Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Poloxamer-based binary hydrogels for delivering tramadol hydrochloride: sol-gel transition studies, dissolution-release kinetics, in vitro toxicity, and pharmacological evaluation
Authors Mendonça dos Santos AC, Santos Akkari AC, Silva Ferreira IR, Maruyama C, Páscoli M, Guilherme VA, de Paula E ![]() , Fraceto L, de Lima R, da Silva Melo P, de Araujo D
, Fraceto L, de Lima R, da Silva Melo P, de Araujo D
Received 6 August 2014
Accepted for publication 1 November 2014
Published 25 March 2015 Volume 2015:10(1) Pages 2391—2401
DOI https://doi.org/10.2147/IJN.S72337
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Ana Claudia Mendonça dos Santos,1 Alessandra Cristina Santos Akkari,1 Iasmin Rosanne Silva Ferreira,2 Cintia Rodrigues Maruyama,3 Monica Pascoli,3 Viviane Aparecida Guilherme,4 Eneida de Paula,4 Leonardo Fernandes Fraceto,5 Renata de Lima,3 Patrícia da Silva Melo,2 Daniele Ribeiro de Araujo1
1Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, Santo André, 2Faculdades Integradas Metropolitanas de Campinas, Campinas, 3Departamento de Biotecnologia, Universidade de Sorocaba, Sorocaba, 4Departamento de Bioquímica, Universidade Estadual de Campinas, Campinas, 5Departamento de Engenharia Ambiental, Universidade Estadual ‘Júlio de Mesquita Filho’, Sorocaba, São Paulo, Brazil
Abstract: In this work, poloxamer (PL)-based binary hydrogels, composed of PL 407 and PL 188, were studied with regard to the physicochemical aspects of sol-gel transition and pharmaceutical formulation issues such as dissolution-release profiles. In particular, we evaluated the cytotoxicity, genotoxicity, and in vivo pharmacological performance of PL 407 and PL 407–PL 188 hydrogels containing tramadol (TR) to analyze its potential treatment of acute pain. Drug–micelle interaction studies showed the formation of PL 407–PL 188 binary systems and the drug partitioning into the micelles. Characterization of the sol-gel transition phase showed an increase on enthalpy variation values that were induced by the presence of TR hydrochloride within the PL 407 or PL 407–PL 188 systems. Hydrogel dissolution occurred rapidly, with approximately 30%–45% of the gel dissolved, reaching ~80%–90% up to 24 hours. For in vitro release assays, formulations followed the diffusion Higuchi model and lower Krel values were observed for PL 407 (20%, Krel =112.9±10.6 µg·h-1/2) and its binary systems PL 407–PL 188 (25%–5% and 25%–10%, Krel =80.8±6.1 and 103.4±8.3 µg·h-1/2, respectively) in relation to TR solution (Krel =417.9±47.5 µg·h-1/2, P<0.001). In addition, the reduced cytotoxicity (V79 fibroblasts and hepatocytes) and genotoxicity (V79 fibroblasts), as well as the prolonged analgesic effects (>72 hours) pointed to PL-based hydrogels as a potential treatment, by subcutaneous injection, for acute pain.
Keywords: micelle, cytotoxicity, genotoxicity, analgesia
Introduction
Tramadol (TR; Figure 1) is a synthetic opioid analgesic chemically related to codeine and according to the World Health Organization is classified as a class II drug for the treatment of moderate intensity pain (eg, postoperative, oncologic, orofacial, neuropathic, ischemic, musculoskeletal disorders, migraine, and osteoarthritis), either as monotherapy or in combination.1 Despite the fact that it is not a new opioid analgesic, TR has been rediscovered in the therapy of acute and chronic pain due to its particular mechanism of action (μ, δ, and κ opioid agonist and α2-adrenergic, 5-HT1A, and 5-HT2A 5-HT3 antagonist),2,3 which evokes a low incidence of adverse effects when compared to classical opioids, such as morphine and fentanyl. However, TR use is associated with a short duration of analgesic effects.4 In this context, the development and pharmacological evaluation of drug delivery systems for TR would be of great interest for studying new formulations with potential clinical use in the treatment of acute and chronic pain. Among the several delivery systems reported in the literature, poloxamer (PL)-based thermoreversible hydrogels have presented promising results in terms of improvement in the biopharmaceutical, pharmacodynamic, and pharmacokinetic properties of the incorporated drugs.
 | Figure 1 Chemical structure of tramadol. |
PLs (Pluronic®) are copolymers consisting of polyethylene oxide (PEO) and polyoxypropylene oxide (PPO) units arranged in a basic structure A–B–A or PEO–PPO–PEO type. Due to differences in the number of PPO and PEO units, PL monomers have the ability to self-assemble in micelles, presenting a hydrophobic core surrounded by a hydrophilic corona.5 At low temperatures, both PEO and PPO units are soluble in water. However, when the temperature rises, the PPO units are dehydrated and aggregate (micellar core), while the hydrophilic PEO units (micellar corona) remain hydrated. Subsequently, these micelles are assembled in different supramolecular structures such as hexagonal, cubic-ordered phases forming the hydrogels’ organization phases. This reversible phenomenon is characterized by a temperature range of sol-gel transition (Tsol-gel), since below this temperature, the systems remain as fluids and favor the development of injectable formulations, but remain as semisolids close to the physiological temperature.6,7
In fact, these features associated with the biocompatibility of PL are essential for the development of delivery systems designed for pain treatment, since there is an influence of copolymer type and concentration, drug incorporation, and also temperature on biopharmaceutical properties (ie, duration of action and reduced uptake from the site of injection in direction to the blood stream). In addition, the use of PL with different hydrophilic–lipophilic balance (HLB), such as PL 407 and PL 188, as binary systems can be an interesting alternative to modulate the biopharmaceutical profile of these formulations.8–11
The use of PL has been reported in the literature specifically for pain treatment. Previous studies showed the increased permeation of lidocaine and ketoprofen incorporated into PL 407 liposomal hydrogels, across the swine dural membrane.12–14 Subsequently, other studies presented the preparation and characterization processes of PL 407 hydrogels containing diclofenac,15 lidocaine, dibucaine, tetracaine,16–18 and bupivacaine19 as well as their pharmacological evaluations in animal models17,20,21 and humans.22,23 However, these formulations were addressed for the use of a single type of PL. Moreover, there are no studies on PL-based in situ thermogelling hydrogel formulations containing TR for subcutaneous injection for the treatment of postoperative pain. Therefore, in this work we present the investigation of PL single-type and binary hydrogels composed of PL 407 and PL 188 and also analyze the sol-gel transition process and pharmaceutical formulation issues such as dissolution-release profiles, cytotoxicity, genotoxicity, and in vivo pharmacological evaluation.
Materials and methods
Materials
Poloxamer 407 (Pluronic® F127, molecular weight: 12,600 Da) and Poloxamer 188 (Pluronic® F68, molecular weight: 8,400 Da) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). TR hydrochloride (attested purity of 98.5%) was donated by Cristália Produtos Químicos Farmacêuticos Ltda. (Itapira, Brazil). HEPES buffer and NaCl were purchased from Sigma-Aldrich Co. All other reagents were of analytical grade. Deionized water (Purelab Option-Q; ELGA LabWater, High Wycombe, UK) was used for all experiments.
