Back to Journals » International Journal of Nanomedicine » Volume 15
Polyanhydride Nanoparticles Induce Low Inflammatory Dendritic Cell Activation Resulting in CD8+ T Cell Memory and Delayed Tumor Progression
Authors Darling R ![]() , Senapati S
, Senapati S ![]() , Christiansen J, Liu L, Ramer-Tait AE
, Christiansen J, Liu L, Ramer-Tait AE ![]() , Narasimhan B
, Narasimhan B ![]() , Wannemuehler M
, Wannemuehler M ![]()
Received 3 May 2020
Accepted for publication 2 July 2020
Published 7 September 2020 Volume 2020:15 Pages 6579—6592
DOI https://doi.org/10.2147/IJN.S261041
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Ross Darling,1 Sujata Senapati,2 John Christiansen,1 Luman Liu,2 Amanda E Ramer-Tait,3 Balaji Narasimhan,2,4 Michael Wannemuehler1,4
1Department of Veterinary Microbiology and Preventative Medicine, Iowa State University, Ames, IA, USA; 2Department of Chemical and Biological Engineering, Iowa State University, Ames, IA, USA; 3Department of Food Science and Technology, University of Nebraska-Lincoln, Lincoln, NE, USA; 4Nanovaccine Institute, Iowa State University, Ames, IA, USA
Correspondence: Michael Wannemuehler; Balaji Narasimhan Email [email protected]; [email protected]
Introduction: Adjuvants and immunotherapies designed to activate adaptive immunity to eliminate infectious disease and tumors have become an area of interest aimed at providing a safe and effective strategy to prevent or eliminate disease. Existing approaches would benefit from the development of immunization regimens capable of inducing efficacious cell-mediated immunity directed toward CD8+ T cell-specific antigens. This goal is critically dependent upon appropriate activation of antigen-presenting cells (APCs) most notably dendritic cells (DCs). In this regard, polyanhydride particles have been shown to be effectively internalized by APCs and induce activation.
Methods: Here, a prophylactic vaccine regimen designed as a single-dose polyanhydride nanovaccine encapsulating antigen is evaluated for the induction of CD8+ T cell memory in a model system where antigen-specific protection is restricted to CD8+ T cells. Bone marrow-derived dendritic cells (BMDCs) are used as an in vitro model system to evaluate the magnitude and phenotype of APC activation. Primary DCs, particularly those with described ability to activate CD8+ T cells, are also evaluated for their in vitro responses to polyanhydride nanoparticles.
Results: Herein, polyanhydride nanoparticles are shown to induce potent in vitro upregulation of costimulatory molecules on the cell surface of BMDCs. In contrast to the classically used TLR agonists, nanoparticles did not induce large amounts of pro-inflammatory cytokines, did not induce characteristic metabolic response of DCs, nor produce innate antimicrobial effector molecules, such as nitric oxide (NO). The polyanhydride nanovaccine results in protective CD8+ T cell responses as measured by inhibition of tumor progression and survival.
Discussion: Together, these results suggest that the use of a polyanhydride-based nanovaccine can be an effective approach to inducing antigen-specific CD8+ T cell memory by providing antigen delivery and DC activation while avoiding overt inflammatory responses typically associated with traditional adjuvants.
Keywords: nanovaccine, metabolism, nitric oxide, T cell memory, inflammation
Introduction
Cytotoxic CD8+ T cells are critical mediators of immunological protection against tumors and intracellular pathogens.1 Although naturally occurring infections are often effective at inducing long-lived CD8+ T cell memory, commonly used adjuvants such as alum, oil-in-water emulsions, or innate immune stimulating Toll-like receptor (TLR) ligands have not proven as successful for the induction of cell-mediated immunity (CMI).2 Many efficacious immunization strategies that induce rapid and robust/durable memory CD8+ T cell responses have either employed a multiple-dose regimen (ie, prime-boost) or treated animals with adoptively transferred DCs pulsed in vitro with antigen.3–5 Single-dose DC vaccines that rapidly induce a large number of memory CD8+ T cells possess the relatively unique property of providing both antigen presentation and co-stimulation without the induction of overt inflammatory responses that result when using more standard antigen-adjuvant combinations or infections.4 Many adjuvants are incorporated into vaccine formulations to induce inflammation and innate immune activation with a focus on the induction of long-lived antibody titers as opposed to CMI.2 While beneficial to antibody tiers, adjuvant-associated proinflammatory cytokines and associated inflammation have been demonstrated to divert an intrinsic CD8+ T cell pathway towards induction of effector memory T cells. It has also been shown that overt inflammatory responses impair effector CD8+ T cell trafficking and function.4,6,7 These blunted T cell responses result in less effective generation of memory or effector T cells.4,6,7 Another common class of adjuvants, TLR agonists, not only induce the production of proinflammatory cytokines and overt inflammation but also induce innate immune effector molecules such as nitric oxide (NO).8–10 Nitric oxide in particular has been described to cause deleterious effects on DC activation via altered metabolism, decreased survival, and impaired co-stimulatory molecule upregulation.11–13 Although effective at generating rapid and elevated numbers of memory CD8+ T cells, the use of DC vaccines also comes with increased costs and the challenges associated with the personalized nature of generating ex vivo DC populations to be used therapeutically.14 Consequently, identification and development of novel, less inflammatory vaccine adjuvants and antigen delivery systems would benefit the induction of favorable outcomes using a more manageable immunization regimen.
To improve vaccines’ ability to induce optimal cytotoxic CD8+ T cell memory, design of vaccine delivery systems that mimic the beneficial effects of DC vaccination (ie, MHC I presentation of antigen) could be used. Polyanhydride particle-based vaccines in particular represent a unique alternative to existing vaccines that employ more traditional adjuvants. Nanoparticles (NPs) consisting of combinations of sebacic acid (SA), 1,6-bis(p-carboxyphenoxy)hexane (CPH), and 1.8-bis(p-carboxyphenoxy)-3,6-dioxaoctante (CPTEG) are readily phagocytosed by DCs and activate them as measured by cytokine and costimulatory molecule upregulation.15 Following administration in vivo, these nanovaccines are also known to persist in the local tissues, thus, facilitating the prolonged release of their encapsulated payload.16,17 When delivered subcutaneously, polyanhydride NPs induce a mild inflammatory response with no evidence of adverse histopathological reactions.16,18 Previous studies have also illustrated the ability of polyanhydride NPs to prophylactically enhance CD8+ T cell memory responses and elicit protection in a tumor challenge model.19 However, the addition of a TLR ligand such as CpG oligodeoxynucleotides (ODN) to the polyanhydride platform decreased relative efficacy indicating that the induction of inflammation adversely affected the generation of effector CD8+ T cell responses.19,20 Considering the unique innate immune stimulatory properties of polyanhydride NPs, the ability of this platform to activate DCs and induce an effective memory CD8+ T cells was tested and compared to the inflammatory TLR agonist CpG ODN. This was evaluated via an ovalbumin (Ova)-expressing tumor model where Ova is expressed cytosolically and antigen-specific protection is restricted to MHCI:CD8+ T cell-mediated immune responses.21–24
Materials and Methods
Materials Synthesis
Chemicals used for the polymer and nanoparticle synthesis, 4-p-hydroxybenzoic acid, tri-ethylene-glycol, and 1,6-dibromohexane were obtained from Sigma Aldrich (St. Louis, MO); dimethylformamide, acetic acid, acetonitrile, acetic anhydride, toluene, methylene chloride, pentane were purchased from Fisher Scientific (Fairlawn, NJ) and 4-p-flourobenzonitrile was purchased from Apollo Scientific (Cheshire, UK).
