrophages treated with PDA NPs and PDA@SCM NPs. *p < 0.05, ***p < 0.001. |
Next, we investigated the phagocytic activity of macrophages on PDA@SCM NPs. FITC-labeled PDA@SCM NPs and FITC-labeled PDA NPs were incubated with an equal number of RAW246.7 macrophages for 12 h and analyzed subsequently by flow cytometry. For PDA@SCM NPs, the percent cellular uptake was 65.9%, which was lower than that for PDA NPs (93.0%). PDA NPs camouflaged by an SCM reduced macrophage phagocytosis significantly compared with that by unmodified PDA NPs (Figure 3C).42 Macrophages produce proinflammatory cytokines upon interaction with NPs.43 TNF-α is a cytokine secreted by activated macrophages. We measured the potential of PDA@SCM NPs for stimulating macrophages by measuring the level of the proinflammatory cytokine TNF-α in cell-culture supernatants. Compared with LPS and PDA NPs, the TNF-α secretion of PDA@SCM NPs was reduced significantly. Compared with the control group, the TNF-α secretion of PDA@SCM NPs increased non-significantly (Figure 3D). These results indicated that an SCM coating could reduce the immune response of macrophages effectively. These data suggested that PDA@SCM NPs had good biocompatibility and the ability to evade the immune system.
Photothermal Properties of PDA@SCM NPs
After coating of SCMs on PDA NPs, we found that PDA NPs and PDA@SCM NPs demonstrated a broad absorption range from 500 nm to 900 nm in the UV–visible absorption spectrum (Figure 4A).44 Absorbance at 808 nm was a linear function of the concentration of PDA@SCM NPs (Figure S3). The photothermal-conversion capacity of PDA@SCM NPs was evaluated by irradiating an aqueous solution of PDA@SCM NP with the 808-nm NIR laser at an intensity of with 0.6 W/cm2. After 5 min of irradiation, the temperature of the water increased by only 3.6°C, and the temperature of PDA NPs and PDA@SCM NPs increased by 30.2°C (Figure 4B). With increasing concentrations of PDA NPs and PDA@SCM NPs, a sharp increase in temperature was observed. In particular, when the concentration of PDA NPs and PDA@SCM NPs was 100 μg/mL, the solution temperature reached 50°C within 5 min of irradiation (the temperature required to kill cancer cells). This finding indicated that the SCM coating did not affect the photothermal property of PDA NPs. Subsequently, the thermal stability of PDA@SCM NPs was evaluated by irradiation with the NIR laser at 0.6 W/cm2 for 5 min and then cooling down to room temperature. Irradiation was undertaken for five cycles. Compared with PDA NPs, the thermal stability of PDA@SCM NPs did not decrease after five irradiation cycles (Figure 4C). Therefore, the excellent light-to-heat conversion efficiency and thermal stability of PDA@SCM NPs indicated the potential of such NPs to be excellent candidates for PTT applications.
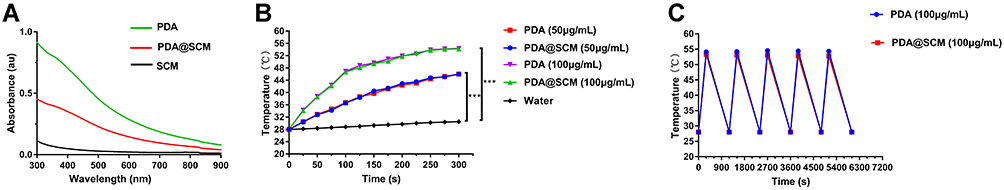 |
Figure 4 Photothermal properties of PDA@SCM NPs. (A) UV–vis absorbance spectra of SCM vesicles, PDA NPs and PDA@SCMs NPs. (B) Photothermal heating curves of PDA NPs and PDA@SCMs NPs in aqueous medium at different concentrations irradiated with an 808-nm laser (0.6 W/cm2, 5 min). (C) Thermal stability of PDA NPs and PDA@SCMs NPs under NIR-laser irradiation over five laser on/off cycles. ***p < 0.001. |
Drug-Loading and Drug-Releasing Capacity of PDA@SCM NPs
