Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Polycythemia Vera (PV): Update on Emerging Treatment Options
Authors Benevolo G ![]() , Vassallo F, Urbino I, Giai V
, Vassallo F, Urbino I, Giai V
Received 7 January 2021
Accepted for publication 21 February 2021
Published 16 March 2021 Volume 2021:17 Pages 209—221
DOI https://doi.org/10.2147/TCRM.S213020
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Giulia Benevolo, Francesco Vassallo, Irene Urbino, Valentina Giai
Division of Haematology, Città della Salute e della Scienza, Turin, Italy
Correspondence: Giulia Benevolo
Hematology, Città della Salute e della Scienza, Corso Bramante 88/90, Turin, 10126, Italy
Email [email protected]
Abstract: Polycythemia Vera (PV) is a chronic myeloproliferative neoplasm characterized by exuberant red cell production leading to a broad range of symptoms that compromise quality of life and productivity of patients. PV reduces survival expectation, primarily due to thrombotic events, transformation to blast phase and post-PV myelofibrosis or to development of second cancers, which are associates with poor prognosis. Current therapeutic first line recommendations based on risk adapted classification divided patients into two groups, according to age (< or > 60 years) and presence of prior thrombotic events. Low-risk patients (age < 60 years and no prior history of thrombosis) should be treated with aspirin (81– 100 mg/d) and phlebotomy, to maintain hematocrit < 45%. High-risk patients (age > 60 years and/or prior history of thrombosis), in addition to aspirin and phlebotomies, should receive cytoreductive therapy in order to reduce thrombotic risk. Nowadays hydroxyurea still remains the cytoreductive agent of first choice, reserving Interferon to young patients or childbearing women. During the last years, ruxolitinib emerged as a new treatment in PV patients, as second line therapy: it appeared especially effective in patients with severe pruritus, symptomatic splenomegaly, or post-PV myelofibrosis symptoms. Currently, in PV treatment, several molecules have been tested or are under investigation. At present, the drug that has shown the most encouraging results is givinostat.
Keywords: polycythemia vera, therapy, ruxolitinib, interferon, givinostat
Overview
Polycythemia Vera (PV) is one of the chronic myeloproliferative neoplasms (MPNs) classified by the World Health Organization (WHO) and is characterized by aberrant hematopoiesis of myeloid lineage with exuberant red cell production and increased release of pro-inflammatory cytokines.1
In PV patients, a broad range of symptoms influence quality of life and productivity. Furthermore, PV reduces survival expectation, primarily due to thrombotic events, transformation to blast phase (BP) and post-PV myelofibrosis (PPV-MF) or to development of second cancers, which are associated with poor prognosis and modest response to therapy.2
Often, PV is suspected when high hemoglobin and hematocrit levels are displayed in blood tests run for other clinical reasons.3
At diagnosis, half of the patients presents hypertension and elevated lactate dehydrogenase, while about a third shows a palpable splenomegaly, pruritus and vasomotor symptoms.4
Quality of life questionnaires reported that mild nonspecific symptoms as fatigue, early satiety, abdominal discomfort, bone pain, sexual dysfunction, excessive sweating and itching could compromise the everyday routine and reduce quality of life.5,6 Moreover, the natural history of the disease may be characterized by worsening of these symptoms.
During the course of the disease, thrombosis affect 29% of PV patients (arterial in 70% of cases),4 while progression to PPV-MF and BP about 9% and 3% of patients, respectively.
Significant associations between age and leukemia evolution or duration of the disease and risk of myelofibrosis7 have been shown. Moreover, an international study demonstrated association between pruritus and superior survival, probably because of an earlier symptom-driven diagnosis.4
The majority (more than 90%) of PV patients exhibit Janus kinase 2 (JAK2) V617F mutation.8 JAK2 V617F is an exon 14 guanine to thymidine somatic mutation; however, around 2% of PV patients do not exhibit JAK2 V617F mutation, but insertions and deletions in exon 12.9
At diagnosis, patients with exon 12 mutations show higher hemoglobin levels and lower platelet and leukocyte counts; however, incidence of thrombosis, of evolution to myelofibrosis or leukemia, and mortality are similar in both groups.10
In the last years, several other genes mutations were identified by next-generation sequencing (NGS). These mutations did not show a diagnostic impact, because of their low specificity and frequency, but might be useful to recognize a subgroup of PV patients with increased risk of leukemic transformation.11
ASXL1, SRSF2, and IDH2 are associated with low survival, as independent prognostic factors;11 a persistently high or a progressive rise of JAK2 V617F allele burden during cytoreductive therapy is the strongest predictor of myelofibrotic transformation12 even if it is more frequent in patients with SF3B1 and IDH1/2 mutation.
In the present review, we address the treatment landscape for PV patients with an update on the emerging therapeutic approaches. We discuss conventional and innovative treatments, focusing on the role they have in current therapeutic strategies and the hopes they carry for the future.
Diagnosis and Management of PV
The 2016 WHO Classification13 (Table 1) revised the previous diagnostic criteria for PV: elevated hemoglobin (Hb) and hematocrit (Hct) levels, bone marrow panmyelosis and JAK2 mutations were identified as major criteria and suppressed erythropoietin (EPO) level as minor criteria. Specifically, the Hb and Hct diagnostic thresholds were reduced to 16.5 g/dL and 49% for men, and 16 g/dL and 48% for women, respectively, allowing identification of a new entity, called “masked PV”, characterized by a worse outcome,14 probably due to missed or delayed diagnoses with a consequent delay in treatment.
 |
Table 1 Diagnostic Criteria: All 3 Major Criteria or the First Two and the Minor One Must Be Present for the Diagnosis |
PV treatment main goals are to control symptoms and prolong survival by preventing thrombosis, massive splenomegaly, and leukemic transformation.
In order to reduce thrombotic risk, treatments should be addressed not only to normalize Hb, Hct and leukocytosis but also to decrease cardiovascular risk factors, such as obesity, diabetes, hypertension, dyslipidemia and smoking. Most importantly, hypertension and its management have a primary role:15 some studies, in fact, suggest that angiotensin converting enzyme inhibitors (ACE) inhibitors could also give a benefit in terms of reducing erythrocytosis.16,17
The cornerstone of thrombotic events prevention is represented by prophylaxis. In the multicenter ECLAP (European Collaboration on Low-Dose Aspirin in Polycythemia Vera) study, low dose aspirin (70–100 mg per day) showed a significant reduction (60%) of combined risk of non-fatal myocardial infarction, non-fatal stroke, pulmonary embolism, major venous thrombosis and death from other cardiovascular causes.18
Furthermore, it was suggested that twice-daily aspirin may be more effective than once daily dose in some cases,19 for example, in patients who are resistant to once daily dosing or considered at higher risk of arterial thrombosis.20
In CYTO-PV21 and PVSG-0122 studies, PV patients were treated with phlebotomy and/or chemotherapy (hydroxyurea-HU and chlorambucil, respectively) and were evaluated for the risk of thrombosis. The phlebotomy group shows longer overall survival; specifically, patients that maintained Hct<45% exhibited a lower rate of cardiovascular death and major thrombotic events than patients with Hct>45%.23 However, another recent study seems not to support prognostic importance of Hct level in preventing thrombo-hemorrhagic complications in PV and requires a more aggressive control of these parameters.24 Nevertheless, it was also demonstrated that antiplatelet therapy, and not cytoreductive treatment, was significantly associated with lower risk of cardiovascular events.7
Current therapeutic first line recommendations based on risk adapted classification divided patients into two groups, according to age and presence of prior thrombotic events.