Polyanhydride Synthesis
Monomers of CPTEG and CPH were synthesized and used in a 20:80 molar ratio to synthesize a 20:80 CPTEG:CPH copolymer via melt polycondensation reaction as previously described.25 The purity and molecular weight of the copolymer was characterized using 1H nuclear magnetic resonance spectroscopy in deuterated chloroform. The molecular weight of the copolymer was about 5.3 kDa, consistent with previous work.26
Nanoparticle Synthesis
20:80 CPTEG:CPH nanoparticles encapsulating 5 wt.% Ova were synthesized using flash nanoprecipitation as previously described.27 Briefly, a solution of 20:80 CPTEG:CPH and ovalbumin in methylene chloride at a concentration of 20 mg/mL was poured into pentane at a solvent to anti-solvent ratio of 1:250. The nanoparticles were then collected using vacuum filtration. The particle morphology and size were examined using scanning electron microscopy. The average particle size of nanoparticles used for these studies was about 200 nm.
Animals
Female BALB/c mice, aged six to eight weeks, were obtained from Charles River Laboratories (Wilmington, MA) for in vitro studies. Female C57BL/6 mice, aged six to eight weeks, were obtained from Charles River and Envigo (Somerset, NJ). Studies involving the use of mice were conducted in accordance with Iowa State University guidelines for the care and use of animals and upon approval of the Institutional Animal Care and Use Committee.
Cell Culture
The E.G7-OVA (ATCC, Manassas, VA) cell line was cultured and maintained in RPMI 1640 (Cat #10-040-CM, Corning) medium containing 2 mM L-glutamine 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol, 0.4 mg/mL G418 (Cat #30-234-CI, Cellgro), 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% fetal bovine serum. The EL4 (ATCC) cell line was culture and maintained in DMEM (Cat #15-013-CV, Corning) supplemented with 2 mM L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10% horse serum.
Bone Marrow DC Generation
Naïve female BALB/c mice were used to generate BMDC cultures. Bone marrow was collected from femurs and tibias. Cells were washed and plated in RPMI 1640 medium supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin, 2 mM glutamine, and 10% FBS in 100 mm petri plates, at a density of 4 x 106 bone marrow cells per plate with 10 mL of medium supplemented with GM-CSF (Peprotech, Rocky Hill, NJ) at 10 ng/mL. On day three of culture, 10 mL of GM-CSF containing medium was added. On days six and eight of culture, 10 mL of culture medium was exchanged for fresh GM-CSF containing medium. DCs were harvested on day 10 of culture by gently rinsing and collecting non-adherent cells.
Splenic DC Isolation
Naïve female BALB/c mice were euthanized to collect spleens. Spleens from eight to ten mice were harvested, pooled, and used to generate a population of single cells. Using these splenocytes, highly enriched populations of DCs were recovered using a pan-DC isolation kit (13–100-875) (Miltenyi Biotec, Auburn, CA) on an autoMACS Pro separator (Miltenyi Biotec) according to manufacturer’s protocol.
BMDC and sDC Stimulation
DCs were plated at 5 x 105 cells/well in a 96-well round bottom tissue culture plate in 200 μL of RPMI 1640 medium supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin, 2 mM glutamine, and 10% FBS. The in vitro treatments consisted of 100 μg/well of 20:80 CPTEG:CPH NPs encapsulating 5 μg of Ova, 5 μg/mL of CpG ODN 1668 (Cat #16E17-MM InvivoGen) with 5 μg Ova, or medium alone (ie, control wells). After the addition of the stimulants, the BMDCs were incubated for 48 h after which supernatants and cells were harvested for cytokine analysis and cell surface marker expression by flow cytometry, respectively.
Nitric Oxide Quantification
Supernatants from stimulated DCs were analyzed for NO indirectly via nitrite concentration by Griess assay. A standard curve was created using two-fold dilutions of sodium nitrite ranging from 100 μM to 0 μM. One hundred microliters of supernatant was incubated with 100 μL of Griess reagents (Cat. No. 03553, Sigma-Aldrich) in a 96-well microtiter plate. Samples were incubated for 15 min at room temperature and read at 540 nm on a SpectraMAX 190 (Molecular Devices, Sunnyvale, CA).
Extracellular Flux Analysis
BMDCs from BALB/c mice were stimulated in RPMI 1640 medium supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin, 2 mM glutamine, and 10% FBS for 18 h with 100 μg of 20:80 CPTEG:CPH encapsulating 5 μg of Ova, 5 μg/mL of CpG ODN with 5 μg Ova, or no stimulation control in 5 mL polypropylene tubes. Treated BMDCs were washed with Seahorse assay media consisting of Agilent Seahorse XF Base medium (Cat #102,353, Agilent, Santa Clara, CA) supplemented with 1 mM sodium pyruvate, 2 mM L-glutamine, and 10 mM glucose with a pH adjusted to 7.4, were seeded into 24-well seahorse plates coated with Cell-Tak (Corning, Corning NY) at a density of 2.5 x 105 cells per well. Metabolic phenotyping was conducted on a Seahorse XFe24 (Agilent). Mitochondrial function was analyzed via mitochondrial stress test according to manufacturer specifications.28 Final concentrations of 1 μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone and antimycin were used (Agilent) and prepared in Seahorse assay medium.
Vaccination
Two antigen doses were evaluated in the in vivo studies. In the high antigen dose (2.0 mg Ova) studies, female C57BL/6 mice were vaccinated with formulations consisting of 250 μg Ova encapsulated in 5 mg of 20:80 CPTEG:CPH (5% loaded) plus 1.75 mg Ova soluble (NP), 2 mg soluble Ova (sOva), or unvaccinated control. In the prime-boost experiment, the mice were vaccinated with formulations consisting of 1.75 mg sOva along with 5 mg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 250 µg Ova with and without a boost (NPx1, NPx2), 2 mg soluble Ova with 5 mg of blank 20:80 CPTEG:CPH polyanhydride nanoparticles (Blank NP x2), or PBS control. Mice that received a booster (indicated by x2) were given the same formulations at half the original dose of antigen and particle mass. In the low antigen dose (100 µg Ova) studies, mice were vaccinated with formulations consisting of 75 µg soluble Ova plus 500 µg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 25 µg Ova (NP), 100 µg soluble Ova (sOva), or PBS control. In the multi-adjuvant experiment, mice received formulations consisting of 75 µg soluble Ova plus 500 µg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 25 µg Ova (NP), 100 µg soluble Ova adjuvanted with 20 µg CpG ODN 1668 (CpG), a combination of 75 µg soluble Ova plus 500 µg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 25 µg Ova adjuvanted with 20 µg CpG ODN (NP+CpG), or PBS control. All formulations were delivered subcutaneously at the nape of the neck.
Tumor Challenge
C57BL/6 mice were challenged subcutaneously on the flank with 2–5 x 106 E.G7 Ova-expressing lymphoma cells or 2.5 x 106 EL4 lymphoma cells that had been washed and suspended in PBS prior to implantation. Tumor growth was monitored three times a week and volumes were calculated by using the equation to determine the volume of an ellipsoid (see below). Mice were removed from study when tumor volume surpassed 1000 mm3. As mice did not succumb to the tumor and were removed based on the criteria of tumor volume, time on study is presented for survival analysis.
Tumor volume (V) = (4/3)π r1r2r3.
Cytokine and Chemokine Analysis
Cytokine quantification was performed using supernatants from the previously described DC stimulations. A Millipore Milliplex cytokine/chemokine panel (MCYTOMAG-70K-32, Burlington, MA) was used to detect cytokines and analyzed on a Bio-Plex 200 System (Bio-Rad, Hercules, CA) according to manufacturer’s specifications.