We evaluated the drug-loading and drug-releasing capabilities of PDA@SCM NPs in vitro. A linear standard curve for determining the SN38 concentration was used to determine the drug-loading efficiency (Figure S4). After loading the hydrophobic drug SN38 onto PDA mediated by π-π stacking interactions, PDA-SN38@SCM NPs were constructed by co-extrusion with SCM vesicles.45 PDA-SN38@SCM NPs had the same absorption peak of SN38 obtained at 365 nm (Figure 5A), thereby indicating that SN38 was loaded on PDA@SCM NPs. The drug-releasing ability of PDA-SN38@SCM NPs was tested at pH 5.0 and 7.4. Within 12 h, PDA-SN38@SCM NPs released ~19.5% of SN38 at pH 5.0, and 5.2% at pH 7.4 (Figure 5B). At pH 5.0, the amount of SN38 released by PDA-SN38@SCM NPs was approximately three times higher than that at pH 7.4 (Figure 5B). The effect of pH on drug release may be due to the protonation of amino groups on the PDA surface under acidic conditions, which weakens the π-π stacking interactions.46 To investigate the photothermal influence upon SN38 release, PDA-SN38@SCM NPs suspended in deionized water were irradiated with an 808-nm NIR laser (0.6 W/cm2) for different intervals. SN38 release was dependent on the duration of NIR irradiation. PDA-SN38@SCM NPs released ~58% of SN38 after NIR irradiation for 30 min. However, ~4.1% of SN38 was released in the absence of irradiation (Figure 5C). Local hyperthermia may disturb the π-π stacking interactions between the drug and PDA NPs, thereby resulting in SN38 release.47 Furthermore, we used TEM to observe the morphology of PDA NPs before and after stimulation by pH and NIR irradiation, and found that the morphology of PDA NPs did not change (Figure S5). This finding indicated that pH-dependent and light-responsive release could regulate the release of intracellular drugs synergistically, thereby leading to improvement in antitumor efficiency and reduction of the side effects of chemotherapy through disturbing π-π stacking interactions between the drug and PDA NPs.
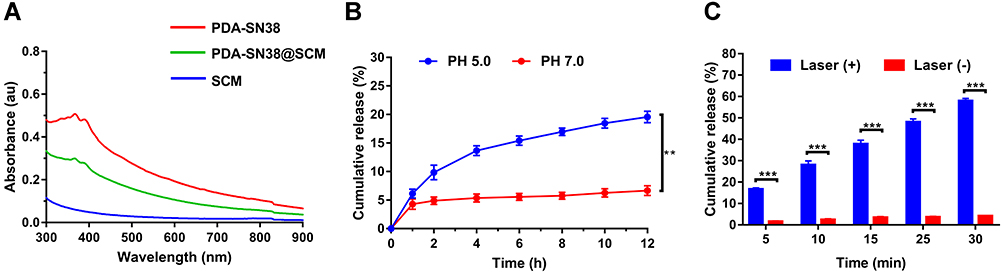 |
Figure 5 Drug-loading and drug-releasing capacity of PDA@SCM NPs. (A) UV–Vis absorption spectra of SCM, PDA-SN38 NPs and PDA-SN38@SCM NPs. (B) SN38 release from PDA-SN38@SCM NPs at different pH within 12 h. (C) SN38 release from PDA-SN38@SCM NPs with or without 808-nm laser irradiation (0.6 W/cm2, 5 min). **p < 0.01, ***p < 0.001. |
Efficacy of PDA-SN38@SCM NPs in Targeting Cancer Cells
The tumor-specific targeting components on the surface of stem cells make them a potent source of cell membranes for coating NPs designed to functionalize cell membranes.48,49 To evaluate the ability of PDA-SN38@SCM NPs in targeting cancer cells in vitro, free SN38, PDA-SN38 NPs, or PDA-SN38@SCM NPs were incubated with MG63 cells for 4 h. CLSM and flow cytometry were used to analyze drug release in cancer cells by measuring the difference between the SN38 content in such cells. Compared with free SN38, MG63 cells treated with PDA-SN38@SCM NPs demonstrated higher fluorescence intensity upon stimulation with the same-intensity laser (Figure 6A). This observation indicated that the presence of SCM on PDA-SN38 NPs led to higher uptake of SN38 by MG63 cells. SN38 exhibits fluorescence by itself, so flow cytometry can be used to measure the content of phagocytic SN38. We found that 91.9% of cancer cells demonstrated the presence of PDA-SN38@SCM NPs, and 73.5% of cancer cells showed the presence of PDA-SN38 NPs (Figure 6B). SCM-derived membrane coatings enhanced the ability of PDA-SN38@SCM NPs to target cancer drugs in vitro, as demonstrated by flow cytometry. The results stated above indicated that the extrusion method not only camouflaged the SCM on the surface of NPs but also mediated the transfer of cancer cell-targeting ability of stem cells to NPs. Hence, PDA-SN38@SCM NPs loaded with anticancer drugs could be used for the transportation of anticancer drugs to cancer cells.
