Back to Journals » Journal of Inflammation Research » Volume 18
PM2.5 Induces Cell-Specific Transcriptomic Alterations in the Lungs of Juvenile Mice
Authors Gui B ![]() , Li Y, Li K, Wang J, Fan W
, Li Y, Li K, Wang J, Fan W ![]() , Li L, Wu Q
, Li L, Wu Q ![]() , Chen H
, Chen H
Received 26 December 2024
Accepted for publication 19 July 2025
Published 25 July 2025 Volume 2025:18 Pages 9951—9966
DOI https://doi.org/10.2147/JIR.S514437
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Biyu Gui,1,* Yu Li,2,* Kuan Li,2 Jianhai Wang,2 Wenping Fan,2 Li Li,1 Qi Wu,1,3 Huaiyong Chen2– 4
1Department of Respiratory Medicine, Haihe Hospital, Tianjin University, Tianjin, People’s Republic of China; 2Tianjin Key Laboratory of Lung Regenerative Medicine, Haihe Hospital, Tianjin University, Tianjin, People’s Republic of China; 3Key Research Laboratory for Infectious Disease Prevention for State Administration of Traditional Chinese Medicine, Tianjin Institute of Respiratory Diseases, Tianjin, People’s Republic of China; 4Key Laboratory of Medical Rescue Key Technology and Equipment, Ministry of Emergency Management, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Huaiyong Chen, Email [email protected] Qi Wu, Email [email protected]
Introduction: Fine particulate matter (PM2.5) is a major environmental pollutant associated with significant respiratory morbidity in children. However, its cell-type-specific effects on the lungs and the underlying molecular mechanisms remain poorly defined.
Methods: This study established a juvenile mouse model of PM2.5 airway exposure to assess transcriptional alterations in lung cells via single-cell RNA sequencing (scRNA-seq). Differentially expressed genes were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Lung histopathology was evaluated through hematoxylin and eosin staining.
Results: Histological staining indicated that PM2.5 inhalation induces structural damage in the lung tissue. ScRNA-seq analysis revealed that macrophages, dendritic cells (DCs), lymphocytes, epithelial cells, and stromal cells in the lungs of juvenile mice exhibited the most prominent differential gene expression following PM2.5 instillation, whereas B cells and endothelial cells showed the least. In particular, GO and KEGG analyses indicated that alveolar macrophages exhibited significant upregulation of oxidative phosphorylation (OXPHOS) pathways and downregulation of antibacterial defense mechanisms. CD209+ DCs showed suppressed antigen presentation and altered energy metabolism, primarily via enhanced OXPHOS. Lymphocytes, including NK and CD4+ T cells, displayed modest dysregulation in ribosomal activity. Among non-immune cells, ciliated cells activated interferon signaling, while adventitial fibroblasts showed increased ribosomal protein translation and calcium ion channel regulation. PM2.5 exposure also reshaped cell-cell communication networks, particularly involving alveolar macrophages and immune cells.
Conclusion: These findings reveal cell-type-specific transcriptomic responses to PM2.5 in juvenile lungs, emphasizing its potential to disrupt immune homeostasis and contribute to pulmonary disease development in children.
Keywords: air pollution, children, macrophages, dendritic cells, lung inflammation
Introduction
Fine particulate matter (PM2.5), as a critical component of air pollution, poses a severe threat to human health. The major sources of PM2.5 in the air include direct emissions from combustion processes such as vehicle exhaust, industrial activities, power plants, residential wood burning, and wildfires. According to the US Environmental Protection Agency (EPA) regulations updated in 2024, the primary annual exposure limit for PM2.5 has been reduced from 12.0 µg/m³ to 9.0 µg/m³, while the 24-hour standard remains at 35 µg/m³. Prolonged exposure to PM2.5 has been demonstrated to significantly increase morbidity and mortality.1 Following inhalation, PM2.5 initially causes damage to lung structure and function. In addition, it can indirectly affect the functions of the cardiovascular system, brain, and other organs.2,3 Therefore, elucidating the effects of PM2.5 on lung cell function can aid in understanding its toxicological mechanisms not only in the lungs but other organs.
The impact of PM2.5 on children’s lung health is a growing concern, as children are more vulnerable to air pollution due to their developing physiology, higher respiratory rates, and greater relative exposure. Studies consistently show that both short-term and long-term exposure to PM2.5 are associated with a range of respiratory health issues in children, including airway inflammation, impaired lung function, and the exacerbation or development of asthma and other respiratory diseases. For instance, research using the NYS Statewide Planning and Research Cooperative System database found that increases in PM2.5 concentrations were linked to higher emergency department visits and hospitalizations for asthma and chronic obstructive pulmonary disease in children.4 Similar findings in rural US households with wood stoves showed that exposure to indoor PM2.5 was associated with a higher incidence of lower respiratory tract infections in children.5 Furthermore, studies from China and India revealed that even PM2.5 levels below national air quality standards are linked to increased respiratory illnesses in children,6,7 highlighting the widespread impact of air pollution on child health. Interventions such as air purification have demonstrated effectiveness in reducing the adverse respiratory effects of PM2.5, underscoring the potential benefits of reducing exposure to improve pediatric lung health.8 Additionally, meta-analyses have shown that acute exposure to PM2.5 leads to reduced lung function in children, with those suffering from asthma being particularly susceptible.9 These findings suggest that PM2.5 impacts children both during the developmental stage and post-developmental stage. However, the toxic effects of PM2.5 on children and the underlying mechanisms have not been fully explored.