Low-risk patients (age <60 years and no prior history of thrombosis) should be treated with aspirin (81–100 mg/d) and phlebotomy, to maintain Hct <45%.18,23
High-risk patients (age >60 years and/or prior history of thrombosis), in addition to aspirin and phlebotomies, should receive cytoreductive therapy (HU) in order to reduce thrombotic risk.21
Although HU is a manageable and well-tolerated drug, during the course of disease, almost one-fourth of patients becomes resistant (11%) or intolerant (13%) to HU therapy.21 Resistance and intolerance to HU have been widely studied but just in the contest of clinical trials25–28 (Table 2).
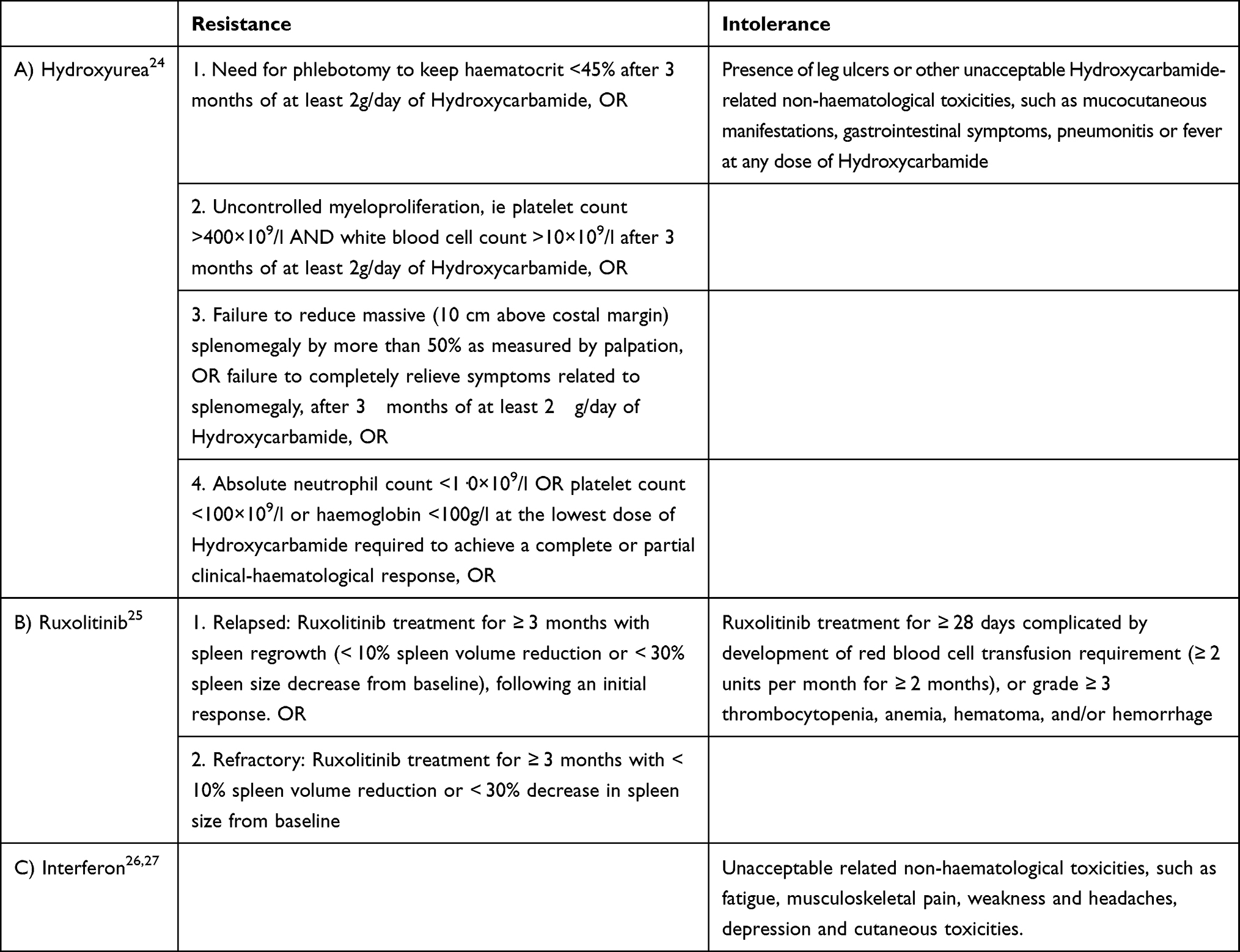 |
Table 2 Criteria for Resistance and Intolerance to the Most Common PV Treatments (Criteria for Ruxolitinib and Interferon Have Been Deduced from Approval Trials) |
Interferon alfa2 (IFN-α2) or peginterferon alfa2 (peg-IFN-α2) and more recently ropeginterferon α-2b (ropeg-INF-α-2b) could be considered for specific subgroups of patients (ie, younger or pregnant patients,29,30 or in those who refuse HU. However, interferons are not currently available in all countries.
In refractory or first-line intolerant patients, ruxolitinib (RUX) can be used; treatments for advanced disease are scarce and chemotherapy, ie, busulfan, may be used as an option.31 An overview of dosage and schedule of most common therapies for PV management are summarized in Table 3.