Flow Cytometry
Splenic DCs and BMDCs were analyzed for costimulatory marker expression using flow cytometry. 5 x 105 DCs were aspirated from a 96-well plate and transferred to FACS tubes. Prior to labeling with specific monoclonal antibodies, Fc receptors on DCs were blocked to prevent non-specific antibody binding by incubating the cells with 100 μg/mL of rat IgG (Sigma Aldrich) and 10 μg/mL of anti-CD16/32 (eBioscience, San Diego, CA). Subsequently, DCs were stained with fluorescently conjugated antibodies specific for CD80 (Biolegend, San Diego, CA, PerCP-Cy5.5, clone 16–10A1), CD86 (eBioscience, FITC, clone GL1), CD40 (eBioscience, APC, clone 1C10), CD11c (Biolegend, APC-Cy7, clone N418), MHCII (eBioscience, AF700, clone M5/114.15.2), and CD8α (Biolegend, BV421, clone 53–67) diluted in PBS containing 0.1% bovine serum albumin (BSA) and 0.1% sodium azide. Cells were fixed using BD stabilizing fixative (BD Bioscience, Franklin Lakes, NJ). Mitochondrial superoxide production was evaluated using live cells stained with MitoSOX Red (Cat # M36008, Invitrogen, Carlsbad, CA) according to manufacturer’s specifications. All data were collected on a FACSCanto II (BD Bioscience, Franklin Lakes, NJ). Data were analyzed using FlowJo (FlowJo LLC, Ashland, OR).
Antibody Responses
Where applicable, immunized mice were bled via saphenous vein 5 weeks post-immunization. Anti-Ova serum IgG titers were measured via indirect ELISA. Costar 3590 96-well EIA/RIA high binding plates (Corning) were coated with 100 µL of Ova (5 µg/mL PBS) or PA (0.5 µg/mL PBS) and incubated overnight at 4°C. Plates were blocked using 2.5% (w/v) powdered skim milk PBS containing 0.05% Tween-20 (PBS-T), that has been heat inactivated at 56°C for hours to inactivate any phosphatase activity, for two hours at room temperature. After three washes using PBS-T, serum samples were titrated across the plate using two-fold serial dilutions, starting at 1:100, in PBS-T and 1% (v/v) normal goat serum. Samples were incubated overnight at 4° C. After three washes in PBS-T, an alkaline phosphatase-conjugated goat anti-mouse IgG (H+L) secondary detection antibody (Cat# 115–005-003, Jackson ImmunoResearch, West Grove, PA) was diluted 1:1000 in PBS-T and added to the wells and allowed to incubate at room temperature for two hours. Plates were washed three times with PBS-T and alkaline phosphatase substrate was added at 1 mg/mL in buffer containing 50 mM sodium carbonate, 2 mM magnesium chloride, and sodium bicarbonate was titrated into the solution in order to achieve a pH of 9.3. Plates were allowed to develop for 30 min and analyzed using the SpectraMAX 190 at a wavelength of 405 nm.
Statistical Analysis
Data generated during flow cytometry assays, metabolic assays, and Griess assays were analyzed via one-way ANOVA with a Tukey, Dunnett, or Sidak post-test for multiple comparisons. Survival data were analyzed using a Log rank (Mantel-Cox) test with a Bonferroni correction for multiple comparisons. All analyses were performed using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA).
Results
High Antigen Dose Polyanhydride Nanoparticle vaccination Reduced Tumor Burden and Increased Time on Study
Previous studies from our laboratories have shown that immunization with a high dose (ie, 2 mg) of Ova in a polyanhydride particle-based vaccine regimen induced OTI CD8+ T cell memory and the ability of these CD8+ T cells to expand upon reencounter with antigen.27 Herein, the ability of polyanhydride nanoparticles to induce efficacious endogenous antigen-specific CD8+ T cell memory and subsequent effector expansion upon challenge was evaluated using a tumor challenge model, where antigen-specific protection is restricted to CD8+ T cell:MHCI recognition. C57BL/6 mice were vaccinated prophylactically with high antigen dose (2.0 mg Ova) formulations consisting of polyanhydride nanoparticles (NP) (characterization data of the NPs are shown in Supplemental Figure 1), sOva alone, or a PBS control. Six weeks later, mice were challenged subcutaneously by implanting Ova-expressing E.G7 tumor cells or EL4 tumor cells (ie, do not express Ova), and tumor progression was tracked. Vaccination with the NP formulation reduced tumor progression in comparison to naïve mice and mice immunized with the sOva alone (Figure 1A). NP vaccination also significantly improved time on study (ie, % of mice with tumors <1000 mm3) compared to control animals (Figure 1C). No significant improvements in survival were observed for mice receiving sOva alone compared to controls.
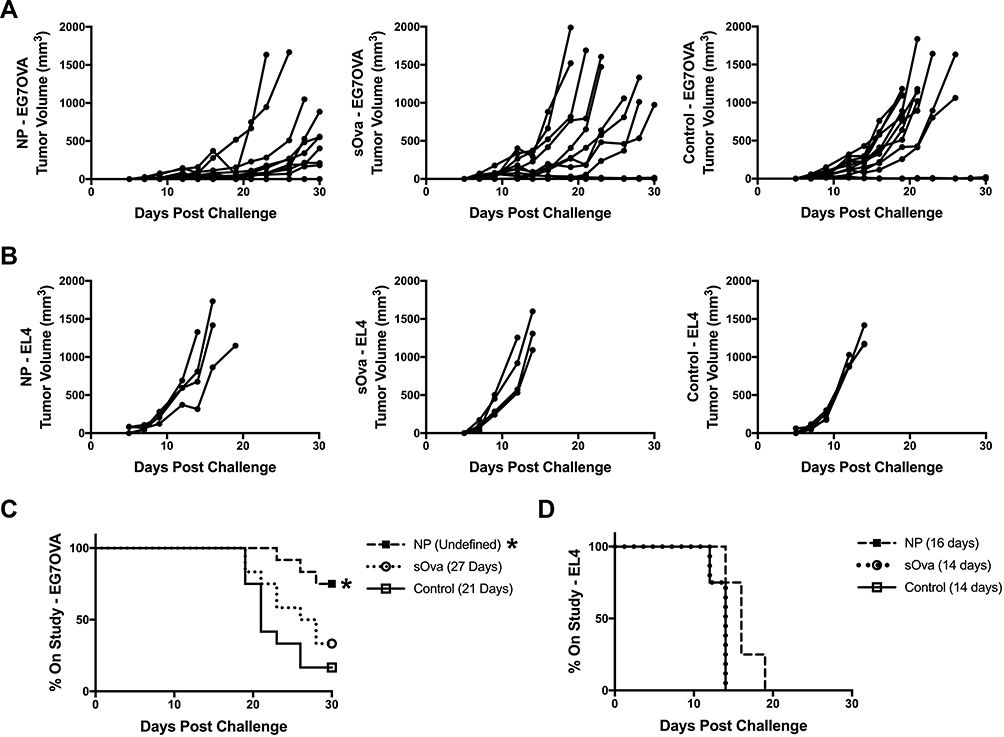 |
Figure 1 Polyanhydride nanoparticles enhance CD8+ T cell memory generation with high dose antigen. C57BL/6 female mice were immunized subcutaneously with formulations consisting of 1.75 mg soluble Ova plus 5 mg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 250 μg Ova (NP); 2 mg soluble Ova (sOva); or PBS (control). Mice were challenged subcutaneously in the flank with (A) EG7OVA Ova-expressing tumor cells (n=12 all groups) or (B) non-Ova expressing EL4 tumor cells (n=4) 40 days post-immunization and tumor volume of individual mice was tracked. Survival of mice receiving (C) EG7OVA tumor or (D) EL4 tumor was evaluated 30 days post-challenge. Significance from PBS control was determined using a Log rank (Mantel-Cox) test with a Bonferroni correction for multiple (2) comparisons. Significance from naïve control is indicated in the treatment group legend as follows *p ≤ 0.025. Median survival of each group is also reported parenthetically. |
To determine whether antigen-specific immune activation was responsible for the observed anti-tumor responses, another set of mice was challenged with the EL4 parent tumor that does not express Ova (Figure 1B and D). Neither the NP formulation nor sOva significantly improved time on study after subsequent tumor challenge (Figure 1B and D). Together, these data demonstrate that the NP formulation is able to induce efficacious antigen-specific, memory CD8+ T cells capable of responding to re-exposure to antigen (ie, tumor implantation).