The mechanisms underlying PM2.5-induced lung injury have been extensively studied, including biochemical reactions such as the activation of oxidases and metabolic enzymes, as well as mitochondrial dysfunction leading to oxidative stress.10 On a single-cell level, several damage mechanisms have been identified in adult animal models. PM2.5 NO3− exposure induces gene expression dysregulation, disrupting circadian rhythms, lysosomal function, and cellular responses to stimuli, thereby affecting respiratory function.11 Transient inflammation, characterized by infiltration of CD11b-positive myeloid cells, has also been observed in the lung. Alveolar type 2 (AT2) cells proliferate in response to PM2.5, and infiltrated neutrophils or macrophages impair the reparative capacity of airway club cells through oxidative stress.12 PM2.5 exposure further promotes the expansion of PD-L1high neutrophils with impaired phagocytic function in the mouse lung, leading to increased susceptibility to bacterial infections.13 Additionally, PM2.5 triggers a Th17-specific inflammatory response, contributing to neutrophilic asthma via epithelial, dendritic, and T cell pathways that promote Th17 differentiation during initial exposure.14 Activation of the IL-17 signaling pathway is linked to the progression of pulmonary fibrosis following sub-chronic PM exposure. Activation of the IL-17 signaling pathway reduces the recruitment of myeloid-derived suppressor cells and downregulates TGF-β, contributing to the progression of lung fibrosis after sub-chronic PM exposure.15 Subway-related PM2.5 induces specific immune responses involving CD4+ and γδ T cells, which are also key contributors to inflammation.16 Prolonged exposure to insoluble PM2.5 particles leads to lung fibrosis by decreasing Nrf2 transcriptional activity and downregulating antioxidant gene expression.17 Organoid technology suggests that lung repair may be compromised because of senescence induced by PM2.5 exposure.18 However, there is still a lack of systematic research on its impact on gene expression in lung cells, including epithelial cells, stromal cells, and immune cells, in children. In particular, the regulatory mechanisms by which PM2.5 affects gene expression in these cells, as well as the associated alterations in signaling pathways, remain critical areas that require further investigation.
Single-cell RNA sequencing (scRNA-seq) offers a powerful platform to delineate transcriptomic changes at cellular resolution, enabling detailed analysis of diverse pulmonary cell populations and their intercellular communication networks. In this study, we employed a juvenile mouse model of PM2.5 airway exposure and used scRNA-seq to investigate the cellular and molecular alterations in the lung. This approach provides critical insights into the early immune and structural responses to PM2.5 in developing lungs, with potential implications for understanding pollutant-driven respiratory diseases in children.
Material and Methods
Animal Model and PM2.5 Administration
C57BL/6 mice were purchased from Beijing SPF Biotechnology Co., Ltd. and housed under a 12-hour light/dark cycle with ad libitum access to food and water in specific pathogen-free facilities at Tianjin Haihe Hospital. Three-week-old male C57BL/6 mice, with body weights ranging from 7.0 to 9.0 g, were randomly assigned to either the PBS or PM2.5 group. Every other day at 9:00 AM, the mice were administered oral treatments: the PBS group received 3 µL/g of PBS, while the PM2.5 group was administered 18 mg/kg of a PM2.5 suspension. Specifically, the PM2.5 was resuspended in PBS to a concentration of 6 µg/µL, and each mouse received 3 µL/g of the suspension, adjusted according to their body weight. Throughout the modeling period, the body weight of each mouse was recorded daily at 9:00 AM.19 PM2.5 was collected as described previously.12 In brief, PM2.5 mixtures were collected from the atmosphere using quartz filters installed in a particulate matter sampler. The chemical composition of the collected samples was characterized using ion chromatography, thermal-optical analysis, high-performance liquid chromatography, and inductively coupled plasma mass spectrometry. Anesthesia was induced using isoflurane prior to each administration. Pulmonary function was measured at 9:00 AM following the final dose, with four mice from each group tested in an alternating sequence. Immediately after pulmonary function measurements, lung tissue samples were collected for paraffin embedding and RNA extraction. Additional lung tissue samples were collected on day 35 of the modeling period for subsequent paraffin embedding and RNA extraction. Mice were sacrificed for analysis the day after the third administration. All animal procedures were conducted in accordance with the guidelines for Ethical Review of Experimental Animal Welfare (GB/T35892-2018) issued by China, and were approved by the Animal Care and Use Committee of Tianjin Haihe Hospital (approval number: 2021HHKT(A)-004).
Pulmonary Function Assessment
Mice were anesthetized with 1% sodium pentobarbital (50 mg/kg, intraperitoneally). After cervical skin incision, the trachea was exposed and intubated. The endotracheal cannula was then connected to the FlexiVent system (SCIREQ) by a certified technician to perform pulmonary function assessment.
Hematoxylin and Eosin (H&E) Staining
Lungs from mice either unexposed or exposed to PM2.5 were collected, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at a thickness of 5 μm. The sections were then stained with hematoxylin and eosin (H&E) according to the manufacturer’s instructions to evaluate structural changes associated with PM2.5 exposure.