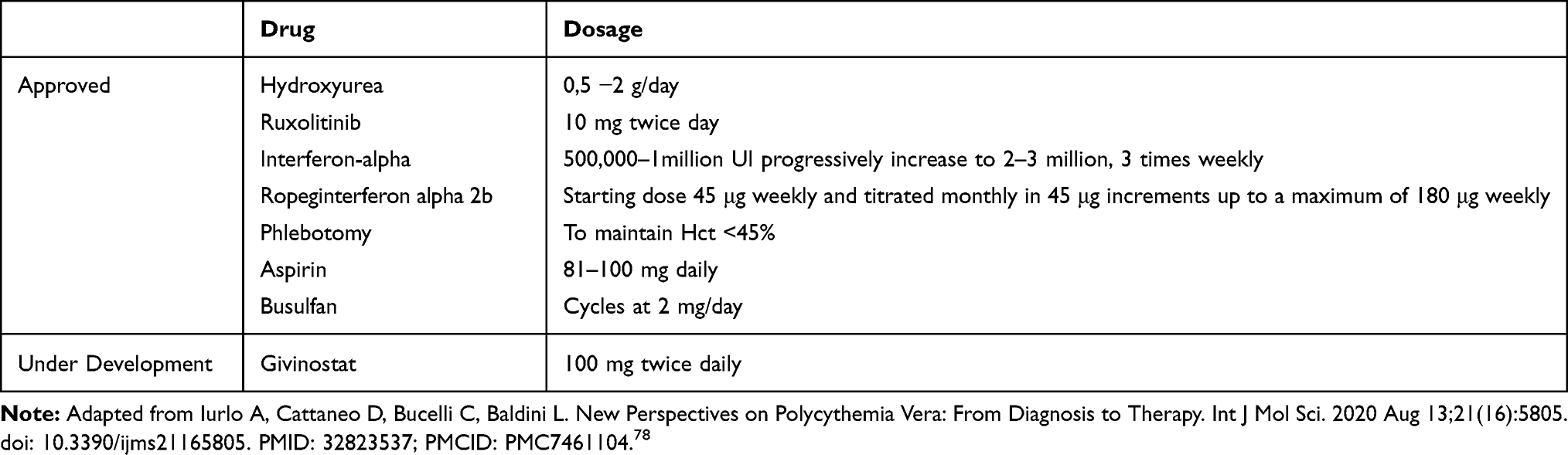 |
Table 3 Dosage and Schedule of Most Common PV Treatments |
Response Assessment
The European LeukemiaNet (ELN) and International Working Group Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) response criteria for PV categorized the disease into complete remission (CR), partial response (PR), no response, and progressive disease (PD)32 (Table 4). Criteria include evaluation of signs and symptoms, peripheral blood counts, absence of vascular events, no disease progression and no bone marrow histological abnormalities. CR is achieved when all these criteria are met; PR is defined if only the first two criteria are met. Assessment of JAK2 V617F allele burden is not routinely used and does not influence treatment decisions.33
 |
Table 4 Disease Response Criteria; Sign and Symptoms are Assessed with the MPN-SAF TSS, Durable Resolution/Remission Means at Least 12 Weeks |
Current Treatment Options
Ruxolitinib
As previously described, PV is characterized by mutations of JAK2 gene, resulting in constitutive activation of JAK/signal transducers and activators of transcription (STAT) pathway. Drugs targeting JAK1/2 pathway were primarily evaluated for PV second line treatment in patients resistant or intolerant to first line available therapies and opened a new MPN therapeutic era.
RESPONSE Trial
RUX is the first JAK1/2 inhibitor evaluated in the international, open-label Phase 3 RESPONSE trial, which included 215 patients with PV resistant or intolerant to HU,34–36 showing phlebotomy-dependence and/or progressive splenomegaly. Patients were randomly assigned to receive the best available treatment (BAT) or RUX (10 mg twice daily), with the opportunity for the ones assigned to BAT to switch to the RUX arm if the response achieved at week 32 was not adequate.
Efficacy
The primary endpoint was both Hct control and a reduction in spleen volume of at least 35% at week 32 and was achieved in 21% of patients in the RUX arm vs 1% in the BAT arm (P<0.001).34 Specifically, Hct control was reached in 60% of patients treated with RUX vs 20% of those receiving BAT; a 35% reduction in spleen volume was achieved in 38% vs 1% of patients in the two groups, respectively. A complete hematologic remission (CHR) was more frequently achieved with RUX (24% vs 9%; P=0.003). Efficacy and safety were finally evaluated after 256 weeks of treatment.37 The treatment was completed by 65.5% of the patients in the RUX group, 1% in the BAT group and 65.3% of those who crossed over to the RUX arm. Of the 70 patients (63.6%) who reached an overall clinical and hematological response at week 32, 21 (30%) had a progression of disease by week 256. Among primary responders to RUX 6 of 25 patients had progressed at the time of the final evaluation. Kaplan-Meier analysis estimated a probability of maintaining a primary response at week 256 of 0.74 (95% CI, 0.51–0.88), while the estimated probability of sustained clinical and hematological response was 0.67 (95% CI, 0.54–0.77) and its median duration was not reached. The estimated probabilities of maintaining Hct control and at least 35% reduction in spleen size were 0.73 (95% CI, 0.60–0.83) and 0.72 (95% CI, 0.34–0.91) respectively. The median overall survival (OS) by intention-to-treat analysis was similar for both arms (hazard ratio [HR], 0.95; 95% CI, 0.38–2.41). The survival rates at 5 years were higher than what was reported previously in this HU resistant/intolerant PV population.35 However, due to the extensive crossover of patients from BAT, the observed HR from this analysis represents a conservative estimate of RUX benefit and warrants further exploration.
Safety
65% of patients treated with RUX completed the study, with only 15% of patients discontinuing the study drug because of an adverse event (AE). Anemia and thrombocytopenia were the main hematological toxicities.34,37 Grade 3 or 4 non-hematologic AEs were in order of frequency infection (3.5%), reactivation of Herpes zoster infection included, increased body weight (0.7%) and thromboembolic events (0.7%). Secondary malignancies rates per hundred patient-years of exposure were 7.0% in the RUX arm vs 4.1% in the BAT arm, and 4.5% in the crossover population.34,37 It is noteworthy that patients in the RUX arm also had longer prior exposure to HU at baseline, which could be an underlying contributing factor for the increased rate of non-melanoma skin cancer (NMSC) observed in the RUX arm.37,38 Instead, no correlation with disease transformation was observed in RUX group.
It is also worth of note that RUX induced molecular response39 with sustained reductions in JAK2 V617F allele burden was observed in patients who were either randomized to RUX or received the drug after crossover from BAT.
RESPONSE-2 Trial
RUX was also evaluated in the RESPONSE-2 trial, a phase 3 study comparing RUX with BAT in PV patients resistant or intolerant to HU without palpable splenomegaly. A total of 149 patients were enrolled. Analysis was performed at week 80 or at discontinuation of the study. Primary endpoint was the proportion of patients achieving Hct control (<45%) at week 28. Secondary endpoints were CHR at week 28, and durability of Hct control and CHR.
Efficacy and Safety
Hct control was reached in 62% of patients in the RUX arm vs 19% of patients in the BAT arm (odds ratio [OR] 7.28 (95% CI 3.43–15.45)). Among patients treated with RUX who obtained Hct response at week 28, the probability of a sustained response up to week 80 was 78%. Durable CHR at week 80 was reported in 24% of patients in the RUX group vs 3% in the BAT group. Safety data on RUX confirmed the safety profile of the drug previously reported.40
Others
PV-related symptoms in patients treated with a stable dose of HU were specifically addressed in the RELIEF study. Patients with a controlled disease but who still reported symptoms were randomized 1:1 to receive RUX or HU with crossover to RUX allowed after week 16. The primary endpoint was the percentage of patients with ≥50% reduction in symptoms and it was achieved in significantly more patients in the RUX arm. A statistically significant reduction in pruritus was also noted in the RUX arm.41
The MAJIC trial is a randomized Phase 2 trial of second-line administration of RUX vs BAT in essential thrombocythemia (ET) and PV resistant or intolerant to first-line treatments. Although the trial includes patients with ET, two sub-analyses were recently presented42,43 that focused on comparing responses between the 2 arms in the PV group. The trial allowed comparisons on longer follow-up, as populations of both arms were followed up until 2.6 years.41,42
A recent systematic review and meta-analysis on RUX for prevention of thrombosis in PV were published by Masciulli et al showing an overall thrombosis annual incidence rate of 4.30% (95% CI, 3.00–5.60); the rate for BAT was 5.51% (95% CI, 3.72–7.30), and the rate for RUX was 3.09% (95% CI, 1.22–4.96). Authors concluded that the number of thrombotic events reported with RUX was consistently lower than that with BAT, but, globally, the difference did not reach significance.44
Interferons
Overview
As known, in MPN, one of the most active downstream transduction pathway is JAK/STAT.45 This pathway overstimulates interferons alpha receptors expression. Due to the hyperactivity of JAK mutant proteins in JAK2 mutated MPN, MPN cells display high rate of interferons alpha receptors.46,47
Also, a possible role of inflammation in the pathogenesis of MPN has been described, as JAK2 V617F mutation increases reactive oxygen species (ROS) levels.48 Moreover, ROS have been shown to inhibit T-cell function, so, for the JAK2 mutated clone is easier to escape from cellular immunity.49
In the last 5–10 years, various studies showed that, in MPN, INF-α2 can sustain long hematological and molecular responses50,51 that could be maintained until 3 years after discontinuation.52 Furthermore, INF-α2 can remodel bone marrow architecture:53 in some patients it completely reverts the bone marrow PV histological features.