Encapsulation of Antigen in Polyanhydride NPs is Critical for Inducing Optimal CD8+ T Cell Memory
Even though the high antigen dose (2.0 mg Ova) vaccine formulation contained a large amount of unencapsulated soluble antigen (1.75 mg), we hypothesized that the portion of antigen encapsulated within the polyanhydride NPs would be more important to the induction of CD8+ T cell memory than the soluble bolus. This hypothesis was tested by vaccinating C57BL/6 mice with a single-dose NP formulation, a prime-boost of the same NP formulation, a prime-boost of blank NPs with no encapsulated antigen (ie, soluble Ova only), or injecting PBS alone (ie, control). Six weeks after their last immunization, all mice were challenged subcutaneously with the Ova-expressing E.G7 tumor, and the tumor progression and time on study for each treatment compared (Figure 2A and B). The only treatments that significantly inhibited tumor progression compared to PBS injections were the single-dose NP (NP x1) and the prime-boost NP (NP x2) regimens. Mice immunized twice with blank NPs (x2) plus sOva tended to have an improved median survival time compared to naïve control mice; however, this difference was not significant.
 |
Figure 2 Encapsulated antigen is crucial to induce antigen-specific CD8+ T cell memory. C57BL/6 female mice were immunized subcutaneously at the base of the neck with formulations consisting of 1.75 mg soluble Ova with 5 mg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 250 μg Ova with (NP x2) and without a boost (NP x1) (n=16); 2 mg soluble Ova with 5 mg of blank 20:80 CPTEG:CPH polyanhydride nanoparticles (Blank NP x2) (n=8); or PBS control (n=16). Mice that received a booster (NP x2) were given the same formulations at half the original dose subcutaneously at the base of the neck 28 days after the primary immunization. Mice were challenged subcutaneously in the flank with E.G7 Ova-expressing tumor cells 42 days after the primary or boost immunization, respectively. (A) Tumor volume of individual mice was tracked. (B) Survival was evaluated 31 days post-challenge. Statistical significance was determined using a Log rank (Mantel-Cox) test with a Bonferroni correction for multiple (6) comparisons. Significance from naïve control group is indicated in the treatment group legend as *p ≤ 0.0083. Median survival of each group is also reported parenthetically. (C) Diagram of vaccination schedule. 1 ° = primary vaccination, 2 ° = primary vaccination, following which all mice received an injection of E.G7 tumor cells as described in Materials and Methods. |
Polyanhydride NPs Upregulated Co-Stimulatory Molecule Expression on DCs but Resulted in Low Levels of Inflammatory Cytokine and Chemokine Production
Previous work on polyanhydride particle formulations illustrated that bone marrow-derived dendritic cells (BMDCs) readily take up and respond to these particles by upregulating co-stimulatory molecules and cytokine secretion similar to other pathogen-mimicking moieties.15,17,29 The low inflammatory response induced by polyanhydride NP vaccines is similar to that induced by DC-based vaccines that support the induction of CD8+ T cell memory.18 Herein, the ability of the 20:80 CPTEG:CPH polyanhydride NP formulation to activate BMDC was compared to the effects of CpG ODN. After 48 h of in vitro stimulation with NPs or CpG ODN, BMDCs significantly (P < 0.05) upregulated the co-stimulatory molecules CD86 and CD40 as compared to non-stimulated BMDCs (Figure 3A). Supernatants from stimulated BMDCs were also evaluated for induction of inflammatory cytokine and chemokine production. Other than MIP2, CXCL1 (KC), and IL-6, the NP formulation induced secretion of markedly less inflammatory cytokines when compared to that induced by CpG ODN (Supplemental Figure 2).
 |
Figure 3 Polyanhydride nanoparticles upregulate costimulatory molecule expression on BMDCs and splenic DC populations. Dendritic cells isolated or generated from naïve BALB/c mice were stimulated for 48 hours with 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating Ova (5% w/w) (NP); CpG ODN plus Ova (CpG); or unstimulated control as described in materials and methods section. (A) BMDCs gated on MHCII+ and CD11c+, (B) the total MHCII+ and CD11c+ population of DCs isolated from the spleens, and (C) the CD8α+ subpopulation of splenic DCs was analyzed for costimulatory expression via flow cytometry. Significance was determined via one-way ANOVA with a Tukey multiple comparison test. P value is indicated as follows *p ≤ 0.05. All graphs represent the treatment average ± SEM. |
Dendritic cell populations isolated from the spleens of naïve mice were also evaluated for their response to stimulation. Splenic DCs (sDC) were stimulated with either the NP formulation or CpG ODN for 48 h. The CD11c+ population of the sDC was evaluated for upregulation of the costimulatory molecules CD86 and CD40 (a gating diagram is shown in Supplemental Figure 3). The NP formulation upregulated CD86 and CD40 expression while CpG ODN only upregulated CD40 as compared to non-stimulated sDC (Figure 3B). The CD8α+ subpopulation of sDC has been previously reported to cross-prime CD8+ T cells.4,30–32 To determine whether NP stimulation activated CD8α+ sDC, we measured co-stimulatory marker expression on CD8α+ sDC. CpG ODN induced only a moderate upregulation of co-stimulatory molecules on CD8α+ sDC; however, NP stimulation induced a marked increase in the expression of CD86, CD80, and CD40 (Figure 3C). These findings suggest that NPs are capable of activating a critically important DC subtype involved in the presentation of antigen to and activation of CD8+ T cells. Supernatants from the total population of stimulated sDC were also evaluated for cytokine production via a multiplex assay. As observed for the BMDCs, a lower magnitude of inflammatory cytokine and chemokine responses were observed for NP stimulated sDC compared to those induced by CpG ODN (Supplemental Figure 4.)
Polyanhydride Nanoparticles Activate BMDCs Without Inducing Innate Effector Molecules or Altering Cellular Metabolism
Upon natural infection or after stimulation with TLR ligands such as CpG ODN, activated APCs produce innate effector molecules such as NO or reactive oxygen species (ROS). Although these molecules provide critical microbicidal activity during acute infections,33,34 NO and ROS can also have inhibitory effects on innate11,13,35 and adaptive13,36–39 immune responses. After 48 h of in vitro stimulation, NO production by BMDC was measured indirectly by assessing nitrite content in culture supernatants. Unlike CpG ODN, which induced high concentrations of NO, stimulation of BMDCs with polyanhydride NPs did not induce significant amounts of NO (Figure 4A). Lower concentrations of mitochondrial superoxide (mROS) were also observed following NP stimulation compared to CpG ODN treatment (Supplemental Figure 5).