Reverse Transcription/Polymerase Chain Reaction (RT/PCR)
Total RNA was extracted from lung tissue using TRIzol reagent (15596026, Invitrogen). RNA concentration was measured using a microplate reader (Multiskan GO, Thermo Fisher Scientific) and adjusted to 80 ng/μL. Reverse transcription was carried out using the HiScript II Reverse Transcriptase kit (R323-01, Vazyme), followed by quantitative PCR using SYBR Green Master Mix (Q711-02, Vazyme). Gene expression levels were calculated using the 2^−ΔCT method, with β-actin serving as the internal reference gene.
Preparation of Single-Cell Lung Suspension
Mice (control and PM2.5-exposed) were euthanized, and their lungs were harvested. The lungs were perfused via the trachea with 4 U/mL elastase (Worthington Biochemical Corporation, Lakewood, NJ) and digested at 37°C for 25 minutes. After digestion, lung tissue was mechanically dissociated into 2 mm³ fragments using a scalpel. The resulting cell suspension was incubated with 5 mL of 0.5% DNase I (Sigma-Aldrich) at 37°C for 15 minutes to degrade DNA released from lysed cells. The suspension was then filtered through a 70 µm cell strainer (FALCON, 352350) to remove larger debris, followed by incubation with red blood cell lysis buffer (Sigma, R7757) for 90 seconds to remove red blood cells. Single-cell suspensions from three individual mice in the PBS- or PM2.5-treated groups were pooled for single-cell RNA sequencing analysis to minimize individual variability.
Single-Cell RNA Sequencing
The cell suspension was quality-checked to assess cell viability (AO/PI fluorescence count > 80% or trypan blue exclusion > 75%) and cell concentration (700–1200 cells/µL). Approximately 20,000 cells were obtained per sample. The cell suspension was loaded onto Chromium microfluidic chips with 3′ (v3) chemistry and barcoded using the 10× Chromium Controller (10X Genomics). RNA from the barcoded cells was reverse-transcribed, and sequencing libraries were constructed using reagents from the Chromium Single Cell 3′ (v3) reagent kit (10X Genomics). Sequencing was performed on an Illumina NovaSeq system (PE150).
Cell Annotation
The sequencing data were processed using the Seurat package (version 4.4.0), with quality control criteria including: a minimum of 200 genes expressed per cell, genes expressed in at least 3 cells, and mitochondrial gene expression accounting for < 10% of total gene expression. Doublets were predicted and removed using the DoubletFinder package (version 2.0.4). Batch effects between the two groups were corrected using the Harmony package (version 1.2.0). Level 1 annotation was conducted after dimensionality reduction and clustering were performed using Seurat with UMAP, with resolution setting of 0.8. And level 2 annotations were conducted using the same approach, with adjustments to the dimensionality and resolution settings of 2.0. Moreover, three type of immune cells, macrophages, dendritic cells and T cells were clustered separately for level 3 annotation. The annotation results were mapped back to the initial clusters.
Single-Cell Analysis
Differential gene expression analysis between two groups was conducted using the FindMarkers function in Seurat, with thresholds set at a fold change > 1.5 and an adjusted p-value < 0.05. Gene set enrichment analysis (GSEA) was performed on the differentially expressed genes (DEGs) using the ClusterProfiler package (v4.6.2), with the enrichGO and enrichKEGG functions for overrepresentation analysis of Gene Ontology Biological Processes (GOBP) and KEGG pathways, respectively. For each cell type, we performed GO and KEGG enrichment analyses and selected terms with an adjusted p-value (P.adjust) < 0.05. Among these, the top 5 or 10 terms ranked by –log(P.adjust) were selected for visualization. GSEA for KEGG pathways was carried out using the gseKEGG function. Cell-cell interactions were analyzed with the CellChat package (version 2.1.2), and results were visualized using the package’s built-in functions.
Data Processing and Statistical Analysis
Data manipulation was conducted using the tidyverse package. Differential expression significance was determined via Wilcoxon test, with a false discovery rate–adjusted p-value <0.05 considered significant.
Data Visualization
Plots were generated using R (v4.2.1), primarily with Seurat (DimPlot, VlnPlot, FeaturePlot) and CellChat (NetVisual_Bubble). Additional figures were created with ggplot2 (v3.5.0) and UpSetR (v1.4.0).
Statistical Analysis
All data analysis was conducted using R (version 4.2.1) and relevant packages tailored for single-cell RNA sequencing (scRNA-seq) data, including Seurat (v4.4.0), CellChat (v2.1.2), and ClusterProfiler (v4.6.2). Differential gene expression (DGE) between control and PM2.5-exposed groups was performed using the FindMarkers function in Seurat. Genes were considered significantly differentially expressed if they met a fold-change threshold of 1.5 and an adjusted p-value of <0.05. To assess the statistical significance of gene expression changes across cell populations, we applied the Wilcoxon rank-sum test, with a false discovery rate (FDR)-adjusted p-value < 0.05 serving as the threshold for significance. These analyses were specifically designed to capture subtle yet biologically meaningful transcriptional shifts in the context of single-cell resolution. All quantitative data are expressed as mean ± standard error of the mean (SEM). Statistical comparisons between groups were conducted using two-tailed Student’s t-test or Wilcoxon rank-sum test, depending on data distribution. Statistical significance was set at p < 0.05 for all analyses.