For many years, the main problem connected with the use of interferon (INF) has been the high rate of discontinuation, due to the numerous AEs.54 Over the last 5–10 years, pegylated (peg) interferons (peg-INF-α2 and ropeg-INF-α-2b) have been developed and studied in MPN: the peg-chain protects the molecule from early degradation and it can be administered weekly or biweekly. Peg-interferons are reported to have a higher tolerability profile27 compared to INF-α2; peg-INF-α2 also shows higher molecular response rates in PV and ET.51,55
Nowadays, INF-α2 and peg-INF-α2 are being recognized as the treatment of choice in early stages of MPN disease: in literature, more than 1000 patients have been enrolled in single arm clinical studies.50,51,54–57
Many clinical trials showed that a majority of PV patients obtained a CHR in the first 6 months of IFN-α2 therapy, together with a reduction of JAK2 V617F allele burden.53,55,58 The decrease of thrombocytosis and the reduction of JAK2 V617F allele burden can contribute also to lower risks of second cancers.59,60
INF-α2 as BAT in RESPONSE Studies
When IFN was chosen as BAT in the RESPONSE studies, only 19% of patients treated with IFN reached Hct control vs 62% of the subjects who received RUX. This is consistent with the data observed with other types of BAT.40,61 In this population of HU-resistant or intolerant patients, RUX consistently outperformed IFN also concerning symptom relief, spleen volume reduction, decrease phlebotomy necessity and improvement in CHR.40,61 Additionally, it was demonstrated that a prior IFN use did not affect toxicity profile and efficacy of RUX. In fact, the proportion of primary responders (who achieved Hct control) in the overall RUX-treated population (60% and 64% in RESPONSE and RESPONSE-2 trial, respectively) was comparable with that in the RUX-treated patients with IFN exposure before randomization (61% and 67%, respectively).40,61
Following crossover to RUX in the RESPONSE studies, patients treated with IFN in the standard arm exhibited clinical improvements in hematologic and spleen responses and an overall reduction in phlebotomy requirements. Furthermore, the rates of the most common toxicities decreased after crossover to RUX, except for infections. Even if the number of IFN-treated patients evaluated in this study is quite small (n = 26), the findings suggest that treatment with IFN following HU resistance or intolerance may not be adequate to fully control PV probably due to advanced disease in patients enrolled in the RESPONSE studies.61,62
However, IFN in the RESPONSE studies was administered in five different forms and at various doses and this could have undermined the response rate confronted to the homogeneous treatment regimens of other trials. Although it is consistent with the low incidence (≈ 9%) of IFN use reported in literature,62 the fact that IFN was chosen rarely as BAT in both studies (14% of BAT-treated patients) may be due to the scarcity of data from randomized phase 3 trials and poor experience with IFN treatment in the clinical setting, probably as a consequence of the lack of IFN approval and reimbursement in PV in some countries.62
Others
Silver et al57 analyzed IFN-α2 treatment in 55 PV patients previously treated with phlebotomies alone or phlebotomies plus HU, with a median follow-up of 13 years. CHR were achieved after 1 or 2 years of therapy. Disease free survival was 10 years. All patients experienced flu-like symptoms (such as fever, muscle aches, chills and fatigue) as main AE but only eight patients had to discontinue the treatment. It is important to note that the 2006 CHR criteria were different from the more recent ones:63 in 2006, CHR was defined, among others, by platelet count <600 × 109/L whereas the actual criteria establish a CHR for platelet count ≤400 × 109/L.
Peg-IFN-α2
Peg-IFN-α2 was studied in a Phase II trial with 7-years of follow-up in 83 ET and PV patients, previously treated with HU, anagrelide or phlebotomies. Overall hematological responses were 80% and molecular responses 63%. 22% of patients discontinued treatment due to therapy toxicity. Rates of transformation to myelofibrosis or acute myeloid leukemia were similar to the cases used as historical control series.50
In PV patients resistant or refractory to hydroxyurea, peg-IFN-α2 can achieve an overall response rate of 60% after 12 months of therapy. Patients with CR showed a significant reduction of JAK2 V617F variant allele fraction compared to PR or NR patients. Treatment was associated with a significant rate of AE: mainly anemia and leucopenia, observed in 25% and 20.3% of patients, respectively. The most frequent non-hematological AE was fatigue (43.8%), followed by headache (31.3%), nausea and pruritus (23.4%).58
MPN-RC 112 was a randomized Phase III clinical trial comparing HU and peg-IFN-α2 as first line treatment in high-risk PV and ET patients. CR rates at 12 and 24 months were similar in the two groups, with no significant differences. Higher AE rates were detected in peg-IFN-α2 arm. Similarly in both arms, a decrease of karyotypical abnormalities and JAK2V617F variant allele fraction (VAF) was shown.64
Being PV a chronic disease, we have to consider the general rule of cancer biology: MPN typical molecular mutations cause genomic instability and this can easily lead to clonal evolution. A new approach, based on the evidence that IFN can induce deep and long molecular remissions (<1% mutated JAK2V617F alleles), suggests that an early treatment could inhibit the malignant clone and improve the long-term outcomes of PV patients, even after years of therapy discontinuation.53 In a recent study, a combination therapy with peg-IFN-α2 and RUX has been evaluated in resistant or intolerant to IFN-α2 therapy PV and myelofibrosis patients. In PV patients, CHR were 44% and PR 9%; JAK2 V617F allele burden decreased in all responders and hematological toxicity was the main AE. Discontinuation rate was about 20%. Further randomized studies are needed to confirm these results.65
Ropeg-INF-α-2b
In a prospective phase I/II study, the mono-peg-IFN-α2b isoform, ropeg-INF-α-2b, administered every two weeks, was studied in 51 PV patients (1/3 of patients was on HU therapy and 2/3 of patients were treated with phlebotomies). The hematological overall response rate was 90% (CR 47%, PR 43%); complete molecular responses (CMR) were seen in 21% of patients, while partial molecular response (PMR) in 47%. There was no correlation between dose levels and response rate or duration, suggesting that ropeg-INF-α-2b can be used at low doses, reducing toxicities. Toxicities were acceptable (only 20% of patients experiences flu-like symptoms).66
Phase III randomized clinical trials, PROUD-PV and its extension study CONTINUATION-PV, compared ropeg-INF-α-2b and HU in 257 PV patients, either new to cytoreduction or HU experienced (but neither intolerant nor complete responders). A hematologic response was observed in 43% (ropeg-INF-α-2b arm) vs 46% (HU arm) of patients at 12 months (PROUD-PV) and in 71% (ropeg-INF-α-2b arm) vs 51% (HU arm) of patients at 36 months (CONTINUATION-PV).67
Need for phlebotomy within 3 months dropped from 86% to 6%. Almost 37% of patients treated with ropeg-INF-α-2b achieved a JAK2 molecular response (PMR or CMR).68 The most frequently reported grade 3 and grade 4 AEs were increased γ-glutamyltransferase (6%) and increased alanine aminotransferase (3%) in the ropeg-INF-α-2b group, and leucopenia (5%) and thrombocytopenia (4%) in the standard therapy group. Ropeg-INF- α-2b related serious AEs occurred in 2% of patients vs 4% in HU group. (stessa ref)
Recently, Barbui et al showed that supplementing phlebotomy with ropeg-INF-α-2b seems to be safe and effective in steadily maintaining Hct values on target even in low-risk PV patients.69
Emerging Treatments
Currently, in PV treatment, several molecules have been tested or are under investigation. These novel agents are evaluated not only to address the standard goals of PV treatment but also with the aim to act directly on the malignant clone in order to arrest or delay disease progression.70 Unfortunately, many trials have failed to reach a significative improvement in terms of response rates and symptoms relief (Table 5), but others are still ongoing and the results will be available in the next years (Table 6). At present, the drug that has shown the most encouraging results is givinostat.