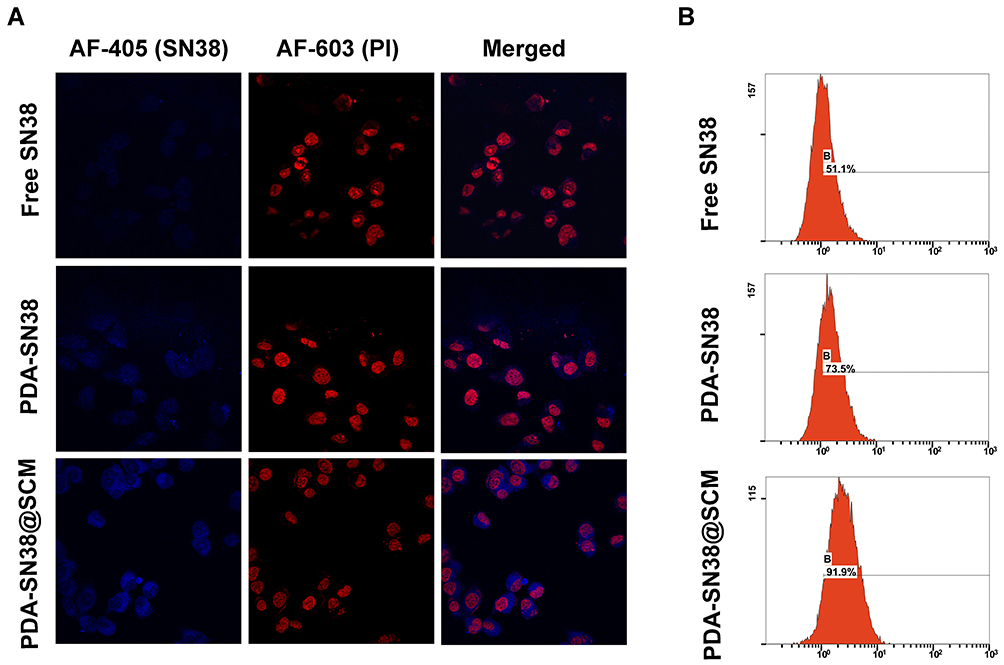 |
Figure 6 Ability of PDA-SN38@SCM NPs to target cancer cells. (A) CLSM images of MG63 cells treated with free SN38, PDA-SN38 NPs or PDA-SN38@SCM NPs for 4 h. Scale bar = 10 μm. (B) Cell-uptake efficiency of different SN38 formulations by flow cytometry. |
Anticancer Performance in vitro
We further examined the anticancer activity of PDA-SN38@SCM NPs in vitro using MTT and calcein AM/PI (live/dead) staining. Virtually no cell death was observed upon treatment with a NIR laser alone or by PDA NPs (Figure 7A). Approximately 47.1% of cells were killed in the absence of NIR irradiation by PDA-SN38@SCM NPs, and 91.1% of cells were killed under NIR irradiation. Compared with treatment by free SN38 or PDA-SN38 NPs, the equivalent concentration of PDA-SN38@SCM NPs exerted a much stronger inhibitory effect in the absence or presence of NIR irradiation. Fluorescent cell staining further confirmed the ability of CPT using PDA-SN38@SCM NPs in ablating cancer cells. The live/dead staining images were consistent with the MTT results. After NIR irradiation, almost all MG63 cells treated with PDA-SN38@SCM NPs were destroyed and stained with red fluorescent PI (Figure 7B). In contrast, cells treated with controls were stained with green fluorescence, indicating the presence of live cells. Taken together, the results shown above indicated that PDA-SN38@SCM NPs exhibited a superior CPT antitumor effect.