Results
Cell-Type-Specific Transcriptomic Responses to PM2.5 Exposure in Juvenile Mouse Lungs
To investigate the pulmonary effects of PM2.5 in developing lungs, juvenile mice were subjected to repeated PM2.5 airway instillation. Lung tissues from exposed and control groups were collected for histological and single-cell RNA sequencing (scRNA-seq) analysis (Figure 1A). Mice exposed to PM2.5 exhibited an approximately 20% greater reduction in body weight from day 1 to day 5 compared to PBS-treated controls (Figure 1B). H&E staining indicated lung inflammation and structural damaged 5 days following PM2.5 exposure (Figure 1C). At this time point, pulmonary function tests demonstrated increased total airway resistance (Rrs) in PM2.5-exposed mice compared to PBS controls (2.17 ± 0.18 vs 2.57 ± 0.20 cmH₂O·s/mL), with a particularly marked increase in large airway resistance (Rn) (0.48 ± 0.03 vs 0.66 ± 0.04 cmH₂O·s/mL) (Figure 1D). In contrast, small airway resistance, as indicated by the tissue damping parameter G, was decreased (19.22 ± 1.51 vs 16.80 ± 0.96 cmH₂O/mL). Additionally, pulmonary function assessment revealed a 0.055 J/L increase in respiratory work per unit volume based on pressure–volume (P–V) loop analysis (1.35 ± 0.02 vs 1.40 ± 0.02 J/L). Peak Expiratory Flow (PEF) decreased to an average of 87.6% of that in PBS controls (17.3 ± 1.21 vs 15.2 ± 0.93 mL/s). Furthermore, forced expiratory flow at 50% of forced vital capacity (FEF50) declined by approximately 3 mL/s (14.70 ± 1.11 vs 11.72 ± 2.02 mL/s). These changes indicate impaired pulmonary mechanics shortly following PM2.5 exposure. By day 35, both body weight and lung histology returned to baseline (Figure S1).
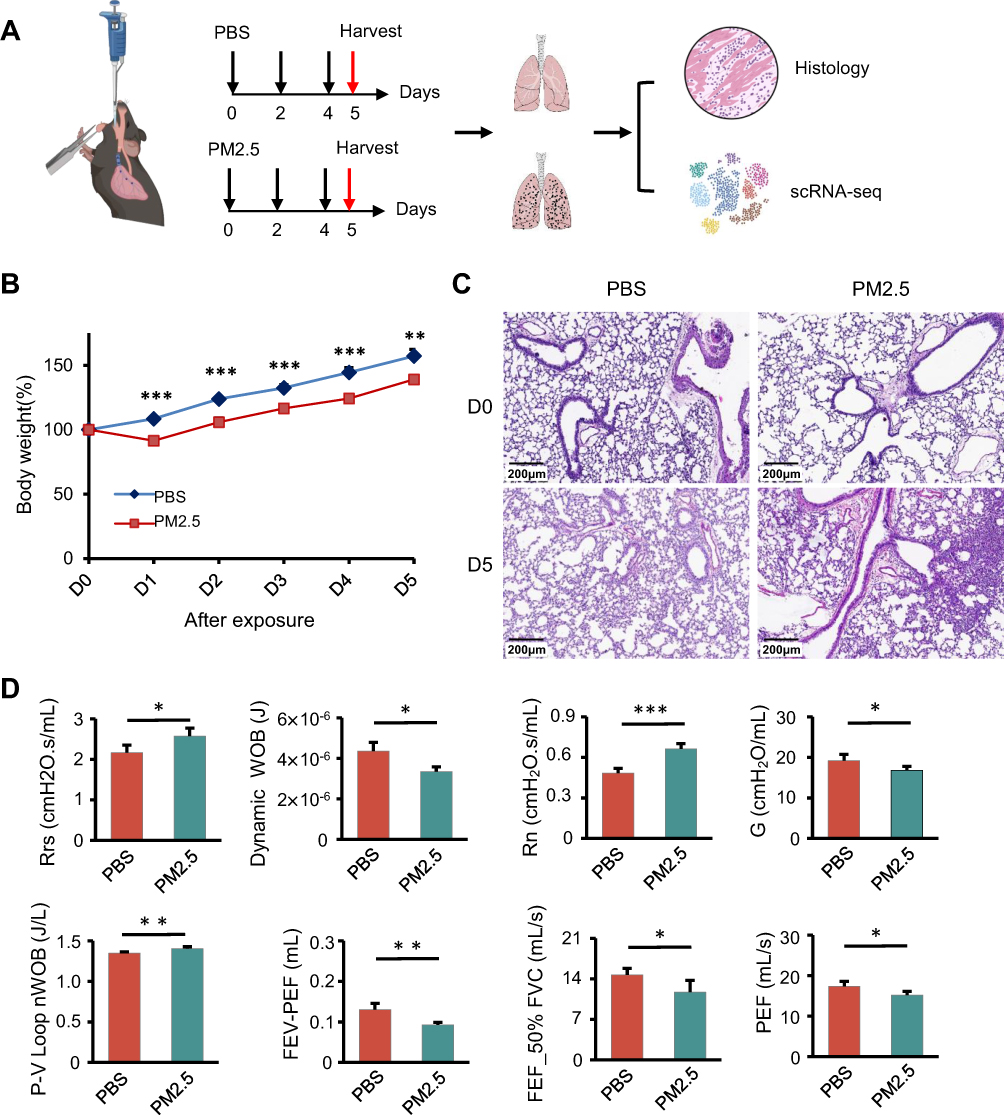 |
Figure 1 PM2.5 exposure induces respiratory dysfunction in juvenile mice. (A) Schematic illustration of the experimental design for establishing the PM2.5 airway instillation model in juvenile mice, incorporating histological analysis and single-cell RNA sequencing (scRNA-seq). (B) Body weight trajectories of juvenile mice in the PBS control group and PM2.5 exposure group over the study period. Representative histological images showing lung tissue alterations following PM2.5 exposure; *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t-test). (C) Representative histological images showing lung tissue alterations following PM2.5 exposure. (D) Lung function