Hydrogels preparation
For hydrogels preparation, PL aqueous solutions (20 mM HEPES buffer with 154 mM NaCl, at pH 7.4) were prepared according to the cold method.24 TR (20 mg·mL−1) was dispersed in different solutions containing PL 407 alone or in binary systems with PL 188 and were kept at 4°C under magnetic stirring (100 rpm). After dissolution, each formulation was equilibrated overnight at 4°C. The PL concentrations were selected in order to obtain a thermoreversible gel at minimum possible final concentration with a maximum final PL concentration of 35% (weight per weight [w/w]). All formulations are presented in Table 1.
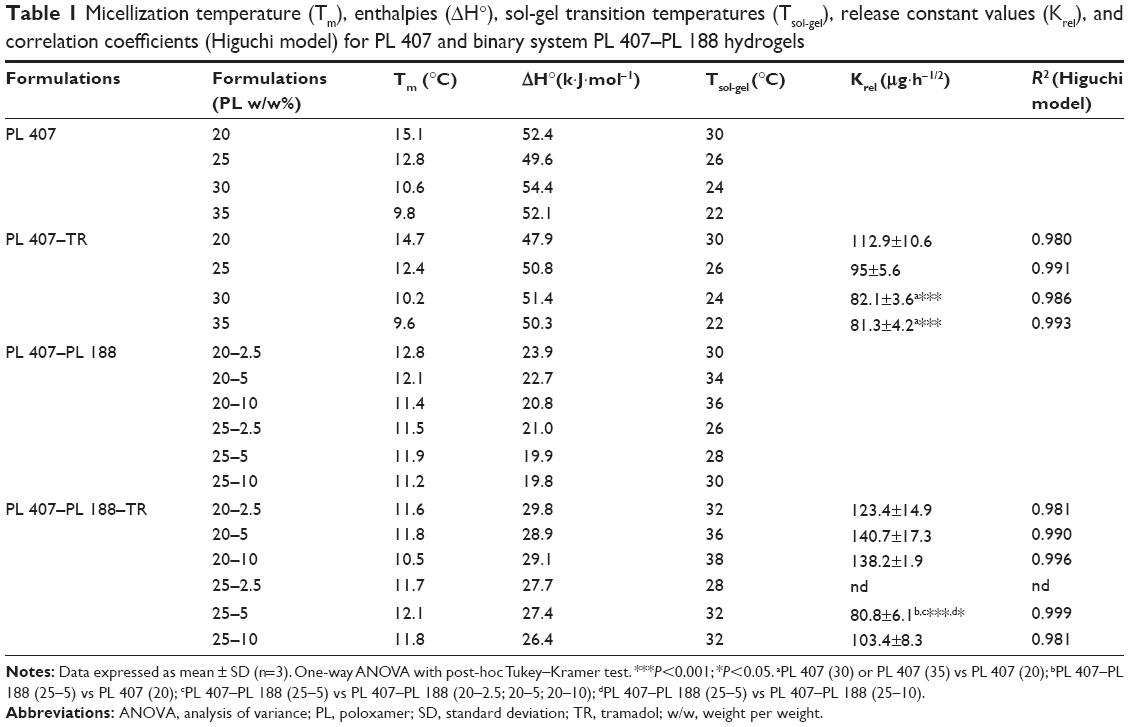 | Table 1 Micellization temperature (Tm), enthalpies (ΔH°), sol-gel transition temperatures (Tsol-gel), release constant values (Krel), and correlation coefficients (Higuchi model) for PL 407 and binary system PL 407–PL 188 hydrogels |
Physicochemical characterization: drug–micelle interaction and sol-gel transition process
Dynamic light scattering was used to determine the polymeric micellar hydrodynamic diameter and the average distribution, in order to study the drug–micelle interaction and its influence on micellar assembly. Measurements were performed using a particle analyzer Zetasizer Nano ZS (Malvern, UK) at a fixed angle of 173° and temperatures of 25°C and 37°C. Micellar solutions (5% weight per volume PL final concentration) were prepared in 20 mM HEPES buffer with 154 mM NaCl, at pH 7.4, and were filtered using a polycarbonate membrane (pore 0.22 μm). The concentration of TR was 20 mg·mL−1 for all PL solutions. Hydrodynamic micellar diameter measurements were determined at least three times for each sample.
For characterization of the hydrogels, differential scanning calorimetry (DSC) analysis was performed to determine the temperature and enthalpy relative to the micellization process and sol-gel phase transition temperature was determined using the tube inversion method. DSC experiments were performed with a TA Instruments (Q-200 DSC; New Castle, DE, USA) apparatus. Fifty milligrams of PL hydrogel (in the presence or absence of TR) were placed in sealed aluminum pans and samples were analyzed according to three successive thermal cycles of heating and cooling (0°C–50°C) at a heating–cooling rate of 5°C per minute with an empty pan as reference. Data were expressed in thermograms represented by heat flux (k·J·mol−1) versus temperature (°C).
For the tube inversion method, vials containing 1 g of hydrogels were incubated in a circulating water bath (Quimis Aparelhos Científicos, Diadema, Brazil) at 0°C for 20 minutes, to allow the samples to equilibrate thermally with their environment.18 After that, the temperature was increased (2°C/10 minutes) from 0°C to 60°C. At each increase, the samples were removed from the water bath (for a maximum of 30 seconds to prevent phase transition due to external temperature) and were inverted to observe the liquid/gel behavior. Samples were classified into three categories: 1) liquid when the sample moved rapidly in the direction of gravity; 2) viscous liquid or soft gel when the sample moved slowly down in the direction of gravity; and 3) hard gel when the sample remained on the bottom of the vial. The temperature at which a sample was classified as hard gel was called the sol-gel point. All experiments were performed in triplicate.
In vitro dissolution and release assays
Hydrogels samples were weighed (1 g) in plastic microtubes with a diameter of 1 cm and incubated in a water bath at 37°C for 12 hours. After that, 1 mL 20 mM HEPES buffer with 154 mM NaCl (pH 7.4) was added to the samples, which were centrifuged at regular intervals from 30 minutes to 24 hours (13,000 g, 10 minutes). Afterward, the supernatant liquid was removed and the remaining hydrogel was weighed. The weight of the remaining hydrogel at each time point was expressed as a percentage of the original mass. All experiments were performed in triplicate.