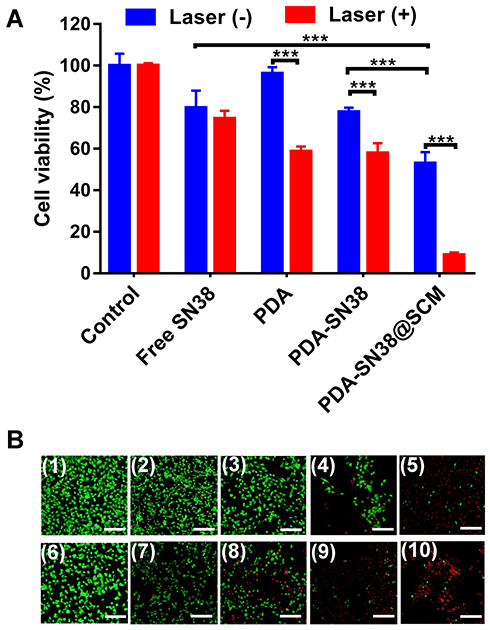 |
Figure 7 Anticancer performance in vitro. (A) Viability of MG63 cells treated with free SN38, PDA-SN38 NPs or PDA-SN38@SCM NPs with or without 808-nm laser irradiation (0.6 W/cm2, 5 min). (B) Live/dead staining of MG63 cells after different treatments. Calcein-AM (green, live cells) and propidium iodide (PI) (red, apoptotic cells). (1) Control, (2) SN38, (3) PDA, (4) PDA-SN38, (5) PDA-SN38@SCM, (6) 808-nm laser irradiation, (7) SN38 plus 808-nm laser irradiation, (8) PDA NPs plus 808-nm laser irradiation, (9) PDA-SN38 NPs plus 808-nm laser irradiation and (10) PDA-SN38@SCM NPs plus 808-nm laser irradiation. Scale bar = 200 μm. ***p < 0.001. |
Pharmacokinetics and Biodistribution
In order to evaluate whether PDA@SCM NPs inherited the long-term blood-circulation ability of natural stem cells and the ability to target tumors in vivo, the blood-retention and distribution of PDA NPs and PDA@SCM NPs in major organs and tumor tissues were determined. Four hours after tail-vein injection, the residual percentage of PDA@SCM NPs in blood was 86.4% (Figure 8A), but the percentage of PDA NPs was only 16.7%. PDA NPs were almost undetectable in blood 24 h after intravenous injection. In contrast, 27.6% PDA@SCM NPs remained in circulation, indicating that PDA@SCM NPs had a longer life in blood. This finding was attributable mainly to the surface camouflage of the SCM that reduced the attachment of biomolecules such as serum proteins to the surface of the micro-nano transport carrier while inhibiting flocculation, opsonization, and subsequent complement activation.50 We further confirmed that the SCM had been translated onto the surface of PDA NPs. In addition, nude mice were euthanized, and then major organs and tumor tissues were collected and subjected to fluorescence analyses to study the biological distribution of NPs in vivo. Ex vivo fluorescence images (Figure 8B) showed the major organs and tumors of two groups 24 h after intravenous injection. The fluorescence intensity of PDA@SCM NPs in the liver, spleen, and lungs was lower than that of PDA NPs, which further confirmed that the modification of the SCM on the surface of PDA NPs reduced NP phagocytosis by body organs. Tumors in the PDA@SCM group elicited a higher intensity of fluorescence signals than those from the PDA group, which could be attributed to cellular membranes having “homing affinity” to tumor sites. In addition, the major organs and tumors were collected and subjected to fluorescence analyses to study the biological distribution of NPs quantitatively. The distribution of PDA@SCM NPs in the liver, spleen, and lungs was lower than that of PDA NPs (Figure 8C), which was consistent with the images shown in Figure 8B. This finding confirmed that SCM modification on the surface of PDA NPs reduced NP phagocytosis by body organs. Compared with PDA NPs, PDA@SCM NPs showed significantly enhanced tumor accumulation. At 24 h after injection, the calculated accumulation of PDA@SCM NPs in the tumor was 12.2% ID/g, whereas accumulation of PDA NPs in the tumor was 5.8% ID/g. This observation indicated that more SCM-camouflaged PDA NPs accumulated at the tumor site after injection into the tail vein, which further verified the tumor-targeting action of SCM-camouflaged PDA NPs.