assessment demonstrates increased airway resistance (Rrs: respiratory system resistance; Rn: Newtonian resistance) and reduced expiratory flow parameters (FEV: forced expiratory volume; PEF: peak expiratory flow; FVC: forced vital capacity; FEF50%FVC: forced expiratory flow at 50% of FVC) following PM2.5 exposure; *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t-test). |
scRNA-seq of lung tissue yielded 20,951 high-quality cells and 16,411 detected genes following quality control and batch correction. UMAP clustering identified 14 major cell types based on canonical markers (Figure 2A and B). Differential gene expression (DGE) analysis revealed that macrophages, dendritic cells (DCs), stromal cells, epithelial cells, and lymphocytes exhibited the most pronounced transcriptomic alterations, while B cells and endothelial cells were minimally affected (Figure 2C). Among all cell types, macrophages displayed the greatest transcriptional responsiveness to PM2.5 exposure.
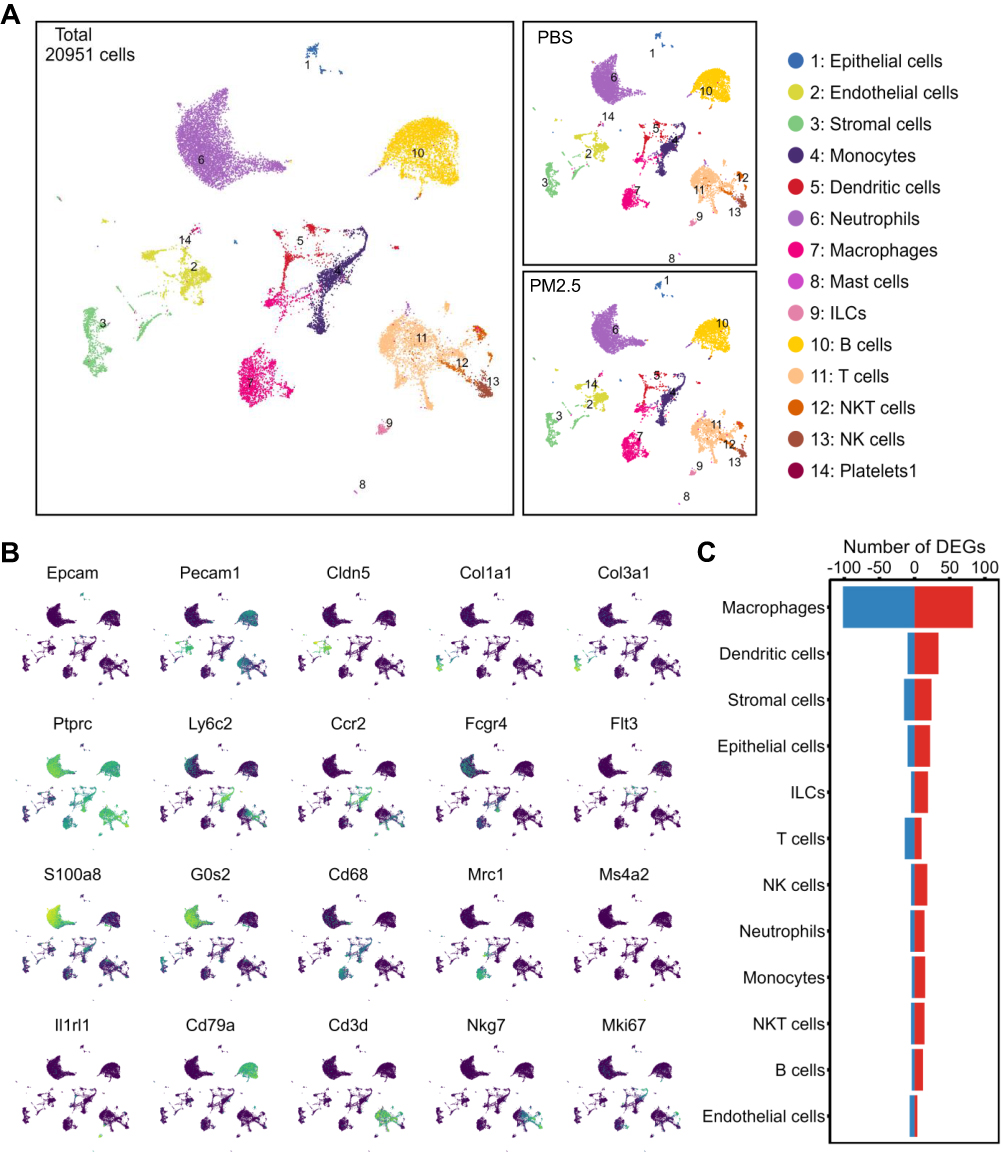 |
Figure 2 Transcriptomic alterations across lung cell types in juvenile mice following PM2.5 exposure. (A) UMAP visualization of 20,951 single cells from juvenile mouse lungs, clustered into 14 major cell types. Middle panels display cell distributions in the PBS and PM2.5 groups, respectively. (B) Feature plots showing expression of marker genes used to annotate major cell clusters; yellow and blue indicate high and low expression levels, respectively. (C) Bar plot summarizing differentially expressed genes (DEGs) in the 14 cell types post PM2.5 exposure. Red and blue bars denote upregulated and downregulated genes, respectively. |
PM2.5 Exposure Induces Metabolic Activation and Suppresses Antibacterial Functions in Alveolar Macrophages
Given their robust transcriptional response, macrophages were further subclustered into 17 subpopulations, ultimately categorized into 7 distinct macrophage subsets based on marker expression (Figure 3A, Figure S2A). Alveolar macrophages (AMs) represented the predominant subtype (Figure S2B). Differential gene analysis revealed that AMs exhibited the most significant upregulation and downregulation of genes following PM2.5 exposure, with most of these genes being unique to AMs (Figure 3B and C).GO enrichment analysis of the DEGs in each macrophage subtype showed that the upregulated genes in AMs were primarily associated with energy