After encountering TLR ligands, murine BMDCs exhibit a profound metabolic shift away from mitochondrial oxidative phosphorylation and towards sustained aerobic glycolysis.40 This extended commitment to aerobic glycolysis is a consequence of the concentration of the innate effector molecule NO. In this context, it must be noted that NO plays an important antimicrobial role during acute infections and in response to TLR-stimulation.11,12,41 Because polyanhydride nanoparticles were observed to activate BMDCs without inducing NO, it was hypothesized that NP stimulation would result in a BMDC activation phenotype that maintained functional mitochondria. After an 18-hour stimulation with NP or CpG, a mitochondrial stress test (MST) was performed on BMDCs and revealed marked differences in metabolic states. CpG ODN stimulation resulted in the expected inhibition of ATP production, loss of spare respiratory capacity, and increased proton leak (Figure 4B and C). In contrast, NP stimulation of BMDCs resulted in an overall maintenance of mitochondrial functionality resembling that of non-stimulated BMDCs (Figures 4B and C). During the MST, the extracellular acidification rate (ECAR) was also measured as an indicator of glycolysis. As expected, the CpG ODN stimulated BMDCs exhibited an elevated glycolytic rate while NP stimulated cells did not (Supplemental Figure 6). Together, these results demonstrate that polyanhydride nanoparticles, while inducing effective DC activation, induced a distinctly less overt activation phenotype that avoid excessive production of innate effector molecules (eg, NO).
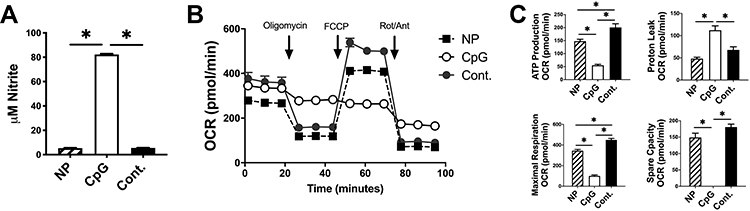 |
Figure 4 Polyanhydride nanoparticles induce a differential metabolic profile of activation and no induction nitric oxide. BMDCs were stimulated with either NPs, CpG ODN, or control as described in materials and methods. (A) Nitric oxide production was measure indirectly via Griess assay as nitrite concentration in the cell supernatant 48 hours after stimulation. BMDCs generated from BALB/c mice were stimulated for 18 hours with, CpG ODN, or non-stimulated control. Stimulated BMDCs were seeded into 24-well seahorse plates coated with Cell-Tak at a density of 2.5 x 105 cells per well and a mitochondrial stress test (MST) was conducted. (B) Kinetic results of the MST oxygen consumption rate (OCR) are shown along with (C) ATP production, proton leak, maximal respiratory capacity, and spare respiratory capacity calculated from the MST. Significance between treatments was determined via one-way ANOVA with a Tukey posttest. P value is indicated as *p ≤ 0.05. All bar graphs and symbols represent the treatment average ± SEM. |
Low Antigen Dose Polyanhydride Nanovaccine Reduced Tumor Burden and Increased Time on Study
Although high doses of antigen and polyanhydride nanoparticles were effective at inducing CD8+ T cell memory upon vaccination, experiments were designed to determine whether lower doses of both Ova and polymer particles could also induce an efficacious immune response. A lower dose of Ova (100 µg) and nanoparticles (500 µg) were administered to mice and the induction of antigen-specific CD8+ T memory was evaluated. C57BL/6 mice were vaccinated prophylactically with formulations consisting of the polyanhydride nanoparticles (NP-Ova), sOva alone, or a PBS control. Six weeks later, mice were challenged subcutaneously with Ova-expressing E.G7 tumor cells and the progression of tumor volume monitored (Figure 5A). The number of mice remaining on the study was evaluated at 30 days post-challenge and only mice immunized with the NP formulation experienced a significant increase in survival compared to the non-vaccinated controls and mice receiving sOva alone (Figure 5B). Together, these results demonstrate that a lower dose NP vaccine regimen had comparable effectiveness as the high dose regimen with respect to the induction of an efficacious immune response.
 |
Figure 5 Polyanhydride nanoparticles induce CD8+ T cell memory with a low antigen dose. C57BL/6 female mice were immunized subcutaneously with formulations consisting of 75 μg soluble Ova plus 500 μg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 25 μg Ova (NP) (n=12); 100 μg soluble Ova alone (sOva) (n=11); or PBS control (n=12). Mice were challenged subcutaneously in the flank with E.G7 Ova-expressing tumor cells 42 days post-immunization and (A) tumor volume of individual mice was tracked. (B) Survival was evaluated 30 days post-challenge. Significance from PBS control was determined using a Log rank (Mantel-Cox) test with a Bonferroni correction for multiple (2) comparisons. Significance from naïve control group is indicated in the treatment group legend as follows *p ≤ 0.025. Median survival of each group is also indicated parenthetically. |
Polyanhydride NPs Provided a More Efficacious Induction of CD8+ T Cell Memory Upon Vaccination Compared to CpG ODN
As previous work described overt CpG-induced inflammatory activation of APCs that diverted CD8+ T cell fate towards effector versus memory phenotypes, the observed low magnitude pro-inflammatory responses induced by polyanhydride NPs on DCs may lead to improved recall responses to challenge.4,7 This led to the hypothesis that the magnitude and/or consequences of inflammation induced by different adjuvants would alter the efficacy of the ensuing immune response. This hypothesis was tested by subcutaneously immunizing C57BL/6 mice with vaccine formulations containing a low dose of Ova (100 µg) as described above: NP, CpG ODN, NP + CpG ODN, or a naïve PBS only control. After implantation of the EG.7 cells, tumor volumes were tracked (Figure 6A) and time on study was compared (Figure 6B). Anti-Ova antibody titers were also evaluated to ensure that mice were effectively vaccinated (Supplemental Figure 7). At 30 days post-tumor challenge, time on study was significantly improved for mice immunized with either the NP + Ova formulation or the combination NP + CpG ODN + Ova formulation compared to unvaccinated control (Figure 6B). Vaccination with CpG ODN + Ova lead to no improvement in survival time compared to unvaccinated controls. These results suggest that polyanhydride nanovaccines containing encapsulated antigen are able to initiate effective T cell memory on their own as well as when co-administered with the pro-inflammatory TLR agonist CpG ODN.
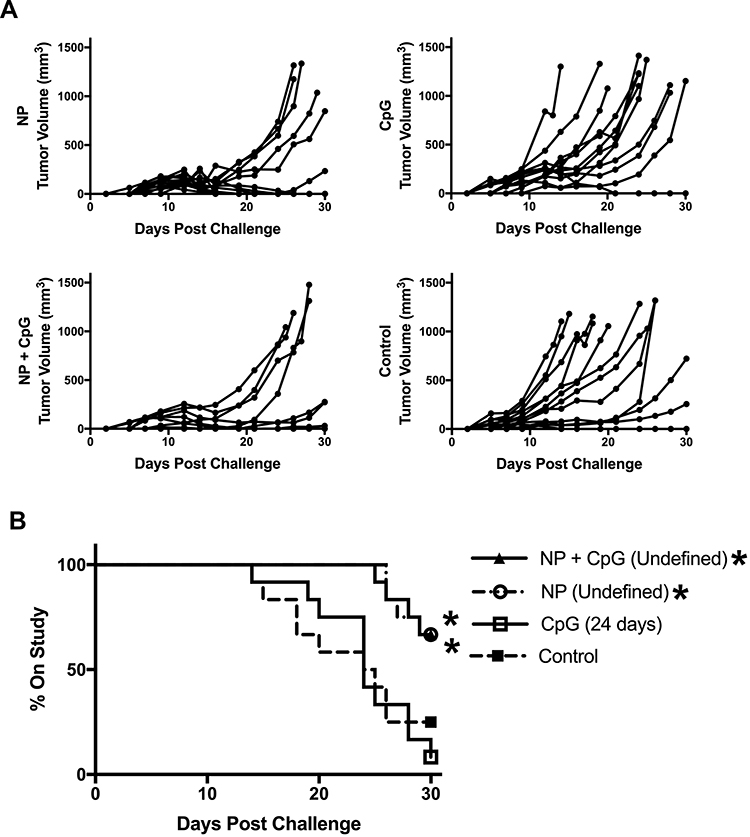 |
Figure 6 Polyanhydride nanoparticles induce effective CD8+ T cell memory as compared to CpG ODN. C57BL/6 female mice (n=12) were immunized subcutaneously with formulations consisting of 75 μg soluble Ova plus 500 μg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 25 μg Ova (NP); 100 μg soluble Ova adjuvanted with 20 μg CpG ODN (CpG); a combination of 75 μg soluble Ova plus 500 μg of 20:80 CPTEG:CPH polyanhydride nanoparticles encapsulating 25 μg Ova plus 20 μg CpG ODN (NP+CpG); or PBS control. Mice were challenged subcutaneously in the flank with E.G7 Ova-expressing tumor cells 42 days post-immunization and (A) tumor volume of individual mice was tracked. (B) Survival was evaluated 30 days post-challenge. Significance from PBS control was determined using a Log rank (Mantel-Cox) test with a Bonferroni correction for multiple (3) comparisons. Significance from naïve control group is indicated in the treatment group legend as follows *p ≤ 0.016. Median survival of each group is also reported parenthetically. |
Discussion
In this work, the ability of a polyanhydride nanovaccine to induce CD8+ T cell memory was investigated. Polyanhydride nanoparticles, encapsulating Ova, enhanced the development of antigen-specific CD8+ T cell memory upon vaccination. The induction of this antigen-specific response occurred without inducing the overt inflammatory responses commonly associated with the use of microbial-associated molecular patterns (MAMPs) that stimulate immune cells via pattern recognition receptors such as TLRs.8 Direct in vivo comparison between the TLR agonist CpG ODN and the polyanhydride nanoparticle formulation illustrates the relative benefits of a low inflammatory vaccine adjuvant formulation on the resulting CD8+ T cell immune response.