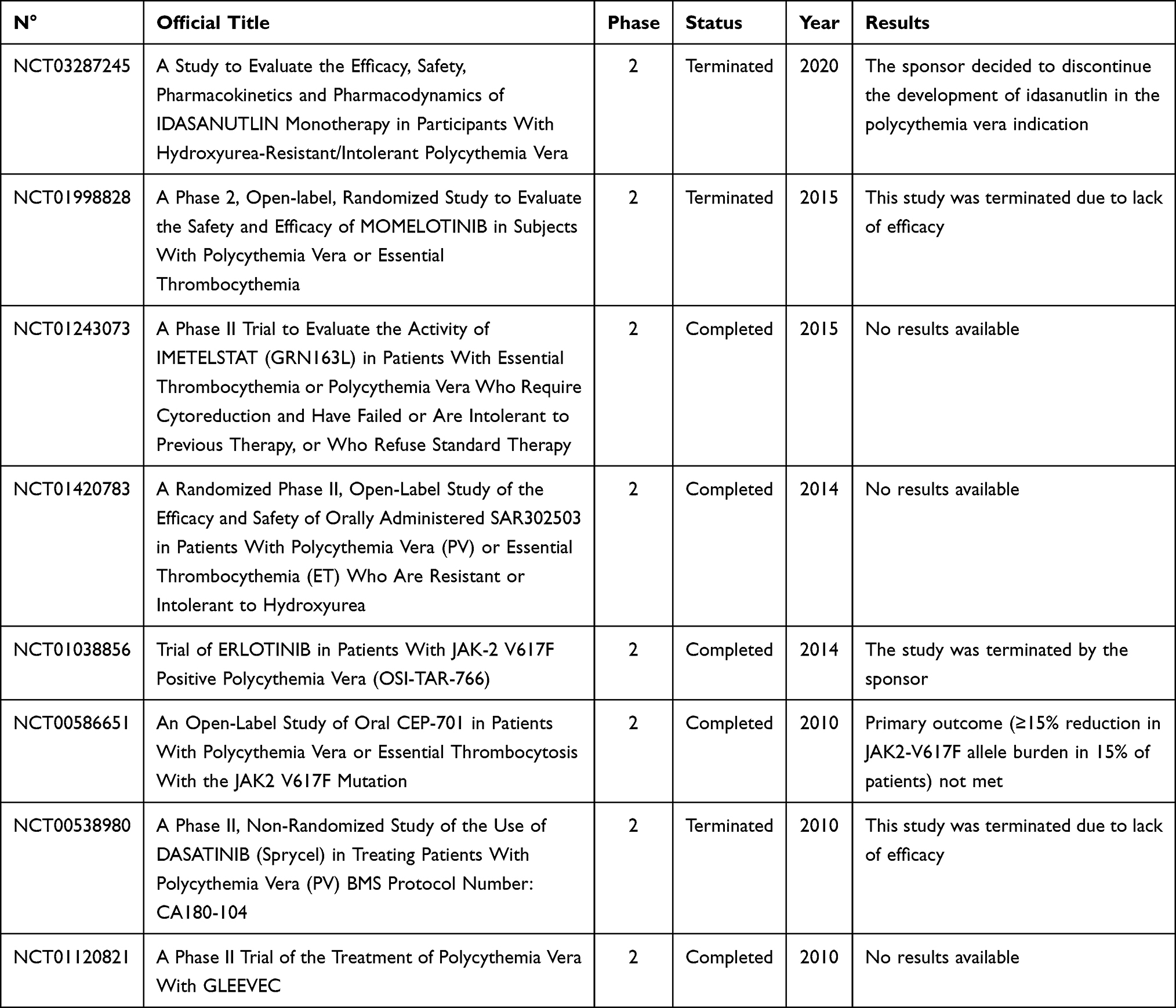 |
Table 5 Failed Clinical Trials |
 |
Table 6 Ongoing Clinical Trials |
Givinostat
Epigenome regulates gene expression; changes to the epigenome can result in changes to the structure of chromatin and changes to the function of the genome. For this reason, it has become in the last years an attractive target in cancer. Histone modifications play a central role in epigenetics of cancer. Particularly, histone deacetylation, catalyzed by histone deacetylases (HDACs), is known to be responsible for silencing tumor suppressor genes expression.71
Givinostat is an orally bioavailable, potent inhibitor of class I and II HDACs. It has been demonstrated that in vitro it induces apoptosis of neoplastic cells and inhibits the synthesis of many pro-inflammatory cytokines. It also acts directly on the JAK2V617F mutated cells reducing their proliferation through a process involving downmodulation of JAK2 protein and inhibition of its downstream signaling. In addition, givinostat promotes the growth of non-mutated over mutated colonies showing the potential to restore normal hematopoiesis in PV patients.72,73
The efficacy and safety of givinostat at a starting dose of 50 mg twice daily were evaluated in a pilot phase IIA study enrolling 29 JAK2 V617F-positive MPN patients, 12 of whom with PV. In the PV cohort, one complete and six partial hematological responses were reached, while 2 patients discontinued treatment. Seven out of ten PV patients (70%) reached a phlebotomy free status; splenomegaly and pruritus were resolved in 70% (7/10) and 90% (9/10) of patients, respectively. Reduction of JAK2 V617F allele burden was also reported. Therapy was safe with no severe toxicities recorded.74 In a subsequent randomized phase II study including 44 PV patients unresponsive to HU, givinostat was evaluated at a dose of 50 mg once daily or twice daily in combination with HU at maximum tolerated dose (MTD). Complete plus partial responses were achieved in 55% vs 50% of patients treated with 50 and 100 mg/day, respectively. Pruritus control was achieved in 64% versus 67% of subjects, respectively. Grade 3 AEs were recorded in low similar percentages (about 4.5%) in both groups, while no patients reported grade 4 AEs.75 Subsequently, a multicenter clinical trial evaluating long-term efficacy and safety of givinostat was performed, allowing PV patients achieving clinical benefit from the drug to continue the treatment. A total of 45 patients were enrolled and received treatment for a median of 4 years, including 32% of patients treated for ≥7 years. Complete and partial hematological remissions were reported in 11% and 89% of patients, respectively. A reduction of Hct below 45% without phlebotomy and a normal spleen size were both observed in 56% of patients; control of pruritus was achieved in 89% of subjects. The overall incidence of thrombosis was 2.3% patients/year. Reduction of JAK2 V617F allele burden was reported in 22% of the subjects. Givinostat was well tolerated also during long-term treatment with only three grade 3 and no grade 4 toxicities observed.76 Recently, a phase Ib/II study was developed with the aim to determine the MTD of givinostat as monotherapy. The recommended dose identified in the first phase of the study was 100 mg twice daily in 4-week cycles. Thirty-five patients were then enrolled in the phase II part; the overall response rate was 80.6%, 3 patients achieved CR after 3 cycles and 1 after 6 cycles. In addition, efficacy in terms of symptoms relief and a significant decrease in JAK2V617F VAF were observed. The safety of givinostat was confirmed, with only two grade 3 toxicities recorded.77 On the basis of the encouraging efficacy and safety data of the early phase trials, a global phase III study that will evaluate the efficacy of givinostat versus HU in untreated high-risk JAK2 V617F PV patients has been planned for the next future.