metabolism, while downregulated genes were involved in inflammatory responses to molecule of bacterial origin and to lipopolysaccharide (Figure 3D and E). These analyses suggest that PM2.5 exposure may impair AMs’ response to bacterial infections and their ability to control inflammation (Figure 3E). To further explore this, we examined the expression of key downregulated genes involved in these inflammatory responses, including Cd14, Fcer1g, Fcgr2b, Lepr, Mertk, and Tlr2 (Figure 3F). Given the critical role of alveolar macrophages (AMs) in immune defense, these processes may be functionally associated with the phagocytic activity of AMs. Notably, we observed significant downregulation of six phagocytosis-related genes—Cd86, Cxcl12, Il18, Nlrp3, Tlr2, and Tnf—in response to PM2.5 exposure (Figure 3G). Gene set enrichment analysis (GSEA) confirmed a shift toward elevated energy metabolism and suppressed innate immune signaling, as reflected by enhanced OXPHOS and downregulated NOD-like receptor (NLR) signaling pathways (Figure 3H). Collectively, these findings suggest that while AMs increase energy production in response to PM2.5, their bactericidal capacity is concurrently impaired, potentially compromising host defense mechanisms in juvenile lungs.
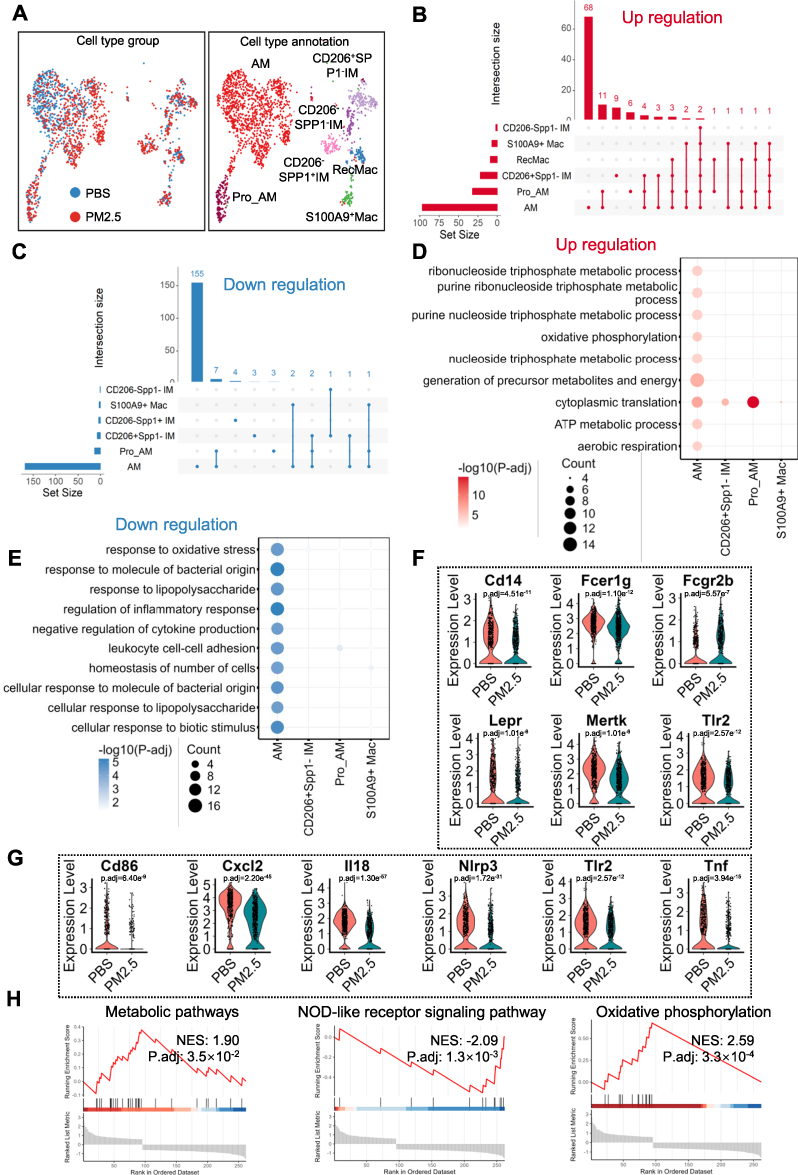 |
Figure 3 PM2.5 exposure enhances energy metabolism while impairing antibacterial responses in alveolar macrophages (AMs). (A) Sub-clustering of macrophages into seven distinct subtypes. Left panel shows distribution across groups; right panel presents final subtype annotations. (B and C) UpSet plots depicting DEGs across AM subtypes. Horizontal bars indicate DEG counts per subtype; matrix plot highlights shared genes; vertical bars display sizes of gene intersections. (B and C) illustrate upregulated and downregulated DEGs, respectively, excluding subtypes without DEGs. (D and E) GO biological process (GOBP) enrichment of DEGs related to energy metabolism and antibacterial activity. Dot size indicates gene count; color intensity reflects –log₁₀ adjusted p-value; magenta and blue represent upregulated and downregulated terms. (F) Violin plots show the expression levels of six representative differentially expressed genes (Cd14, Fcer1g, Fcgr2b, Lepr, Mertk, and Tlr2) in alveolar macrophages (AMs) from PBS-treated and PM2.5-exposed mice. (G) Violin plots show the expression levels of six representative phagocytosis-related genes (Cd86, Cxcl12, Il18, Nlrp3, Tlr2, and Tnf) in AMs from PBS-treated and PM2.5-exposed mice. (H) GSEA results for KEGG pathways show enhanced oxidative phosphorylation and suppressed NOD-like receptor signaling post PM2.5 exposure. |