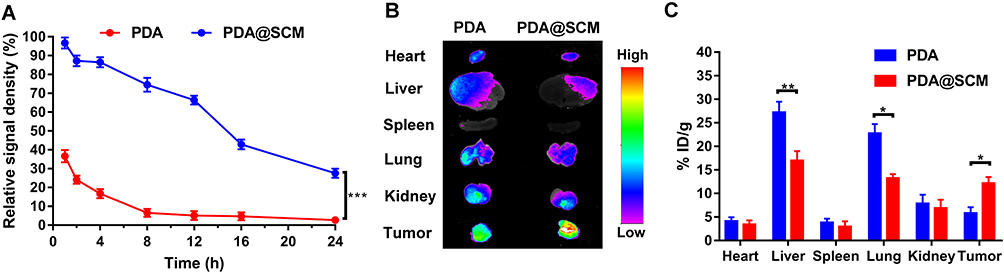 |
Figure 8 Pharmacokinetics and biodistribution. (A) Pharmacokinetics of PDA NPs and PDA@SCM NPs over a span of 24 h. (B) Representative ex vivo image showing biodistributions of PDA NPs and PDA@SCMs NPs in major organs and tumor tissues from each group 24 h after intravenous injection of PDA and PDA@SCM. (C) In vivo study the biological distribution of PDA NPs and PDA@SCM NPs quantitatively in major organs and tumor tissues at 24 h after intravenous injection. *p < 0.05, **p < 0.01, ***p < 0.001. |
Antitumor Activity in vivo
We evaluated the antitumor ability of PDA-SN38@SCM NPs in combination with CPT upon irradiation with an 808-nm laser in vivo. Tumor-bearing mice were divided randomly into eight groups and treated by intravenous injection composed of different formulations. After treatment, the tumor size in mice of different groups was measured every 2 days for 14 days. Compared with that by the control group, treatment with free SN38 and 808-nm laser irradiation exhibited no antitumor effect (Figure 9A and B). Mice treated with PDA NPs plus laser irradiation, PDA-SN38, and PDA-SN38 plus laser irradiation could suppress tumor growth to some extent. Mice treated with PDA-SN38@SCM NPs could reduce tumor volume by 47.0%. When PDA-SN38@SCM NPs were used in combination with 808-nm laser irradiation, the tumor volume decreased by 60.0%. The mean tumor weight in PDA-SN38@SCM NPs was notably smaller than that in the other seven groups (Figure 9C). In addition, there was no significant difference in weight change between groups (Figure S6). Morphology features such as inflammation, necrosis, and apoptosis of cells did not appear in the tissue sections of various organs (Figure S7). The results stated above indicated that PDA-SN38@SCM NPs under laser irradiation enhanced tumor-targeting capability effectively due to stem-cell coating, and exhibited low toxicity and effective inhibition of growth of malignant bone tumors.