To avoid the direct dissolution of PL in the receptor medium, TR released was investigated to determine if the hydrogel is not expected to be quickly dissolved in physiological fluids, such as the subcutaneous fluid (since the interstitial volume medium is limited).5 A membrane diffusion model (Franz-type cells, 1.76 cm2 permeation area, Automatized Microette Plus®; Hanson Research Corporation, Chatsworth, CA, USA) with artificial membrane (cellulose acetate sheets, molecular weight cut-off 1,000 Da; Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA) as a barrier was used to evaluate the in vitro release of TR from PL hydrogels. The donor compartment was filled with 1 g of the hydrogel formulations containing 20 mg·mL−1 TR, while the receptor compartment was filled with 20 mM HEPES buffer with 154 mM NaCl buffer, pH 7.4, at 37°C, under constant magnetic stirring (350 rpm). A solution of 20 mg·mL−1 TR was used as control for total drug release. Aliquots from the receptor compartment were withdrawn (1 mL) and analyzed by ultraviolet–visible (UV–Vis) spectrophotometry (271 nm, y=0.03141+0.00983x, R2=0.99971, limit of quantification (LQ) =0.19 μg·mL−1; limit of detection (LD) =0.063 μg·mL−1, n=6, by an analytical curve previously obtained). Data were expressed as a percentage of TR released for each sample (n=6 replicates/experiment).
Animals
Male Wistar rats (250–300 g) and male Swiss mice (30–35 g) were obtained from the CEMIB-UNICAMP (Centro de Bioterismo, Universidade Estadual de Campinas, Campinas, Brazil). Protocols were approved by the UNICAMP Institutional Animal Care and Use Committee (protocols 2618-1/2012 and 2619-1/2012), which follows the recommendations of the Guide for the Care and Use of Laboratory Animals. All animals were housed in standard cages and were maintained under a 12/12-hour controlled light–dark cycle at 23°C±2°C, with four animals per cage with food and water ad libitum.
Cytotoxicity and genotoxicity assays
Cell viability tests were performed using fibroblasts (V79 cells from Chinese hamster lungs) and rat hepatocyte primary cell cultures. V79 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum with 100 UI·mL−1 penicillin and 100 μg·mL−1 streptomycin sulfate (pH 7.2–7.4, humidified atmosphere at 37°C and 5% CO2), seeded with 2×104 cells/well (96-well plates) and cultured for 48 hours. For primary cell cultures, hepatocytes were extracted from the rat liver using the perfusion technique, according to the method described previously25 with minor modifications. Initially, animals were anesthetized with alpha-chloralose (50 mg·kg−1) and urethane (1 g·kg−1) and the liver was perfused with Hank’s buffer for 20 minutes at 25 mL·min−1. After that, perfusion was maintained with a collagenase–Hank’s buffer (0.05% collagenase and 1 mM CaCl2) for 20 minutes at 15 mL·min−1 flow rate. Then, the collected hepatocytes were suspended in DMEM supplemented with 50 UI·mL−1 streptomycin sulfate, 50 μg·mL−1 penicillin, 0.2% bovine serum albumin, 0.1 IU·mL−1 bovine insulin, 1 μM dexamethasone, and 1% dimethyl sulfoxide in 10% fetal bovine serum. Cells were plated at 6×105 cells in 96-well culture plates.
Cells were incubated for 24 hours with TR (0.1–4 mg·mL−1), PL 407 isolated or in a binary system PL 407–PL 188 (0.1–40 mg·mL−1) before and after TR incorporation (n=6 replicates/concentration). After that, the number of viable cells was evaluated by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction test and neutral red (NR) uptake.
The first method is based on the reduction of MTT to formazan by mitochondrial dehydrogenases. The formazan crystals were dissolved in a 1 N HCl–isopropyl alcohol mixture (1:24 volume per volume) and shaken for 20 minutes. After that, the dye-containing solution was removed and the absorbance was determined at 570 nm.26 For the NR uptake test, the capability of viable cells to accumulate in lysosomes is measured. After cells were treated with different formulations, 0.2 mL of the 50 μg·mL−1 NR solution was added and incubated for 3 hours (in order to allow uptake of the dye by the lysosomes of viable cells). Subsequently, DMEM was removed and the cells were washed with a mixture of 1% formaldehyde and 1% CaCl2. Then, a solution of 1% acetic acid and 50% ethanol was added to remove the dye. After brief shaking, the absorbance was measured at 540 nm.27
Genotoxicity assays were also performed in V79 fibroblasts from Chinese hamster lungs by the comet assay single cell gel electrophoresis (SCGE).28 The cells were exposed to the formulations at 4 mg·mL−1 TR concentration for 1 hour. After exposure, 15 μL of treated cells were mixed in 120 μL agarose at a low melting point. The mixture was placed on a slide previously covered with agarose and was placed in lysing solution for 40 minutes. After this, the slides were transferred to a electrophoresis solution for 20 minutes before analysis was performed at 4°C for 30 minutes (voltage: 25 V, amperage: 300 mA). Slides were then washed in neutralization solution for 5 minutes, and dried at room temperature. They were then hydrated, and the material contained in the slides was stained with silver nitrate for 30 minutes. The analyses were performed in an optical microscope and counting was carried out on three slides per treatment. The DNA damage was classified as class 0–4. Class 0 denotes no DNA damage and class 4 signifies the maximum DNA damage. The damage index (DI) was calculated by dividing the score of the slides by the number of cells present. The relative index of each treatment analyzed was obtained by dividing the DI relative to the different treatments (ie, formulations) per DI obtained for the control group (ie, no treated cells).
Pharmacological evaluation: in vivo analgesic effects
The analgesic effects were evaluated using the tail-flick test. First, the animal was placed in a horizontal acrylic restraint and fixed on an analgesimeter with a portion of the tail, 5 cm from its tip, exposed to heat from a projector lamp (55°C±1°C, 150 W), for the baseline measurement (ie, normal response to the noxious stimulus). The time for normal response (in seconds) was recorded by a control switch and a timer simultaneously activated. The timer stops immediately when the rat tail flicks. The formulations (TR or PL hydrogels) were injected subcutaneously (0.06 mL, at 40 mg·kg−1 TR) on the dorsal surface of the cervical region. The evaluation was started 10 minutes after the injection of the formulations. The baseline values for each animal were considered as the time for normal response recovery (time for recovery or Trec). A 30-second cut-off period was used to avoid thermal injury. All experiments were performed by the same observer. Data were expressed as percentage of maximum possible effect (MPE%; equation 1), duration of the analgesic effect (minutes), and area under the efficacy curve (AUEC) for each experimental group.
MPE = (test latency time – baseline/cut-off – baseline) ×100 | (1) |
Statistical analysis
Data were expressed as percentage or mean ± standard deviation (SD) and were analyzed by one-way analysis of variance (ANOVA) with the Tukey–Kramer post-hoc test using Graph Pad InStat (GraphPad Software, Inc., La Jolla, CA, USA) or Origin (v6.0; Microcal™ Software, Inc., Northampton, MA, USA) programs. Statistical differences were defined as P<0.05.
Results and discussion
Physicochemical characterization: drug–micelle interaction and sol-gel phase transition studies
The hydrodynamic diameter determination was used as an important parameter for monitoring the formation of mixed micelles composed of PL 407 and PL 188 and also to study the micellar system before the supramolecular changes due to the sol-gel transition, in the presence or absence of TR.
Initially, no significant changes were observed on micellar hydrodynamic diameter for PL 407 and PL 407–PL 188 systems after TR incorporation. For the system composed of isolated PL 407, the micellar diameter was 30 nm at 25°C and represented 86% of the population analyzed against a small size population of 12 nm with 14% distribution. However, micellar diameters of ~20 nm (with a 98.5% mean distribution) were observed when the temperature was raised to 37°C. At this point, a transition was detected from a unimodal to a bimodal distribution, with differences on polydispersity values from 0.27 at 25°C to 0.19 at 37°C.