PM2.5 Exposure Impairs Antigen Presentation in CD209+ Type 2 Dendritic Cells
To further elucidate dendritic cell (DC) responses to PM2.5, a total of 712 DCs were reclustered into eight distinct clusters, subsequently annotated into six functional subtypes based on established markers (Figure 4A, Figure S3A). Among these, CD209+ type 2 DCs (DC2s) constituted the most abundant population (Figure S3B).
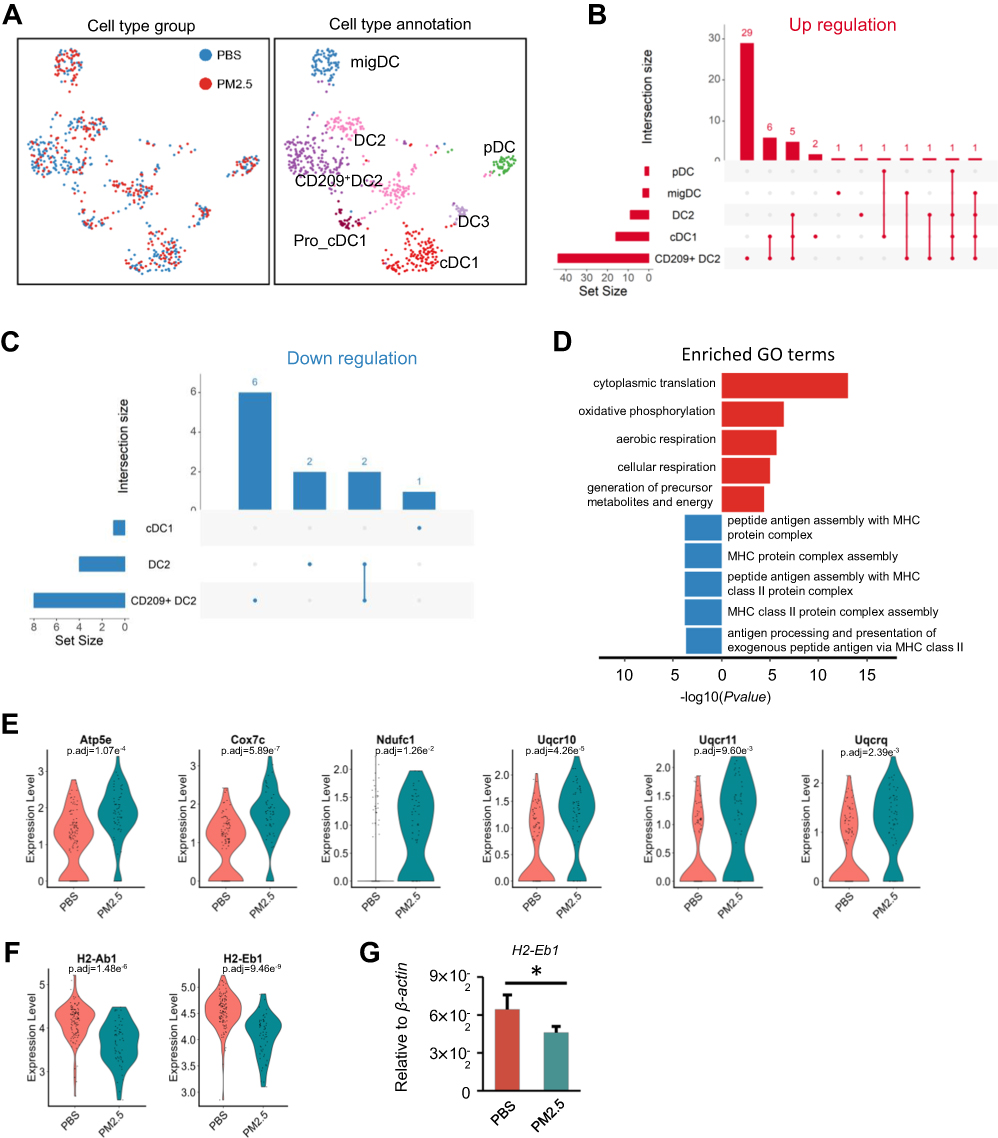 |
Figure 4 PM2.5 exposure disrupts antigen presentation in CD209+ dendritic cells (DCs). (A) Further subdivision of DCs into seven subtypes, with left and right panels showing distribution and final annotation, respectively. (B and C) UpSet plots showing the extent and overlap of DEGs across DC subtypes, with CD209+ DCs exhibiting the highest DEG counts. (D) Bar plot displaying GOBP enrichment of upregulated and downregulated genes related to antigen presentation in CD209+ DCs. (E) Violin plots display the expression levels of six representative OXPHOS-related DEGs (Atp5e, Cox7c, Ndufc1, Uqcr10, Uqcr11, and Uqcrq) in CD209⁺ DC2s. The upregulation of these genes suggests enhanced mitochondrial metabolic activity in response to PM2.5 stimulation. (F) Violin plot of H2-Eb1 and H2-Eb1 expression in CD209+ DC2 from PM2.5 and PBS groups. (G) Quantitative PCR analysis of H2-Eb1 mRNA in lung tissues of PM2.5-exposed vs control mice. Statistical significance was assessed by two-tailed Student’s t-test (*p < 0.05). |
Differential expression analysis revealed that CD209+ DC2s exhibited the highest number of DEGs among DC subtypes, with a predominance of upregulated genes (Figure 4B and C). GO enrichment analyses of the DEGs in CD209+ DC2s showed significant upregulation in OXPHOS pathways (Figure 4D). Six representative upregulated DEGs, including Atp5e, Cox7c, Ndufc1, Uqcr10, Uqcr11, and Uqcrq, were further highlighted (Figure 4E), underscoring the potential metabolic reprogramming of CD209⁺ DC2s in response to PM2.5 exposure. Furthermore, pathways associated with antigen presentation via MHC class II in CD209+ DC2s were downregulated (Figure 4D). Two key differentially