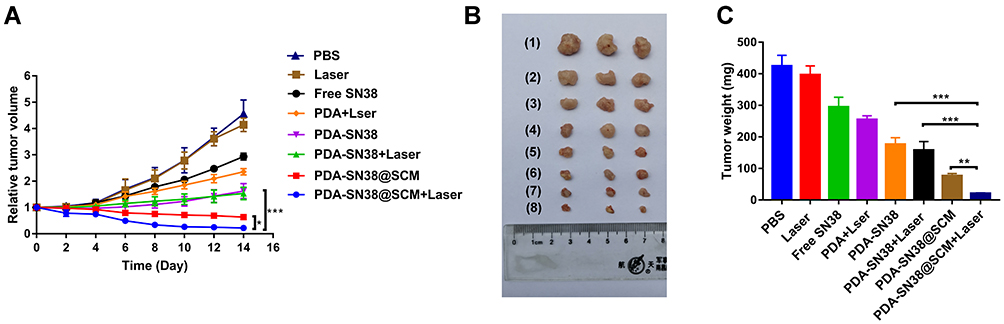 |
Figure 9 Antitumor activity in vivo. (A) Tumor growth curves of different groups of mice after treatment (day-14). Tumor volumes were normalized to their initial sizes. Antitumor effect of different treatments: (1) PBS; (2) 808-nm laser irradiation; (3) SN38; (4) PDA NPs plus 808-nm laser irradiation; (5) PDA-SN38 NPs; (6) PDA-SN38 NPs plus 808-nm laser irradiation; (7) PDA-SN38@SCM NPs and (8) PDA-SN38@SCM NPs plus 808-nm laser irradiation. (n = 3, mean ± SD). (B) Photographs of representative mice after different treatments. (C) Tumor weight of each group after treatment (day-14). *p < 0.05, **p < 0.01, ***p < 0.001. |
Conclusion
We designed a novel multimodal therapy platform by coating SCMs onto PDA-SN38 NPs for CPT of tumors. The obtained PDA-SN38@SCM NPs exhibited the biological function of natural SCMs, demonstrated good biocompatibility and stability, along with tumor-targeting abilities. The resulting PDA-SN38@SCM NPs showed pH- and photo-responsive SN38 release. In vitro and in vivo results under NIR light irradiation showed that a combination of CPT of PDA-SN38@SCM NPs demonstrated low toxicity and efficacy in tumor treatment. Our results indicated that PDA-SN38@SCM NPs demonstrate an extraordinary synergistic CPT effect and could be a promising strategy for treatment of malignant bone tumors.
Abbreviations
NP, nanoparticle; CPT, chemo-photothermal therapy; SCM, PDA@SCM NPs, stem cell membrane (SCM)-camouflaged polydopamine nanoparticles; SN38, 7-ethyl-10-hydroxycamptothecin; PDA-SN38@SCM NPs, stem cell membrane-coated polydopamine nanoparticles encapsulating SN38; CLSM, confocal laser scanning microscopy; DDSs, drug-delivery systems; EPR, enhanced permeability and retention; PEG, polyethylene glycol; PLGA, poly(lactic-co-glycolic acid); RBC, nanoparticles with red blood cell; MSC, mesenchymal stem cells; PTT, photothermal therapy; UCMSC, umbilical-cord mesenchymal stem cell; PDA NPs, polydopamine nanoparticles.
Acknowledgments
This study was financially supported by the Science and Technology Development Plan Projects of Jilin Province (Grant No. 20200201558JC), the Bethune project of Jilin University (Grant No. 2018A03), and the Health Technology Innovation Project of Jilin Province (Grant No. 2016J070).
Disclosure
The authors declare no conflicts of interest.
References
1. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37.
2. Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: a review. Biomaterials. 2016;85:152–167. doi:10.1016/j.biomaterials.2016.01.061
3. Taghizadeh B, Taranejoo S, Monemian SA, et al. Classification of stimuli-responsive polymers as anticancer drug delivery systems. Drug Deliv. 2015;22(2):145–155. doi:10.3109/10717544.2014.887157
4. Dadwal A, Baldi A, Kumar Narang R. Nanoparticles as carriers for drug delivery in cancer. Artif Cells, Nanomed Biotechnol. 2018;46(sup2):295–305. doi:10.1080/21691401.2018.1457039
5. Alshaer W, Hillaireau H, Fattal E. Aptamer-guided nanomedicines for anticancer drug delivery. Adv Drug Deliv Rev. 2018;134:122–137. doi:10.1016/j.addr.2018.09.011
6. Schroeder A, Heller DA, Winslow MM, et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2011;12(1):39–50.
7. Barenholz Y. Doxil®–the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi:10.1016/j.jconrel.2012.03.020
8. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(5):16014.
9. Tavares AJ, Poon W. Effect of removing Kupffer cells on nanoparticle tumor delivery. Proc Natl Acad Sci. 2017;114(51):E10871–e10880.