Similar results were also observed for PL binary systems PL 407–PL 188. However, larger mean hydrodynamic diameters (~50–75 nm) were detected for all copolymers proportions, which were associated with higher polydispersity values (0.5–0.6) at both temperatures. The high mean hydrodynamic diameter and polydispersion values for binary system PL 407–PL 188 could be due to the formation of mixed micelles with more hydrated corona composed of copolymers presenting different ethylene oxide:propylene oxide (EO:PO) ratios: 3:1 and 5:1 for PL 407 and PL 188, respectively, when compared to PL 407 micelles.
The mean micellar hydrodynamic diameter determination can provide important information about the effects of changes in PL concentrations, micellar composition, the addition of TR and temperature on different formulations. The interactions between lidocaine, naproxen, salicylic acid, or pentobarbital with PL 407 micelles were also studied by other authors,29 showing that the presence of charge on the incorporated molecule chemical structure influences the partitioning into micelles. These observations may also be applied to our systems, since all formulations were prepared at physiological pH (7.4), below the pKa of TR (9.41). These conditions promote the presence of ionized TR species and their possible interaction with the hydrated micellar corona region.
Table 1 presents the micellization temperatures (Tm) and enthalpy variation (ΔH°) relative to the micellization process. In addition, the sol-gel transition temperature (Tsol-gel) is also presented for all formulations. DSC analysis showed that the Tm were fairly similar for all formulations, at the same PL final concentration, especially for PL 407 hydrogels. However, Tm was decreased by ~5°C for the high PL 407 concentration (30% w/w). On the other hand, for the binary system PL 407–PL 188, Tm variation was discrete (~0.5–1.6°C) when compared to PL 407 hydrogels.
For the PL 407 systems, high ΔH° values were observed according to the final copolymer concentration, even after TR incorporation. However, for the binary system PL 407–PL 188, the enthalpy for micelle formation changed after the addition of PL 188, since ΔH° values were reduced at high PL 407 and PL 188 concentration. In addition, as observed after comparisons among the systems, there is a greater interference of the TR on self-assembly and micelle formation in the presence of PL 188, since ΔH° values were higher than those observed before TR incorporation.
In fact, the addition of more hydrophilic polymers (such as PL 188 with HLB =29) induces conformational changes in the micellar arrangement of the more hydrophobic copolymer PL 407 (HLB =22) as well as increases the hydration of the micellar corona (due to the larger number of EO units). This justifies the broader endothermic peak and the minor enthalpy variations observed during the micellization process for the binary systems.30–33 Moreover, comparisons among PL 407 and its binary systems with PL 188 showed an increase in enthalpy variation values, possibly due to dehydration of units of the PPO micellar core that was induced by the presence of a hydrophilic drug such as TR hydrochloride into the PL 407–PL 188 systems.
After DSC studies, Tsol-gel was determined for all formulations. Comparative analysis among the formulations composed of PL 407 showed lower Tsol-gel with higher copolymer concentrations; this favors micelle formation and their self-assembly in gel structure. In addition, it was possible to compare the effect of the association between 30% PL 407 (Tsol-gel =24°C) and PL 407–PL 188 binary systems at 20:10, 25:5, or 25:10 (% w/w), showing that the presence of PL 188 increased the Tsol-gel in a range of 8°C–12°C, when compared to PL 407 formulations.
Results from Table 1 show that the formulations with Tsol-gel values considered for subcutaneous use were 20% PL 407 (Tsol-gel =30°C), binary systems PL 407–PL 188 at 20:2.5, 20:5, 20:10, 25:5, and 25:10 with Tsol-gel ranging from 32°C–38°C, after TR incorporation. For this reason, those formulations were selected for in vitro dissolution and release studies.
In vitro dissolution and release profiles