ASH2020 News
At the last ASH meeting data from a phase II study with idasanutlin on 16 HU-resistant/intolerant patients were presented. At week 32, 8 of 16 pts (50.0%) achieved a CHR, overall response rates per modified ELN response criteria in patients with baseline splenomegaly, patients without baseline splenomegaly and in all patients were 69.2% (9 of 13), 66.7% (2 of 3) and 68.8% (11 of 16), respectively; moreover, 6 of 14 pts (42.9%) had a ≥ 50% reduction in the Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF-TTS). However, the low-grade gastrointestinal toxicity profile of idasanutlin was not effectively mitigated with antiemetic prophylaxis and led to frequent discontinuations, greatly limiting the possible use of idasanutlin in these patients.75
A phase II trial, evaluating PTG-300, a hepcidin-mimetic, in 13 PV patients and ≥3 phlebotomies with or without concurrent cytoreductive therapy, showed efficacy in reversing iron deficiency and eliminating the need for phlebotomy in both low and high-risk patients.76
Conclusions
In conclusion, the main aims in the treatment of PV patients are the reduction of thrombosis events and the decrease of symptoms burden, both crucial for impairing patient’s quality of life. Modern therapies for PV, while not curative, can relieve symptoms and prolong survival. As summarized in Figure 1, treatment options should be based on age and history of thrombosis.
Hydroxyurea remains still nowadays the cytoreductive agent of first choice, reserving Interferon to young patients or childbearing women, since it is an appropriate therapeutic option but not available in many countries.
During the last years, ruxolitinib emerged as a new treatment in PV patients, as second line therapy: it appeared especially effective in patients with severe pruritus, symptomatic splenomegaly, or post-PV myelofibrosis symptoms.
Many emerging treatments have been evaluated in the last years, especially for HU resistant patients. Unfortunately, conclusive results are still lacking. At present, givinostat seems to be the most promising novel drug and a phase III trial is ready to start in the near future. Givinostat complete clinical trials results will be available in the next years.
There are still open questions about more consolidated PV treatments, for example, studies comparing IFN and ruxolitinib might be of interest but are lacking. Furthermore, the evolving response criteria make it difficult to compare the results of clinical trials conducted in different periods of time. For all these reasons, further efforts are needed to define the best current therapeutic approach in PV patients and to find the most promising novel drugs that will be available for the future.
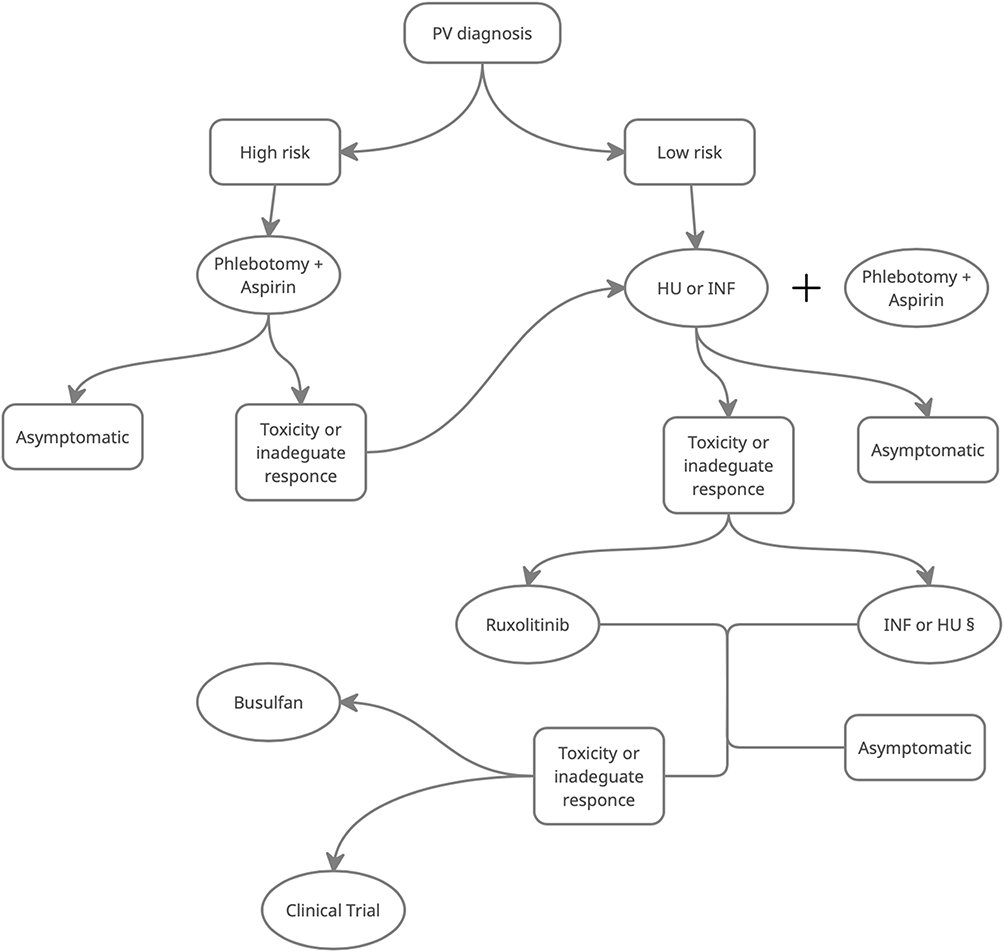 |
Figure 1 Flow chart indicating suggest approach to patients with diagnosis of PV. §Use the one which was not used as first line. Abbreviations: INF, interferon; HU, hydroxyurea; low risk, age<60 and no history of prior thrombosis; high risk, age >60 and positive history of prior thrombosis |
Disclosure
Dr Giulia Benevolo reports honoraria from Novartis, Takeda, Celgene, Amgen, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. McMullin MF, Harrison CN, Ali S, et al. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Br J Haematol. 2019.