10. Cai H, An X, Cui J, et al. Facile hydrothermal synthesis and surface functionalization of polyethyleneimine-coated iron oxide nanoparticles for biomedical applications. ACS Appl Mater Interfaces. 2013;5(5):1722–1731. doi:10.1021/am302883m
11. Yang X, Liu X, Liu Z, Pu F, Ren J, Qu X. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer gated nanovehicles. Adv Mater (Deerfield Beach, Fla). 2012;24(21):2890–2895. doi:10.1002/adma.201104797
12. Mu X, Zhang F, Kong C, et al. EGFR-targeted delivery of DOX-loaded Fe(3)O(4)@ polydopamine multifunctional nanocomposites for MRI and antitumor chemo-photothermal therapy. Int J Nanomedicine. 2017;12:2899–2911. doi:10.2147/IJN.S131418
13. Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56(11):1649–1659. doi:10.1016/j.addr.2004.02.014
14. Szebeni J, Bedőcs P, Urbanics R, et al. Prevention of infusion reactions to PEGylated liposomal doxorubicin via tachyphylaxis induction by placebo vesicles: a porcine model. J Control Release. 2012;160(2):382–387. doi:10.1016/j.jconrel.2012.02.029
15. Mohamed M, Abu Lila AS, Shimizu T, et al. PEGylated liposomes: immunological responses. Sci Technol Adv Mater. 2019;20(1):710–724. doi:10.1080/14686996.2019.1627174
16. Neun BW, Barenholz Y, Szebeni J, Dobrovolskaia MA. Understanding the role of anti-PEG antibodies in the complement activation by doxil in vitro. Molecules. 2018;23(7).
17. Fang RH, Kroll AV, Gao W, Zhang L. Cell membrane coating nanotechnology. Adv Mater. 2018;30(23):e1706759.
18. Zhen X, Cheng P, Pu K. Recent advances in cell membrane-camouflaged nanoparticles for cancer phototherapy. Small. 2019;15(1):e1804105.
19. Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108(27):10980–10985. doi:10.1073/pnas.1106634108
20. Jiang Q, Liu Y, Guo R, et al. Erythrocyte-cancer hybrid membrane-camouflaged melanin nanoparticles for enhancing photothermal therapy efficacy in tumors. Biomaterials. 2019;192:292–308. doi:10.1016/j.biomaterials.2018.11.021
21. Xuan M, Shao J, Dai L, He Q, Li J. Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv Healthcare Mater. 2015;4(11):1645–1652. doi:10.1002/adhm.201500129
22. Hu CM, Fang RH, Wang KC, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526(7571):118–121. doi:10.1038/nature15373
23. Gao C, Lin Z, Wu Z. Stem-cell-membrane camouflaging on near-infrared photoactivated upconversion nanoarchitectures for in vivo remote-controlled photodynamic therapy. ACS Appl Mater Inter. 2016;8(50):34252–34260.
24. Li X, Zhao X, Pardhi D, et al. Folic acid modified cell membrane capsules encapsulating doxorubicin and indocyanine green for highly effective combinational therapy in vivo. Acta Biomater. 2018;74:374–384. doi:10.1016/j.actbio.2018.05.006
25. Wang D, Dong H. Erythrocyte-cancer hybrid membrane camouflaged hollow copper sulfide nanoparticles for prolonged circulation life and homotypic-targeting photothermal/chemotherapy of melanoma. ACS Nano. 2018;12(6):5241–5252.
26. Rao L, Bu LL, Cai B, et al. Cancer cell membrane-coated upconversion nanoprobes for highly specific tumor imaging. Adv Mater (Deerfield Beach, Fla). 2016;28(18):3460–3466. doi:10.1002/adma.201506086
27. Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi:10.1038/nnano.2012.212
28. Choi MR, Stanton-Maxey KJ, Stanley JK, et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7(12):3759–3765. doi:10.1021/nl072209h
29. Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16(9):1035–1041. doi:10.1038/nm.2198
30. Hao X, Xu B, Chen H, et al. Stem cell-mediated delivery of nanogels loaded with ultrasmall iron oxide nanoparticles for enhanced tumor MR imaging. Nanoscale. 2019;11(11):4904–4910. doi:10.1039/C8NR10490E
31. Kaneti L, Bronshtein T, Malkah Dayan N, et al. Nanoghosts as a novel natural nonviral gene delivery platform safely targeting multiple cancers. Nano Lett. 2016;16(3):1574–1582. doi:10.1021/acs.nanolett.5b04237
32. Yang N, Ding Y, Zhang Y, et al. Surface functionalization of polymeric nanoparticles with umbilical cord-derived mesenchymal stem cell membrane for tumor-targeted therapy. ACS Appl Mater Inter. 2018;10(27):22963–22973.