In order to investigate the mechanisms involved with TR in vitro release, membrane-free dissolution assays were carried out to simulate the contact between the hydrogels and the interstitial liquid after subcutaneous injection to determine whether the hydrogel dissolution associated with the PL unimers solubilization could control the drug release.5 As observed in Figure 2A, in general, hydrogel dissolution occurred rapidly, with approximately 30%–45% of the gel dissolved until it reached ~80%–90% at 24 hours. This fact is possibly due to the relatively high aqueous solubility of PL 407 and PL 188 (even considering the differences between HLB values for both copolymers used as base polymer for the formulations) and the low viscosity of the gel formed. However, these are important and relatively conserved features for the design of formulations for parenteral administration (eg, subcutaneous route)7 and must be considered for the future clinical application of the hydrogel on pain treatment, since it is interesting that the dissolution rate is fast enough to allow drug release to the site of action, especially for post- or intraoperative analgesia.
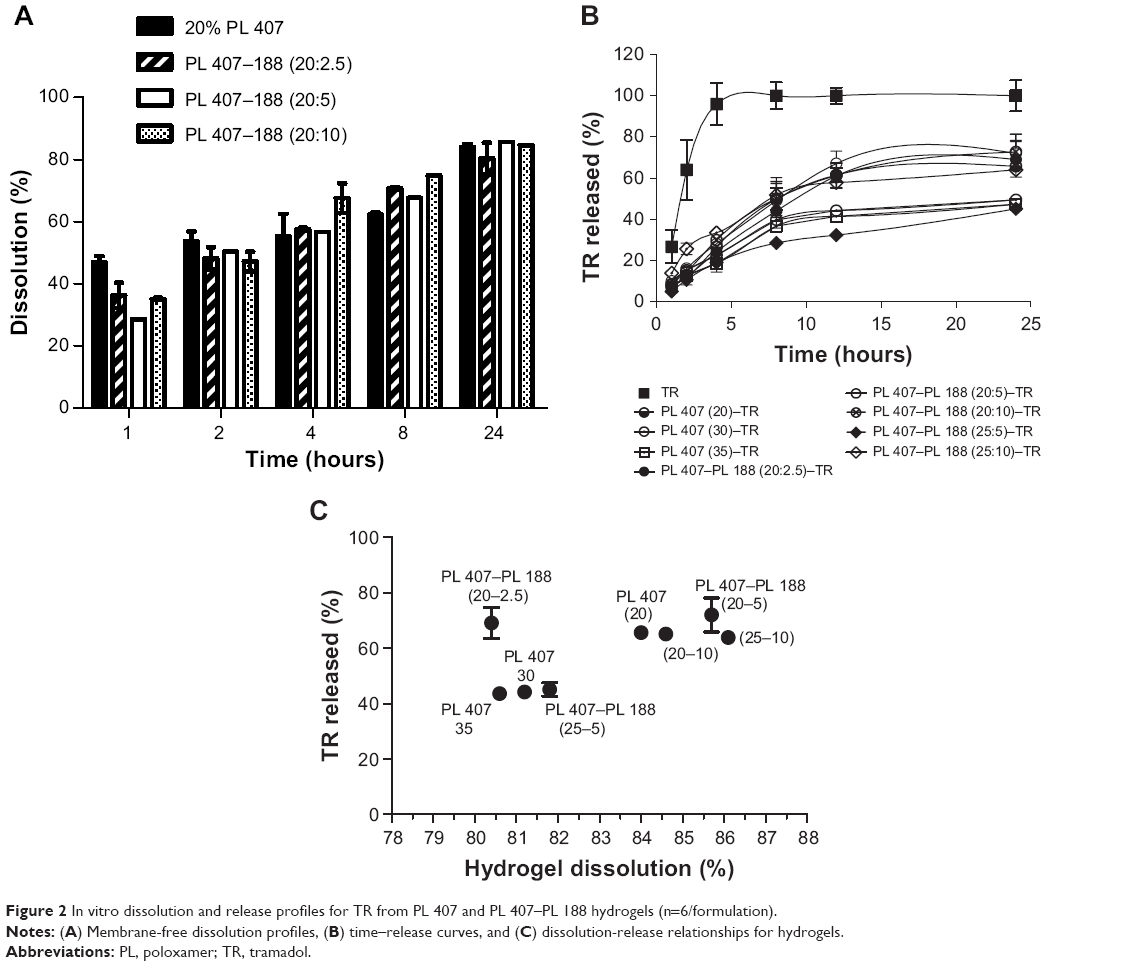 | Figure 2 In vitro dissolution and release profiles for TR from PL 407 and PL 407–PL 188 hydrogels (n=6/formulation). |
Figure 2B shows the TR release profiles from the different hydrogels. For the TR solution, the drug release was progressive and reached total release at 4 hours, with 100% of the TR released. On the other hand, the formulation 20% PL 407 and its binary systems with PL 188 (20%–2.5%; 20%–5%; 20%–10%; 25%–5%; and 25%–10%) showed regular release profiles over 24 hours, with low release percentages for 20% PL 407 (65.6%±1.4%); PL 407–PL 188, 20%:2.5% (69.1%±5.6%); PL 407–PL 188, 20%:5% (72.0%±6.2%); PL 407–PL 188, 20%:10% (72.6%±8.6%); PL 407–PL 188, 25%:5% (45.1%±2.5%); and PL 407–PL 188, 25%:10% (63.9%±2.2%) when compared to TR solution (P<0.001).
Release constant (Krel) values were then calculated using the Higuchi model, where the drug release rate is linear as a function of the root square of time, according to Fick’s laws of diffusion:34
Qt = KHt1/2 |
where Q is the drug concentration released against time, KH is the release constant, and t is time.
The calculated Krel values are presented in Table 1. Statistical differences were observed for all formulations when compared to the TR solution (417.9±47.5 μg·h−1/2, P<0.001), since hydrogels prolonged TR release for more than 24 hours with percentage release ranging from 40% to 60%.
The dissolution and release percentages were compared at 24 hours (Figure 2C) considering different mechanisms, such as possible hydrogel erosion by interstitial fluid at the administration site (subcutaneous route) and the diffusion of the drug through the hydrogels until reaching the systemic circulation.34,35 According to the lower Krel values observed among the hydrogels, the formulations PL 407 (20%), PL 407 (30%), PL 407 (35%) control groups and the binary systems PL 407–PL 188 (25%–5% and 25%–10%) were selected for in vivo pharmacological evaluation and further comparisons with the TR solution.
Cytotoxicity and genotoxicity assays
Figure 3A and B show the cell viability percentage after treatment with different TR concentrations from 0.1–4 mg·mL−1. In general, TR reduced the V79 cell viability percentages for the MTT reduction and NR uptake tests; more pronounced cytotoxic effects were observed for higher TR concentrations (30% and 40% of viable cells with 2 mg·mL−1 TR and 4 mg·mL−1 TR, respectively). On the other hand, low hepatocyte viability percentages were observed from 1–4 mg·mL−1 (60%–55% of viable cells). These results were used in order to maintain the same TR concentrations for cell viability tests in the hydrogel formulations. The drug:PL (TR:PL) proportion was maintained as 1:10 (w/w%) or 1:200 molar ratio for all hydrogels into the cell culture wells for a final volume of 0.1 μL. Hydrogel cell viability curves presented similar profiles for V79 fibroblasts and hepatocytes for both the MTT and NR tests (Figure 4A and B).
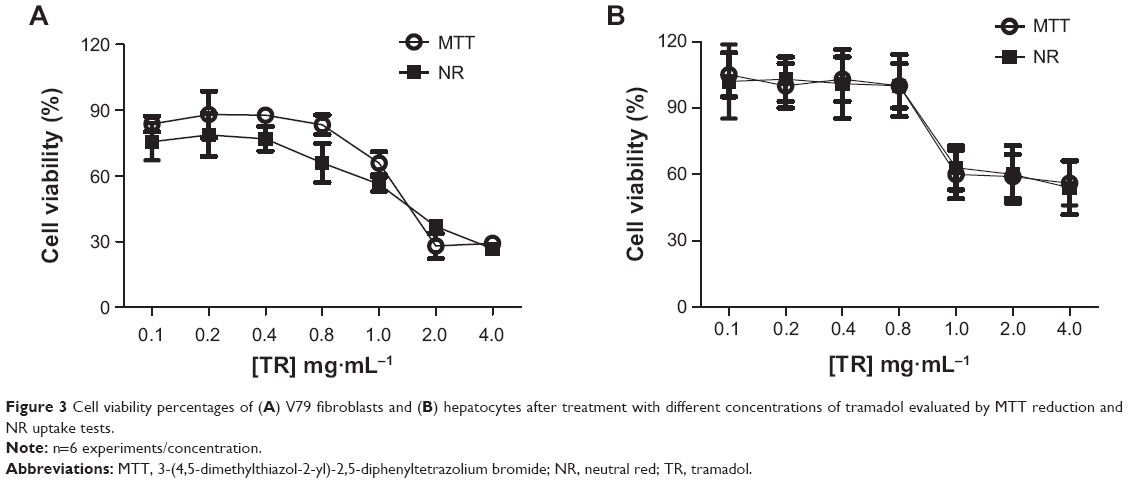 | Figure 3 Cell viability percentages of (A) V79 fibroblasts and (B) hepatocytes after treatment with different concentrations of tramadol evaluated by MTT reduction and NR uptake tests. |
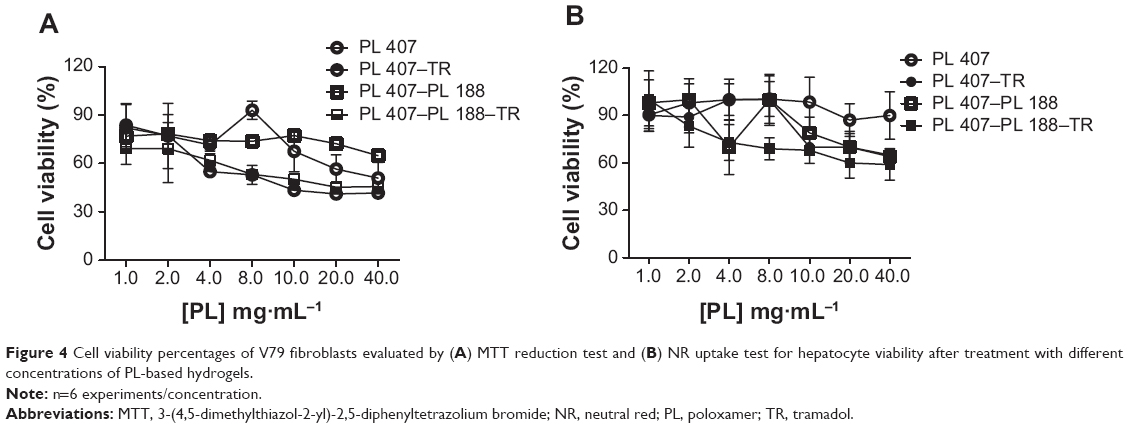 | Figure 4 Cell viability percentages of V79 fibroblasts evaluated by (A) MTT reduction test and (B) NR uptake test for hepatocyte viability after treatment with different concentrations of PL-based hydrogels. |
Figure 5 summarizes comparisons of the cytotoxic effects for all formulations with respect to the maximum TR concentration tested (4 mg·mL−1). PL hydrogels did not induce pronounced cytotoxic effects and hydrogels reduced the TR cytotoxic effects by 2.0- and 1.8-fold for V79 fibroblasts and hepatocytes, respectively (P<0.001). In another analysis, the TR concentration was considered for genotoxicity studies. Results showed no significant genotoxic effects for all formulations tested (Figure 6).