Previously, inclusion of a TLR agonist in the nanoparticle vaccine formulation negatively affected the ability 20:80 CPTEG:CPH polyanhydride NPs to induce an immune response that inhibited tumor progression.19,20 Inflammatory adjuvants, while effective at enhancing antibody production and expansion of terminally differentiated effector CD8+ T cells, often fail to induce optimal T cell memory populations among other less than optimal outcomes.1,6,39,42,43 Vaccination strategies that rapidly induce high numbers of CD8+ T cell memory, such as DC vaccination or antigen-coated particle formulations, share an important quality.4,42,44,45 The common feature of these strategies is that they induce CD8+ T cell memory more effectively when the antigen is delivered in the absence of overt inflammation (eg, DC-based vaccines).45,46
One component of the overt inflammatory responses induced by pathogens or TLR-based adjuvants is the production of microbicidal innate immune effector molecules such as NO.47,48 Although NO is important for the innate immune response to overcome acute pathogenic infections,49 it has negative autocrine or paracrine effects on APCs. When NO is produced by BMDCs, it inhibits the electron transport chain (ie, oxidative phosphorylation) leading to a dependence on aerobic glycolysis for ATP production and cellular survival.12,40 This sustained dependence on aerobic glycolysis, while not necessary for activation, has been described to provide the necessary energy demands for BMDC survival when stimulated via a TLR agonist.50 Previous studies have also indicated that mitochondrial functional deficiencies are associated with suboptimal antigen processing and presentation by APCs.51 In the present studies, the ability of the NP formulations to avoid adverse impacts on mitochondrial function in APCs and a reliance on sustained glycolysis (Figure 4) likely contributed to a DC phenotype that more effectively promoted the induction of antigen-specific, memory CD8+ T cells (Figures 1 and 5). Furthermore, production of high amounts of NO by DCs can lead to decreased co-stimulatory capacity and survival.12,52 Nitric oxide production by DCs and other innate immune cells has also been implicated in the direct inhibition of B cell (ie, antibody production) and T cell function (ie, effector and memory phenotypes).36,38,53,54 Pharmacological inhibition of NO production can prevent some of these deleterious effects on the adaptive immune response.36,52,55 Compared to the robust stimulation of DCs by TLR ligands, the low inflammatory activation phenotype of DCs following polyanhydride particle stimulation likely contributed to the enhanced anti-tumor immune response demonstrated in these studies (Figures 3 and 4).
Previous intervention strategies targeting the inflammatory response to enhance CD8+ T cell memory have focused on interfering with chemokine actions and chemokine effects on lymphocytes.56–59 Other strategies have inhibited the expansion of effector cells at strategic timepoints after immunization, such as dosing with the mTOR inhibitor rapamycin to interfere with the metabolic requirements of expanding effector cells in order to shift differentiation toward memory development.52,60,61 Although these approaches have positively affected DC activation and memory CD8+ T cell induction, the systemic effects of using these inhibitors may have unintended consequences. Reducing the magnitude of the inflammatory response associated with vaccine adjuvants may be able to provide many of these same benefits without the necessary post-vaccination intervention with inhibitors such as rapamycin. Together, these studies along with the current observations suggest that induction of effective adaptive immune memory following vaccination can be induced using adjuvant formulations that provide optimal innate activation of APCs with a “just-right” amount of inflammation.62
Polyanhydride nanoparticle adjuvants have been shown to be endocytosed by APCs, provide innate immune activation, and induce co-stimulatory molecule upregulation. These stimulatory capabilities of polyanhydride nanoparticles have contributed to the successful generation of long-lived protective antibody responses to various encapsulated protein antigens.15,17,63–65 In the present work, it was shown that a 20:80 CPTEG:CPH polyanhydride nanoparticle formulation induced antigen-specific CD8+ T cell memory capable of providing protection against a tumor challenge distant from the vaccination site at both high (Figure 1) and low dosages of antigen (Figure 5). This immune-enhancing effect was associated with the ability to activate DCs, particularly the CD8α+ subpopulation of DCs associated with cross-priming CD8+ T cells (Figure 3). Such activation was accomplished in the absence of deleterious vaccination site inflammation, which can lead to dysfunctional CD8+ T cell trafficking and effector functions. Further, it was demonstrated that the delivery of empty nanoparticles with soluble antigen did not provide the same benefits as particulate formulations including encapsulated antigen. This finding suggests that there is a critical role for antigen encapsulation in the capability of polyanhydride NP formulations to induce CD8+ T cell memory (Figure 2). The benefits of antigen encapsulation with a single-dose regimen that provide comparable results of prime-boost formulations were also described (Figure 2). Direct in vivo comparison of polyanhydride nanoparticles with the TLR agonist CpG ODN, illustrates the effectiveness of the low inflammatory nature at work to provide significantly greater survival and decreased tumor burden while the CD8+ T cell memory induced by CpG ODN vaccination failed to do so (Figure 6). Related to its phlogistic potential, the addition of CpG ODN to vaccine formulations has been shown to limit the effectiveness of the polyanhydride nanovaccine administered by the parenteral route.4,7,20 This suggests that the nature of the innate inflammatory phenotype induced by a vaccine plays a key role in the outcome of CD8+ T cell response. It is also of note that, in this study, the addition of CpG ODN to the polyanhydride nanoparticle formulation did not have any inhibitory impact. This observation suggests that the encapsulation of antigen and persistence of the polyanhydride particles allows the host to overcome the apparent negative impact of the acute inflammatory response induced by CpG ODN at the time of immunization (Figure 6).
In conclusion, these results indicate that the beneficial effects of polyanhydride nanoparticles arise, in part, from their ability to activate DCs and upregulate costimulatory molecule expression without overt induction of inflammatory cytokines, chemokines, and effector molecules (eg, NO) that are often associated with a pathogenic infection or TLR ligand administration. The use of adjuvants that closely mimic the immunological activation similar to that of a natural infection has been successful in generating protective antibody mediated immunity, but many of these adjuvants induce relatively poor CD8+ T cell responses. By virtue of their low inflammatory activation properties (ie, selectively pathogen mimicking), the use of polyanhydride nanovaccines is emerging as a strategy to more effectively generate CD8+ T cell memory through the absence of adverse innate inflammatory effects.