2. Spivak JL. How I treat polycythemia vera. Blood. 2019;134(4):341–352. doi:10.1182/blood.2018834044
3. Passamonti F. How I treat polycythemia vera. Blood. 2012;120(2):275–284. doi:10.1182/blood-2012-02-366054
4. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. doi:10.1038/leu.2013.163
5. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30(33):4098–4103. doi:10.1200/JCO.2012.42.3863
6. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118(2):401–408. doi:10.1182/blood-2011-01-328955
7. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224–2232. doi:10.1200/JCO.2005.07.062
8. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi:10.1016/S0140-6736(05)71142-9
9. Pietra D, Li S, Brisci A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111(3):1686–1689. doi:10.1182/blood-2007-07-101576
10. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. 2011;117(10):2813–2816. doi:10.1182/blood-2010-11-316810
11. Tefferi A, Lasho TL, Guglielmelli P, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21–30. doi:10.1182/bloodadvances.2016000216
12. Senín A, Fernández-Rodríguez C, Bellosillo B, et al. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann Hematol. 2018;97(3):443–451. doi:10.1007/s00277-017-3193-5
13. Arber DA, Orazi A, Hasserjian R, et al. The 2016revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
14. Barbui T, Thiele J, Gisslinger H, et al. Masked polycythemia Vera (mPV): results of an international study. Am J Hematol. 2014;89(1):52–54. doi:10.1002/ajh.23585
15. Barbui T, Vannucchi AM, Carobbio A, et al. The effect of arterial hypertension on thrombosis in low-risk polycythemia vera. Am J Hematol. 2017;92(1):E5–E6. doi:10.1002/ajh.24583
16. Vrsalovic MM, Pejsa V, Veic TS, et al. Bone marrow renin-angiotensin system expression in polycythemia vera and essential thrombocythemia depends on JAK2 mutational status. Cancer Biol Ther. 2007;6(9):1430–1432. doi:10.4161/cbt.6.9.4568
17. Vlahakos DV, Marathias KP, Agroyannis B, Madias NE. Posttransplant erythrocytosis. Kidney Int. 2003;63(4):1187–1194. doi:10.1046/j.1523-1755.2003.00850.x
18. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–124. doi:10.1056/NEJMoa035572
19. Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. 2012;119(15):3595–3603. doi:10.1182/blood-2011-06-359224
20. Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am J Hematol. 2019;94(1):133–143. doi:10.1002/ajh.25303
21. Marchioli R, Finazzi G, Specchia G, Masciulli A, Mennitto MR, Barbui T. The CYTO-PV: a large-scale trial testing the intensity of CYTOreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis. 2011;2011:1–9. doi:10.1155/2011/794240
22. Berk PD, Goldberg JD, Donovan PB, Fruchtman SM, Berlin NI, Wasserman LR. Therapeutic recommendations in polycythemia vera based on Polycythemia Vera Study Group protocols. Semin Hematol. 1986.
23. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. doi:10.1056/NEJMoa1208500
24. Di Nisio M, Barbui T, Di Gennaro L, et al. The haematocrit and platelet target in polycythemia vera. Br J Haematol. 2007;136(2):249–259. doi:10.1111/j.1365-2141.2006.06430.x
25. Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010;148(6):961–963. doi:10.1111/j.1365-2141.2009.08019.x
26. Harrison CN, Schaap N, Vannucchi AM, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017.
27. Samuelsson J, Hasselbalch H, Bruserud O, et al. A phase II trial of pegylated interferon α-2b therapy for polycythemia vera and essential thrombocythemia: feasibility, clinical and biologic effects, and impact on quality of life. Cancer. 2006;106(11):2397–2405. doi:10.1002/cncr.21900
28. Jabbour E, Kantarjian H, Cortes J, et al. PEG-IFN-α-2b therapy in BCR-ABL-negative myeloproliferative disorders: final result of a phase 2 study. Cancer. 2007;110(9):2012–2018. doi:10.1002/cncr.23018
29. Yazdani Brojeni P, Matok I, Garcia Bournissen F, Koren G. A systematic review of the fetal safety of interferon alpha. Reprod Toxicol. 2011;33(3):265–268. doi:10.1016/j.reprotox.2011.11.003
30. Finazzi G, Ruggeri M, Rodeghiero F, Barbui T. Second malignancies in patients with essential thrombocythaemia treated with busulphan and hydroxyurea: long-term follow-up of a randomized clinical trial. Br J Haematol. 2014;110(3):577–583. doi:10.1046/j.1365-2141.2000.02188.x
31. Alvarez-Larrán A, Martínez-Avilés L, Hernández-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol. 2014;93(12):2037–2043. doi:10.1007/s00277-014-2152-7
32. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778–4781. doi:10.1182/blood-2013-01-478891
33. Mesa RA, Jamieson C, Bhatia R, et al. Myeloproliferative neoplasms, version 2.2018 featured Updates to the NCCN guidelines. JNCCN J Natl Compr Cancer Netw. 2017;15(10):1193–1207. doi:10.6004/jnccn.2017.0157
34. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. doi:10.1056/NEJMoa1409002
35. Kiladjian -J-J, Verstovsek S, Griesshammer M, et al. Results from the 208-Week (4-Year) Follow-up of RESPONSE Trial, a Phase 3 Study Comparing Ruxolitinib (Rux) with Best Available Therapy (BAT) for the Treatment of Polycythemia Vera (PV). Blood. 2017.
36. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101(7):821–829. doi:10.3324/haematol.2016.143644
37. Kiladjian JJ, Zachee P, Hino M, et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. 2020.
38. Antonioli E, Guglielmelli P, Pieri L, et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am J Hematol. 2012;87(5):552–554. doi:10.1002/ajh.23160
39. Vannucchi AM, Verstovsek S, Guglielmelli P, et al. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol. 2017;96(7):1113–1120. doi:10.1007/s00277-017-2994-x
40. Griesshammer M, Saydam G, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythemia vera without splenomegaly: 80-week follow-up from the RESPONSE-2 trial. Ann Hematol. 2018;97(9):1591–1600. doi:10.1007/s00277-018-3365-y
41. Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxycarbamide therapy versus switching to ruxolitinib in patients with polycythaemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Br J Haematol. 2017;176(1):76–85. doi:10.1111/bjh.14382
42. Harrison C, McMullin MF, Panchal A, et al. Ruxolitinib compared with best available therapy for polycythaemia vera patients resistant or intolerant to hydroxycarbamide in MAJIC-an investigator-led randomised trial. HemaSphere. 2018.