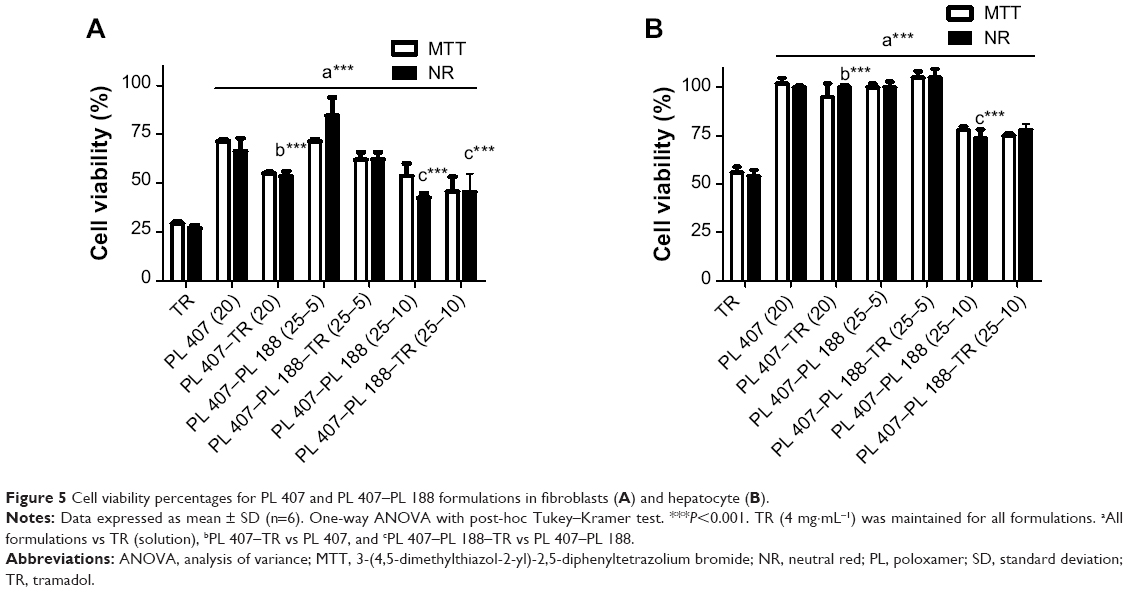 | Figure 5 Cell viability percentages for PL 407 and PL 407–PL 188 formulations in fibroblasts (A) and hepatocyte (B). |
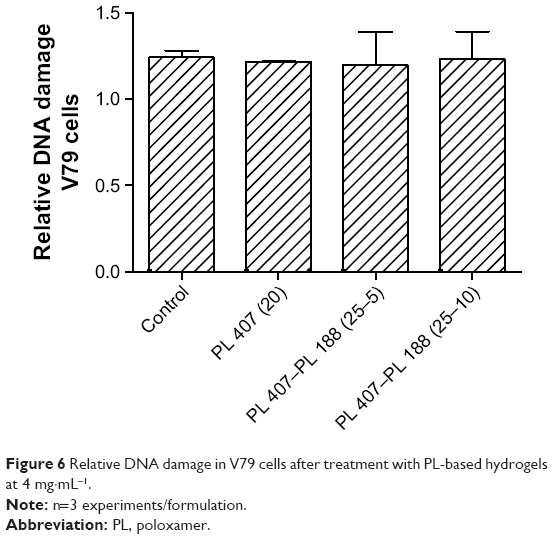 | Figure 6 Relative DNA damage in V79 cells after treatment with PL-based hydrogels at 4 mg·mL−1. |
In general, treatment of fibroblasts and hepatocytes with different concentrations of TR reduced the viability percentage determined by the MTT reduction and NR uptake tests. On the other hand, treatment of cells with PL 407 and the binary system PL 407–PL 188 showed that TR incorporation into PL hydrogels reduced the cytotoxic effects evoked by the drug. Further, the addition of PL 188 to formulations did not evoke significant cytotoxic effects in relation to the PL 407 hydrogels. These results are in agreement with previous reports that showed a structural relationship between the copolymers and their cytotoxic effect, since lipophilic copolymers induce more pronounced fluidizing effects in cell membranes and particularly on the mitochondrial membrane (by PO unit insertion into the lipid bilayer) in different cell types such as endothelial cells from the blood–brain barrier vessels36 and lung carcinoma cells.37
Pharmacological evaluation: in vivo analgesic effects
Figure 7 presents the time curve of MPE% obtained after treatment with the TR solution and different hydrogels. In general, all hydrogels induced prolonged analgesic effects when compared to the TR solution. Moreover, the time for hydrogel formation was less than 20 seconds (data not shown), suggesting that a depot hydrogel was readily formed after subcutaneous injection. Overall, the binary hydrogels (PL 407–PL 188) induced prolonged duration of analgesia (>72 hours) and more pronounced AUEC when compared to the TR solution and 20% PL 407 (P<0.001; Table 2).