33. Liu Y, Ai K, Liu J, Deng M, He Y, Lu L. Dopamine-melanin colloidal nanospheres: an efficient near-infrared photothermal therapeutic agent for in vivo cancer therapy. Adv Mater (Deerfield Beach, Fla). 2013;25(9):1353–1359. doi:10.1002/adma.201204683
34. Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat Commun. 2018;9(1):1074.
35. Hu W, Bai X. Upper critical solution temperature polymer-grafted hollow mesoporous silica nanoparticles for near-infrared-irradiated drug release. J Mater Chem B. 2019;7(38):5789–5796.
36. Zhang Z, Wang J, Chen C. Near-infrared light-mediated nanoplatforms for cancer thermo-chemotherapy and optical imaging. Adv Mater (Deerfield Beach, Fla). 2013;25(28):3869–3880. doi:10.1002/adma.201301890
37. Wang X, Wang C, Wang X, Wang Y, Zhang Q, Cheng Y. A polydopamine nanoparticle-knotted poly(ethylene glycol) hydrogel for on-demand drug delivery and chemo-photothermal therapy. Chem Mater. 2017;29(3):1370–1376. doi:10.1021/acs.chemmater.6b05192
38. Liu Y, Ai K, Lu L. Polydopamine and its derivative materials: synthesis and promising applications in energy, environmental, and biomedical fields. Chem Rev. 2014;114(9):5057–5115. doi:10.1021/cr400407a
39. Toledano Furman NE, Lupu-Haber Y, Bronshtein T, et al. Reconstructed stem cell nanoghosts: a natural tumor targeting platform. Nano Lett. 2013;13(7):3248–3255. doi:10.1021/nl401376w
40. Liu Y, Zhao J, Jiang J, Chen F, Fang X. Doxorubicin delivered using nanoparticles camouflaged with mesenchymal stem cell membranes to treat colon cancer. Int J Nanomedicine. 2020;15:2873–2884. doi:10.2147/IJN.S242787
41. Peng LH, Zhang YH, Han LJ, et al. Cell membrane capsules for encapsulation of chemotherapeutic and cancer cell targeting in vivo. ACS Appl Mater Interfaces. 2015;7(33):18628–18637. doi:10.1021/acsami.5b05065
42. Dobrovolskaia MA, McNeil SE. Understanding the correlation between in vitro and in vivo immunotoxicity tests for nanomedicines. J Control Release. 2013;172(2):456–466. doi:10.1016/j.jconrel.2013.05.025
43. Elsabahy M, Wooley KL. Cytokines as biomarkers of nanoparticle immunotoxicity. Chem Soc Rev. 2013;42(12):5552–5576. doi:10.1039/c3cs60064e
44. Jiang Q, Luo Z, Men Y, et al. Red blood cell membrane-camouflaged melanin nanoparticles for enhanced photothermal therapy. Biomaterials. 2017;143:29–45. doi:10.1016/j.biomaterials.2017.07.027
45. Wang Y, Huang Q, He X, et al. Multifunctional melanin-like nanoparticles for bone-targeted chemo-photothermal therapy of malignant bone tumors and osteolysis. Biomaterials. 2018;183:10–19. doi:10.1016/j.biomaterials.2018.08.033
46. Ho CC, Ding SJ. The pH-controlled nanoparticles size of polydopamine for anti-cancer drug delivery. J Mater Sci Mater Med. 2013;24(10):2381–2390. doi:10.1007/s10856-013-4994-2
47. Lin M, Gao Y, Hornicek F, et al. Near-infrared light activated delivery platform for cancer therapy. Adv Colloid Interface Sci. 2015;226(Pt B):123–137. doi:10.1016/j.cis.2015.10.003
48. Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96(21):1593–1603. doi:10.1093/jnci/djh299
49. Lenna S, Bellotti C. Mesenchymal stromal cells mediated delivery of photoactive nanoparticles inhibits osteosarcoma growth in vitro and in a murine in vivo ectopic model. J Exp Clin Cancer Res. 2020;39(1):40.
50. Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. J Control Release. 2015;220(Pt B):600–607. doi:10.1016/j.jconrel.2015.07.019
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.