Abbreviations
APC, antigen-presenting cell; BMDC, bone marrow-derived dendritic cell; CMI, cell-mediated immunity; CPH, 1,6-bis(p-carboxyphenoxy) hexane; CPTEG, 1.8-bis(p-carboxyphenoxy)-3,6-dioxaoctante; DC, dendritic cell; ECAR, extracellular acidification rate; mROS, mitochondrial reactive oxygen species; MST, mitochondrial stress test; NO, nitric oxide; NP, nanoparticle; OCR, oxygen consumption rate; ODN, oligodeoxynucleotide; Ova, ovalbumin; ROS, reactive oxygen species; sOva, soluble ovalbumin; TLR, Toll-like receptor.
Acknowledgments
This work was supported by the Margaret B. Barry Cancer Research Award, the Iowa State University Nanovaccine Institute, and the National Institutes of Health (U01 CA213862). B.N. acknowledges the Vlasta Klima Balloun Faculty Chair.
Author Contributions
All authors made substantial contributions to conceptualization and design, acquisition of data, and analysis and interpretation of data. All authors took part in drafting the article and revising it critically for important intellectual content. All authors gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
Balaji Narasimhan and Michael Wannemuehler are co-founders of ImmunoNanoMed Inc., a start-up with business interests in the development of nano-based vaccines against infectious diseases. Balaji Narasimhan and Michael Wannemuehler have patents: US Patent No. 7,858,093 and US 8,173,104 B2 issued (Controlled-Release Immunogenic Formulations to Modulate Immune Response). The authors report no other possible conflicts of interest for this work.
References
1. Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8(2):107–119. doi:10.1038/nri2251
2. Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33(4):492–503. doi:10.1016/j.immuni.2010.10.002
3. Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25(2):98–104. doi:10.1016/j.it.2003.11.009
4. Badovinac VP, Messingham KAN, Jabbari A, Haring JS, Harty JT. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med. 2005;11(7):748–756. doi:10.1038/nm1257
5. Dillman RO, Cornforth AN, Nistor GI, McClay EF, Amatruda TT, Depriest C. Randomized phase II trial of autologous dendritic cell vaccines versus autologous tumor cell vaccines in metastatic melanoma: 5-year follow up and additional analyses. J Immunother Cancer. 2018;6(1):1–10. doi:10.1186/s40425-018-0330-1
6. Hailemichael Y, Dai Z, Jaffarzad N, et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat Med. 2013;19(4):465–472. doi:10.1038/nm.3105
7. Pham N-L-L, Badovinac VP, Harty JT, et al. A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J Immunol. 2009;183(4):2337–2348. doi:10.4049/jimmunol.0901203
8. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30(1):16–34. doi:10.3109/08830185.2010.529976
9. Pindado J, Balsinde J, Balboa MA. TLR3-dependent induction of nitric oxide synthase in raw 264.7 macrophage-like cells via a cytosolic phospholipase A2/cyclooxygenase-2 pathway. J Immunol. 2007;179(7):4821–4828. doi:10.4049/jimmunol.179.7.4821
10. Lowenstein CJ, Padalko E. iNOS (NOS2) at a glance. J Cell Sci. 2004;117(14):2865–2867. doi:10.1242/jcs.01166
11. Everts B, Amiel E, Van Der Windt GJW, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 2012;120:1422–1431. doi:10.1182/blood-2012-03-419747
12. Amiel E, Everts B, Fritz D, et al. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J Immunol. 2014;193(6):2821–2830. doi:10.4049/jimmunol.1302498
13. Thwe PM, Amiel E. The role of nitric oxide in metabolic regulation of dendritic cell immune function. Cancer Lett. 2018;412:236–242. doi:10.1016/j.canlet.2017.10.032
14. Janikashvili N, Larmonier N, Katsanis E. Personalized dendritic cell-based tumor immunotherapy. Immunotherapy. 2010;2(1):57–68. doi:10.2217/imt.09.78
15. Torres MP, Wilson-Welder JH, Lopac SK, et al. Polyanhydride microparticles enhance dendritic cell antigen presentation and activation. Acta Biomater. 2011;7(7):2857–2864. doi:10.1016/j.actbio.2011.03.023
16. Ross KA, Haughney SL, Petersen LK, Boggiatto P, Wannemuehler MJ, Narasimhan B. Lung deposition and cellular uptake behavior of pathogen-mimicking nanovaccines in the first 48 hours. Adv Healthc Mater. 2014;3(7):1071–1077. doi:10.1002/adhm.201300525
17. Petersen LK, Ramer-Tait AE, Broderick SR, et al. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials. 2011;32(28):6815–6822. doi:10.1016/j.biomaterials.2011.05.063
18. Huntimer L, Ramer-Tait AE, Petersen LK, et al. Evaluation of biocompatibility and administration site reactogenicity of polyanhydride-particle-based platform for vaccine delivery. Adv Healthc Mater. 2013;2(2):369–378. doi:10.1002/adhm.201200181
19. Joshi VB, Geary SM, Carrillo-Conde BR, Narasimhan B, Salem AK. Characterizing the antitumor response in mice treated with antigen-loaded polyanhydride microparticles. Acta Biomater. 2013;9(3):5583–5589. doi:10.1016/j.actbio.2012.11.001
20. Wafa EI, Geary SM, Goodman JT, Narasimhan B, Salem AK. The effect of polyanhydride chemistry in particle-based cancer vaccines on the magnitude of the anti-tumor immune response. Acta Biomater. 2017;50:417–427. doi:10.1016/j.actbio.2017.01.005
21. Shrikant P, Mescher MF. Control of syngeneic tumor growth by activation of CD8+ T cells: efficacy is limited by migration away from the site and induction of nonresponsiveness. J Immunol. 1999;162(5):2858–2866.
22. Zhou F, Rouse BT, Huang L. Prolonged survival of thymoma-bearing mice after vaccination with a soluble protein antigen entrapped in liposomes: a model study. Cancer Res. 1992;52(22):6287–6291.
23. Becerra JC, Arthur JF, Landucci GR, Forthal DN, Theuer CP. CD8+ T-cell mediated tumor protection by Pseudomonas exotoxin fused to ovalbumin in C57BL/6 mice. Surgery. 2003;133(4):404–410. doi:10.1067/msy.2003.112