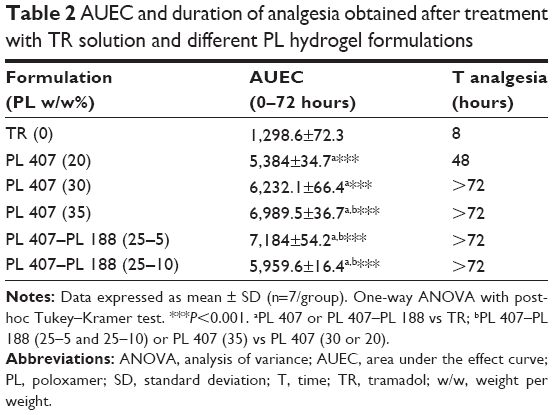 | Table 2 AUEC and duration of analgesia obtained after treatment with TR solution and different PL hydrogel formulations |
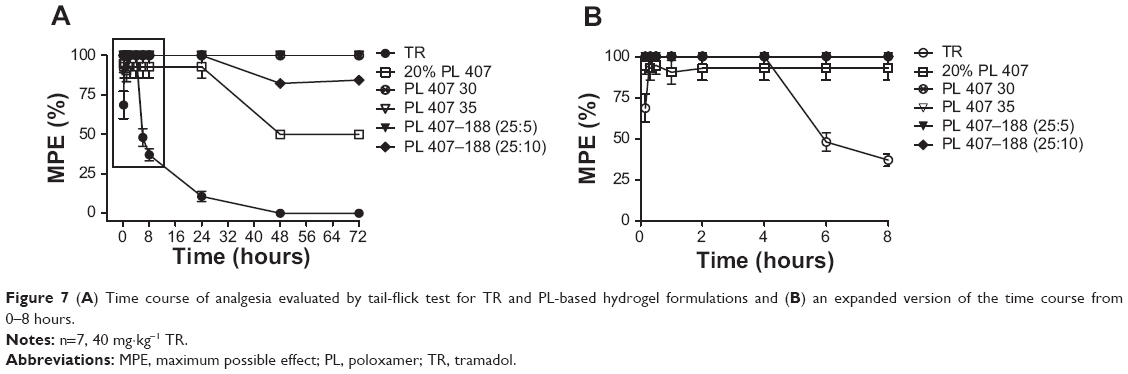 | Figure 7 (A) Time course of analgesia evaluated by tail-flick test for TR and PL-based hydrogel formulations and (B) an expanded version of the time course from 0–8 hours. |
TR can be administered by different routes, including oral, subcutaneous, epidural, intrathecal, intravenous, and intramuscular. After oral administration, TR is rapidly absorbed, with high plasma concentrations achieved at approximately 2 hours; the maximum analgesic effect is found between 1 and 4 hours after administration, but it persists for only 3–6 hours.1 However, because of the short duration of action, TR reinjections are required (orally or by continuous infusion) every 6 hours to obtain a prolonged analgesic effect.38 For this reason, the development of new pharmaceutical formulations that aim to control the drug release rate can reduce fluctuations in drug plasma levels and extended intervals between the daily administrations. This provides a significant advantage over commercially available immediate-release formulations. Therefore, the modified release formulations containing TR have been recommended for the treatment of both acute and chronic pain, and may be a possible solution with respect to inadequate analgesia and low patient compliance to the treatment.1,38
In this study, we proposed the use of thermosensitive hydrogel formulations for TR controlled release by subcutaneous route. In fact, subcutaneous TR has been demonstrated as an easy alternative to oral, intravenous, or intramuscular routes for postsurgical pain or local infiltrative analgesia.39–41 Several studies have reported the development of new TR formulations such as hydroxypropyl methylcellulose tablets containing semipermeable membranes42 and ethylcellulose microparticles43 for oral route. Other examples include chitosan-based adhesives and carbohydrate hydrogels for skin delivery44,45 and injectable polyhydroxybutyrate microspheres.38 However, none of them aimed to develop a hydrogel for subcutaneous use. Finally, even though this is a single-dose study, our results showed that the incorporation of TR into PL hydrogels promoted an increase in duration of analgesic effects with the possibility for reapplying every 48–72 hours at lower doses. This would significantly reduce systemic toxic effects and provides a hydrogel delivery system for subcutaneous TR.
Conclusion
Results obtained in this study allowed the design of PL-based hydrogel preformulations for controlled release of TR by subcutaneous use for the potential future treatment of postoperative pain. Physicochemical characterization showed that the formation of binary systems composed of PL 407 and PL 188 alters the micellization and sol-gel transition processes. The dissolution/release relationships and analgesic activity assays demonstrated that PL 407 and its binary systems with PL 188 are effective hydrogels for controlling and prolonging TR release for 48–72 hours after subcutaneous injection; this could reduce the number of administrations and possible adverse effects. Finally, the formulations tested here reduced the cytotoxicity compared to TR and no significant genotoxic effects were observed. These results demonstrate important features for the development of new drug carriers and their purpose as new therapeutic systems.
Acknowledgments
This work was supported by Fundҫão de Amparo à Pesquisa do Estado de São Paulo (FAPESP; grants 2006/00121-9, 2010/11475-1, 2010/13088-5) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grants 487619/2012-9, 300952/2010-4, 309612/2013-6).
Disclosure
The authors report no conflicts of interest in this work.
References
Coluzzi F, Mattia C. Chronic non-cancer pain: focus on once-daily tramadol formulations. Ther Clin Risk Manag. 2007;3(5):819–829. | ||
Desmeules JA, Piguet V, Collart L, Dayer P. Contribution of monoaminergic modulation to the analgesic effect of tramadol. Br J Clin Pharmacol. 1996;41(1):7–12. | ||
Xie H, Dong ZQ, Ma F, Bauer WR, Wang X, Wu GC. Involvement of serotonin 2A receptors in the analgesic effect of tramadol in mono-arthritic rats. Brain Res. 2008;1210:76–83. | ||
Nicholson B. Benefits of extended-release opioid analgesic formulations in the treatment of chronic pain. Pain Pract. 2009;9(1):71–81. | ||
Dumortier G, Grossoid JL, Agnely F, Chaumeil JC. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res. 2006;23(12):2709–2728. | ||
Yu L, Ding J. Injectable hydrogels as unique biomedical materials. Chem Soc Rev. 2008;37(8):1473–1481. | ||
Klouda L, Mikos AG. Thermoresponsive hydrogels in biomedical applications. Eur J Pharm Biopharm. 2008;68(1):34–45. | ||
Ricardo NM, Pinho ME, Yang Z, Atwood D, Booth C. Controlling the gelation of aqueous micellar solutions of ethylene-oxide-based block copoly(oxyalkylene)s. Int J Pharm. 2005;300(1–2):22–31. | ||
Harrison WJ, Aboulgassem GJ, Elathrem FA, et al. Micelles and gels of mixed triblock copoly(oxyalkylene)s in aqueous solution. Langmuir. 2005;21(14):6170–6178. | ||
Chaibundit C, Ricardo NM, Costa Fde M, Yeates SG, Booth C. Micellization and gelation of mixed copolymers P123 and F127 in aqueous solution. Langmuir. 2007;23(18):9229–9236. | ||
Newby GE, Hamley IW, King SM, Martin CM, Terril NJ. Structure, rheology and shear alignment of Pluronic block copolymer mixtures. J Colloid Interface Sci. 2009;329(1):54–61. | ||
Paavola A, Kilpeläinen I, Yliruusi J, Rosenberg P. Controlled release injectable liposomal gel of ibuprofen for epidural analgesia. Int J Pharm. 2000;199(1):85–93. | ||
Paavola A, Yliruusi J, Kajimoto Y, Kalso E, Wahlström T, Rosenberg P. Controlled release of lidocaine from injectable gels and efficacy in rat sciatic nerve block. Pharm Res. 1995;12(12):1997–2002. | ||