43. Curto-Garcia N, Baxter J, Harris E, et al. S1607 molecular analysis in Majic PV correlation with clinical endpoints. HemaSphere. 2019;3(S1):740. doi:10.1097/01.HS9.0000564676.68330.b5
44. Masciulli A, Ferrari A, Carobbio A, Ghirardi A, Barbui T. Ruxolitinib for the prevention of thrombosis in polycythemia vera: a systematic review and meta-analysis. Blood Adv. 2020;4(2):380–386. doi:10.1182/bloodadvances.2019001158
45. Levine RL, Wernig G. Role of JAK-STAT signaling in the pathogenesis of myeloproliferative disorders. Hematology Am Soc Hematol Educ Program. 2006;2006(1):233–239. doi:10.1182/asheducation-2006.1.233
46. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–386. doi:10.1038/nri1604
47. Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (80-). 1994;264(5164):1415–1421. doi:10.1126/science.8197455
48. Marty C, Lacout C, Droin N, et al. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia. 2013;27(11):2187–2195. doi:10.1038/leu.2013.102
49. Bjørn ME, Hasselbalch HC. The role of reactive oxygen species in myelofibrosis and related neoplasms. Mediators Inflamm. 2015;2015:1–11. doi:10.1155/2015/648090
50. Mascarenhas JO, Prchal JT, Rambaldi A, et al. Interim analysis of the Myeloproliferative Disorders Research Consortium (MPD-RC) 112 Global Phase III Trial of Front Line Pegylated Interferon Alpha-2a vs. hydroxyurea in high risk polycythemia vera and essential thrombocythemia. Blood. 2016;128(22):479. doi:10.1182/blood.V128.22.479.479
51. Kiladjian JJ, Chomienne C, Fenaux P. Interferon-α therapy in bcr-abl-negative myeloproliferative neoplasms. Leukemia. 2008;22(11):1990–1998. doi:10.1038/leu.2008.280
52. Stauffer Larsen T, Iversen KF, Hansen E, et al. Long term molecular responses in a cohort of Danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk Res. 2013;37(9):1041–1045. doi:10.1016/j.leukres.2013.06.012
53. Larsen TS, Møller MB, De Stricker K, et al. Minimal residual disease and normalization of the bone marrow after long-term treatment with alpha-interferon2b in polycythemia vera. A report on molecular response patterns in seven patients in sustained complete hematological remission. Hematology. 2009;14(6):331–334. doi:10.1179/102453309X12473408860587
54. Hasselbalch HC, Larsen TS, Riley CH, Jensen MK, Kiladjian -J-J. Interferon-alpha in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Status and perspectives. Curr Drug Targets. 2011.
55. Quintás-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27(32):5418–5424. doi:10.1200/JCO.2009.23.6075
56. Kiladjian -J-J, Giraudier S, Cassinat B. Interferon-alpha for the therapy of myeloproliferative neoplasms: targeting the malignant clone. Leukemia. 2016;30(4):776–781. doi:10.1038/leu.2015.326
57. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-α. Cancer. 2006;107(3):451–458. doi:10.1002/cncr.22026
58. Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112(8):3065–3072. doi:10.1182/blood-2008-03-143537
59. Frederiksen H, Farkas DK, Christiansen CF, et al. Survival of patients with chronic myeloproliferative neoplasms and new primary cancers: a population-based cohort study. Lancet Haematol. 2015;2(7):e289–e296. doi:10.1016/S2352-3026(15)00092-7
60. Frederiksen H, Farkas DK, Christiansen CF, Hasselbalch HC, Sørensen HT. Chronic myeloproliferative neoplasms and subsequent cancer risk: a Danish population-based cohort study. Blood. 2011;118(25):6515–6520. doi:10.1182/blood-2011-04-348755
61. Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 2017;18(1):88–99. doi:10.1016/S1470-2045(16)30558-7
62. Kiladjian -J-J, Guglielmelli P, Griesshammer M, et al. Efficacy and safety of ruxolitinib after and versus interferon use in the RESPONSE studies. Ann Hematol. 2018;97(4):617–627. doi:10.1007/s00277-017-3225-1
63. Savona MR, Malcovati L, Komrokji R, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015;125(12):1857–1865. doi:10.1182/blood-2014-10-607341
64. Mascarenhas J, Kosiorek HE, Prchal JT, et al. Results of the Myeloproliferative Neoplasms - Research Consortium (MPN-RC) 112 randomized trial of Pegylated Interferon Alfa-2a (PEG) versus hydroxyurea (HU) therapy for the treatment of high Risk Polycythemia Vera (PV) and high risk essential thrombocythemia. Blood. 2018;132(Supplement 1):577. doi:10.1182/blood-2018-99-111946
65. Mikkelsen SU, Kjær L, Bjørn ME, et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med. 2018;7(8):3571–3581. doi:10.1002/cam4.1619
66. Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, et al. Ropeginterferon alfa-2b, a novel IFNα-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015;126(15):1762–1769. doi:10.1182/blood-2015-04-637280
67. Gisslinger H, Klade C, Georgiev P, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020;7(3):e196–e208. doi:10.1016/S2352-3026(19)30236-4
68. Gisslinger H, Klade C, Georgiev P, et al. Final results from PROUD-PV a randomized controlled Phase 3 trial comparing Ropeginterferon Alfa-2b to hydroxyurea in polycythemia vera patients. Blood. 2016;128(22):475. doi:10.1182/blood.V128.22.475.475
69. Barbui T, Vannucchi AM, De Stefano V, et al. Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): a multicentre, randomised phase 2 trial. Lancet Haematol. 2021. doi:10.1016/S2352-3026(20)30373-2
70. Tremblay D, Mascarenhas J. Novel therapies in polycythemia vera. Curr Hematol Malig Rep. 2020;15(2):133–140. doi:10.1007/s11899-020-00564-7
71. Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32(3):157–165. doi:10.1016/j.ctrv.2005.12.006
72. Chifotides HT, Bose P, Verstovsek S. Givinostat: an emerging treatment for polycythemia vera. Expert Opin Investig Drugs. 2020;29(6):525–536. doi:10.1080/13543784.2020.1761323
73. Guerini V, Barbui V, Spinelli O, et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2V617F. Leukemia. 2008;22(4):740–747. doi:10.1038/sj.leu.2405049
74. Rambaldi A, Dellacasa CM, Finazzi G, et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010. doi:10.1111/j.1365-2141.2010.08266.x
75. Finazzi G, Vannucchi AM, Martinelli V, et al. A phase II study of Givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol. 2013;161(5):688–694. doi:10.1111/bjh.12332
76. Finazzi G, Iurlo A, Martino B, et al. A long-term safety and efficacy study of givinostat in patients with polycythemia vera: the first 4 years of treatment. Blood. 2017;130(17):1873–1874. doi:10.1182/blood-2017-08-802165
77. Rambaldi A, Iurlo A, Vannucchi AM, et al. Safety and efficacy of the maximum tolerated dose of givinostat in polycythemia vera: a two-part Phase Ib/II study. Leukemia. 2020;34(8):2234–2237. doi:10.1038/s41375-020-0735-y
78. Iurlo A, Cattaneo D, Bucelli C, Baldini L. New Perspectives on Polycythemia Vera: From Diagnosis to Therapy. Int J Mol Sci. 2020;21(16):5805. PMID: 32823537; PMCID: PMC7461104. doi:10.3390/ijms21165805
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.