24. ATCC. E.G7-OVA Product Information. Available from: https://www.atcc.org/Products/All/CRL-2113.aspx.
25. Torres MP, Vogel BM, Narasimhan B, Mallapragada SK. Synthesis and characterization of novel polyanhydrides with tailored erosion mechanisms. J Biomed Mater Res A. 2006;76(1):102–110. doi:10.1002/jbm.a.30510
26. Ross KA, Loyd H, Wu W, et al. Structural and antigenic stability of H5N1 hemagglutinin trimer upon release from polyanhydride nanoparticles. J Biomed Mater Res - Part A. 2014;102(11):4161–4168. doi:10.1002/jbm.a.35086
27. Huntimer LM, Ross KA, Darling RJ, et al. Polyanhydride nanovaccine platform enhances antigen-specific cytotoxic T cell responses. Technology. 2014;02(02):171–175. doi:10.1142/S2339547814500162
28. Pelgrom LR, van der Ham AJ, Everts B. Analysis of TLR-induced metabolic changes in dendritic cells using the Seahorse XF(e)96 extracellular flux analyzer. Methods Mol Biol. 2016;1390:273–285. doi:10.1007/978-1-4939-3335-8_17
29. Petersen LK, Xue L, Wannemuehler MJ, Rajan K, Narasimhan B. The simultaneous effect of polymer chemistry and device geometry on the in vitro activation of murine dendritic cells. Biomaterials. 2009;30(28):5131–5142. doi:10.1016/j.biomaterials.2009.05.069
30. den Haan JMM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192(12):1685–1696. doi:10.1084/jem.192.12.1685
31. Belz GT, Smith CM, Eichner D, et al. Cutting edge: conventional CD8 + dendritic cells are generally involved in priming CTL immunity to viruses. J Immunol. 2004;172(4):1996–2000. doi:10.4049/jimmunol.172.4.1996
32. Iyoda T, Shimoyama S, Liu K, et al. The CD8 + dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med. 2002;195(10):1289–1302. doi:10.1084/jem.20020161
33. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19(1):59–70. doi:10.1016/S1074-7613(03)00171-7
34. Cobb JP, Hotchkiss RS, Swanson PE, et al. Inducible nitric oxide synthase (iNOS) gene deficiency increases the mortality of sepsis in mice. Surgery. 1999;126(2):438–442. doi:10.1016/S0039-6060(99)70189-3
35. Simioni PU, Fernandes LG, Tamashiro WM. Downregulation of L-arginine metabolism in dendritic cells induces tolerance to exogenous antigen. Int J Immunopathol Pharmacol. 2017;30(1):44–57. doi:10.1177/0394632016678873
36. Sammicheli S, Kuka M, Di Lucia P, et al. Inflammatory monocytes hinder antiviral B cell responses. Sci Immunol. 2016;1(4):1–11. doi:10.1126/sciimmunol.aah6789
37. Powell TJ, Jenkins CD, Hattori R, MacPherson GG. Rat bone marrow-derived dendritic cells, but not ex vivo dendritic cells, secrete nitric oxide and can inhibit T-cell proliferation. Immunology. 2003;109(2):197–208. doi:10.1046/j.1365-2567.2003.01639.x
38. Yamamoto K, Akbar SM, Masumoto T, Onji M. Increased nitric oxide (NO) production by antigen-presenting dendritic cells is responsible for low allogeneic mixed leucocyte reaction (MLR) in primary biliary cirrhosis (PBC). Clin Exp Immunol. 1998;114(1):94–101. doi:10.1046/j.1365-2249.1998.00696.x
39. Case AJ, McGill JL, Tygrett LT, et al. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge. Free Radic Biol Med. 2011;50(3):448–458. doi:10.1016/j.freeradbiomed.2010.11.025
40. Krawczyk CM, Holowka T, Sun J, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742–4749. doi:10.1182/blood-2009-10-249540
41. Everts B, Amiel E, Huang S-C-C, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15(4):323–332. doi:10.1038/ni.2833
42. Butler NS, Nolz JC, Harty JT. Immunologic considerations for generating memory CD8 T cells through vaccination. Cell Microbiol. 2011;13(7):925–933. doi:10.1111/j.1462-5822.2011.01594.x
43. Souza CK, Rajão DS, Sandbulte MR, et al. The type of adjuvant in whole inactivated influenza a virus vaccines impacts vaccine-associated enhanced respiratory disease. Vaccine. 2018;36(41):6103–6110. doi:10.1016/j.vaccine.2018.08.072
44. Pham N-L-L, Pewe LL, Fleenor CJ, et al. Exploiting cross-priming to generate protective CD8 T-cell immunity rapidly. Proc Natl Acad Sci. 2010;107(27):12198–12203. doi:10.1073/pnas.1004661107
45. Dong H, Wen Z-F, Chen L, et al. Polyethyleneimine modification of aluminum hydroxide nanoparticle enhances antigen transportation and cross-presentation of dendritic cells. Int J Nanomed. 2018;13:3353–3365. doi:10.2147/INJ.S164097
46. Mi Y, Hagan T, Vincent BG, et al. Emerging nano-/microapproaches for cancer immunotherapy. Adv Sci. 2019;6:e1801847. doi:10.1002/advs.201801847
47. Lee M, Rey K, Besler K, Wang C, Choy J. Immunobiology of nitric oxide and regulation of inducible nitric oxide synthase. Results Probl Cell Differ. 2017;62:181–207. doi:10.1007/978-3-319-54090-0_8
48. Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298(2):249–258. doi:10.1042/BJ2980249
49. Wei XQ, Charles IG, Smith A, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375(6530):408–411. doi:10.1038/375408a0
50. Everts B, Pearce EJ. Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Front Immunol. 2014;5(MAY):203. doi:10.3389/fimmu.2014.00203
51. Chougnet CA, Thacker RI, Shehata HM, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol. 2015;195(6):2624–2632. doi:10.4049/jimmunol.1501006
52. Lawless SJ, Kedia-Mehta N, Walls JF, et al. Glucose represses dendritic cell-induced T cell responses. Nat Commun. 2017;8:15620. doi:10.1038/ncomms15620
53. Xu L, Huang Y, Yang J, et al. Dendritic cell-derived nitric oxide is involved in IL-4-induced suppression of experimental allergic encephalomyelitis (EAE) in Lewis rats. Clin Exp Immunol. 1999;118(1):115–121. doi:10.1046/j.1365-2249.1999.01029.x
54. Schouppe E, Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA. Modulation of CD8+T-cell activation events by monocytic and granulocytic myeloid-derived suppressor cells. Immunobiology. 2013;218(11):1385–1391. doi:10.1016/j.imbio.2013.07.003
55. Darling RJ, Senapati S, Kelly SM, Kohut ML, Narasimhan B, Wannemuehler MJ. STING pathway stimulation results in a differentially activated innate immune phenotype associated with low nitric oxide and enhanced antibody titers in young and aged mice. Vaccine. 2019;37(20):2721–2730. doi:10.1016/j.vaccine.2019.04.004
56. Hu JK, Kagari T, Clingan JM, Matloubian M. Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc Natl Acad Sci. 2011;108(21):E118–E127. doi:10.1073/pnas.1101881108
57. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317(5):620–631. doi:10.1016/j.yexcr.2010.12.017.CXCR3
58. Li R, Zhang N, Tian M. Temporary CXCR3 and CCR5 antagonism following vaccination enhances memory CD8 T cell immune responses. Mol Med. 2016;22(1):1. doi:10.2119/molmed.2015.00218
59. Kurachi M, Kurachi J, Suenaga F, et al. Chemokine receptor CXCR3 facilitates CD8+ T cell differentiation into short-lived effector cells leading to memory degeneration. J Exp Med. 2011;208(8):1605–1620. doi:10.1084/jem.20102101
60. Araki K, Turner AP, Shaffer VO, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–112. doi:10.1038/nature08155
61. Araki K, Youngblood B, Ahmed R. The role of mTOR in memory CD8 T-cell differentiation. Immunol Rev. 2010;235(1):234–243. doi:10.1111/j.0105-2896.2010.00898.x
62. Zeng H. mTOR signaling in immune cells and its implications for cancer immunotherapy. Cancer Lett. 2017;408:182–189. doi:10.1016/j.canlet.2017.08.038
63. Ulery BD, Kumar D, Ramer-Tait AE, Metzger DW, Wannemuehler MJ, Narasimhan B. Design of a protective single-dose intranasal nanoparticle-based vaccine platform for respiratory infectious diseases. PLoS One. 2011;6(3):e17642. doi:10.1371/journal.pone.0017642
64. Wagner-Muñiz DA, Haughney SL, Kelly SM, Wannemuehler MJ, Narasimhan B. Room temperature stable PspA-based nanovaccine induces protective immunity. Front Immunol. 2018;9(MAR):325. doi:10.3389/fimmu.2018.00325
65. Ross KA, Loyd H, Wu W, et al. Hemagglutinin-based polyanhydride nanovaccines against H5N1 influenza elicit protective virus neutralizing titers and cell-mediated immunity. Int J Nanomed. 2015;10(1):229–243. doi:10.2147/IJN.S72264
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.