Paavola A, Yliruusi J, Rosenberg P. Controlled release and dura mater permeability of lidocaine and ibuprofen from injectable poloxamer-based gels. J Control Release. 1998;52(1–2):169–178. | ||
Park YJ, Yong CS, Kim HM, et al. Effect of sodium chloride on the release, absorption and safety of diclofenac sodium delivered by poloxamer gel. Int J Pharm. 2003;263(1–2):105–111. | ||
Ricci E, Bentley MV, Farah M, Bretas RE, Marchetti JM. Rheological characterization of Poloxamer 407 lidocaine hydrochloride gels. Eur J Pharm Sci. 2002;17(3):161–167. | ||
Ricci EJ, Lunardi LO, Nanclares DM, Marchetti JM. Sustained release of lidocaine from Poloxamer 407 gels. Int J Pharm. 2005;288(2):235–244. | ||
Sharma PK, Reilly MJ, Bhatia SK, Sakhitab N, Archambault JD, Bhatia SR. Effect of pharmaceuticals on thermoreversible gelation of PEO-PPO-PEO copolymers. Colloids Surf B Biointerfaces. 2008;63(2):229–235. | ||
Lee JW, Lim TH, Park JB. Intradiscal drug delivery system for the treatment of low back pain. J Biomed Mater Res A. 2010;92(1):378–385. | ||
Chen PC, Kohane DS, Park YJ, Bartlett RH, Langer R, Yang VC. Injectable microparticle-gel system for prolonged and localized lidocaine release. II. In vivo anesthetic effects. J Biomed Mater Res A. 2004;70(3):459–466. | ||
Yin QQ, Wu L, Gou ML, Qian ZY, Zhang WS, Liu J. Long-lasting infiltration anaesthesia by lidocaine-loaded biodegradable nanoparticles in hydrogel in rats. Acta Anaesthesiol Scand. 2009;53(9):1207–1213. | ||
Daniels S, Reader S, Berry P, Goulder M. Onset of analgesia with sodium ibuprofen, ibuprofen acid incorporating poloxamer and acetaminophen – a single dose, double-blind, placebo-controlled study in patients with post-operative dental pain. Eur J Clin Pharmacol. 2009;65(4):343–353. | ||
Fisher A, Watling M, Smith A, Knight A. Pharmacokinetic comparisons of three nasal fentanyl formulations; pectin, chitosan and chitosan-poloxamer 188. Int J Clin Pharmacol Ther. 2010;48(2):138–145. | ||
Schmolka IR. Artificial skin. I. Preparation and properties of pluronic F-127 gels for treatment of burns. J Biomed Mater Res. 1972;6(6):571–582. | ||
Guillouzo A. Use of isolated and cultures hepatocytes for xenobiotic metabolism and cytotoxicity studies. In: Guillouzo A, Guguen-Guillouzo C, editors. Research in Isolated and Cultures Hepatocytes. London, UK: John Libbey Eurotext; 1986:313–332. | ||
Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Meth. 1986;89(2):271–277. | ||
Lee JK, Kim DB, Kim JI, Kim PY. In vitro cytotoxicity tests on cultured human skin fibroblasts to predict skin irritation potential of surfactants. Toxicol In Vitro. 2000;14(4):345–349. | ||
Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184–191. | ||
Valero M, Dreiss CA. Growth, shrinking, and breaking of pluronic micelles in the presence of drugs and/or beta-cyclodextrin, a study by small-angle neutron scattering and fluorescence spectroscopy. Langmuir. 2010;26(13):10561–10571. | ||
Zhang Y, Lam YM, Tan WS. Poly(ethyleneoxide)-poly(propylene oxide)-poly(ethylene oxide)-g-poly(vinylpyrrolidone): association behavior in aqueous solution and interaction with anionic surfactants. J Colloid Interface Sci. 2005;285(1):74–79. | ||
Wei Z, Hao J, Yuan S, et al. Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. Int J Pharm. 2009;376(1–2):176–185. | ||
Kabanov AV, Batrakova EV, Alakov VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Rel. 2002;82(2–3):189–212. | ||
Trong LC, Djabourov M, Ponton A. Mechanisms of micellization and rheology of PEO-PPO-PEO triblock copolymers with various architectures. J Colloid Interface Sci. 2008;328(2):278–287. | ||
Nie S, Hsiao WL, Pan W, Yang Z. Thermoreversible Pluronic F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: in vitro drug release, cell cytotoxicity, and uptake studies. Int J Nanomedicine. 2011;6:151–166. | ||
Ju C, Sun J, Zi P, Jin X, Zhang C. Thermosensitive micelles-hydrogel hybrid system based on poloxamer 407 for localized delivery of paclitaxel. J Pharm Sci. 2013;102(8):2707–2717. | ||
Batrakova EV, Li S, Alakhov VY, Elmquist WF, Miller DW, Kabanov AV. Sensitization of cells overexpressing multidrug-resistant proteins by pluronic P85. Pharm Res. 2003;20(10):1581–1590. | ||
Batrakova EV, Li S, Brynskikh AM, et al. Effects of pluronic and doxorubicin on drug uptake, cellular metabolism, apoptosis and tumor inhibition in animal models of MDR cancers. J Control Release. 2010; 143(3):290–301. | ||
Salman MA, Sahin A, Onur MA, Oge K, Kassab A, Aypar U. Tramadol encapsulated into polyhydroxybutyrate microspheres: in vitro release and epidural analgesic effect in rats. Acta Anaesthesiol Scand. 2003; 47(8):1006–1012. | ||
Buhari S, Hashim K, Yong Meng G, Mustapha NM, Gan SH. Subcutaneous administration of tramadol after elective surgery is as effective as intravenous administration in relieving acute pain and inflammation in dogs. Scientific World Journal. 2012;2012:564939. | ||
Dooney NM, Sundararajan K, Ramkumar T, et al. Pharmacokinetics of tramadol after subcutaneous administration in a critically ill population and in a healthy cohort. BMC Anesthesiol. 2014;14:33. | ||
Jabalameli M, Hazegh P, Talakoub R. Preemptive subcutaneous tramadol for post-operative pain in lower abdomen surgeries: a randomized double blinded placebo-control study. Adv Biomed Res. 2013;2:68. | ||
Kumar P, Singh S, Mishra B. Development and biopharmaceutical evaluation of extended release formulation of tramadol hydrochloride based on osmotic technology. Acta Pharm. 2009;59(1):15–30. | ||
Morales ME, Ruiz MA, López G, Gallardo V. Development of oral suspensions of microparticles of ethylcellulose with tramadol. Drug Dev Ind Pharm. 2010;36(8):885–892. | ||
Ammar HO, Ghorab M, El-Nahhas SA, Kamel R. Design of a transdermal delivery system for aspirin as an antithrombotic drug. Int J Pharm. 2006;327(1–2):81–88. | ||
Kamel R, Mahmoud A, El-Feky G. Double-phase hydrogel for buccal delivery of tramadol. Drug Dev Ind Pharm. 2012;38(4):468–483. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.