expressed genes (DEGs) involved in this process, H2-Eb1 and H2-Ab1, were identified and are shown in (Figure 4F). Consistent with these findings, we detected that H2-Eb1 mRNA was deregulated in lung tissue, as confirmed by RT-PCR analysis (Figure 4G). These findings indicate that PM2.5 exposure leads to metabolic activation but impairs the antigen-presenting capabilities of CD209+ DC2s in juvenile lungs, potentially compromising their role in initiating adaptive immune responses.
PM2.5 Exposure Disrupts Ribosomal Function in CD4+ T and NK Cells
Lymphocytes, integral to adaptive immunity, exhibited relatively minor alterations compared to myeloid immune cells following exposure to PM2.5. Re-clustering of 3,890 lymphocytes, including T cells, NK cells, and NKT cells, resulted in the identification of 19 distinct clusters. Based on characteristic surface markers, six subtypes of T/NK lymphocytes were delineated (Figure 5A, Figure S4A). The analysis also incorporated innate lymphoid cells (ILCs) and B cells, revealing that B cells, CD4+ T cells, and CD8+ T cells were the predominant subtypes (Figure S3B). Differential gene expression analysis of these lymphocyte subsets demonstrated that NK cells exhibited the highest number of upregulated genes, whereas CD4+ T cells showed a greater extent of downregulation (Figure 5B and C). GO enrichment analysis of the differential genes in CD4+ T cells indicated that both upregulated and downregulated genes were primarily involved in ribosomal GOBP (Figure 5D), while GSEA of KEGG pathway further showed that CD4+ T cells displayed downregulation in ribosomal-related processes (Figure 5E). In line with this, six representative downregulated DEGs involved in cytoplasmic translation—Rpl9, Rpl15, Rpl19, Rps3, Rps4x, and Rps7—were identified and illustrated (Figure 5F). In contrast, upregulated genes in NK cells were enriched in ribosomal GOBP (Figure 5G), as exemplified by four representative differentially expressed genes (DEGs): Rpl37a, Rpl41, Rps28, and Rps21 (Figure 5H). On the other hand, the downregulated genes in NK cells were associated with sex-related GO biological processes (Figure 5G), including two key DEGs—Ubb and H3f3b (Figure 5I). These findings suggest that PM2.5 exposure disrupts protein synthesis pathways in both CD4⁺ T cells and NK cells in juvenile mice, potentially affecting their immune functions and metabolic homeostasis.
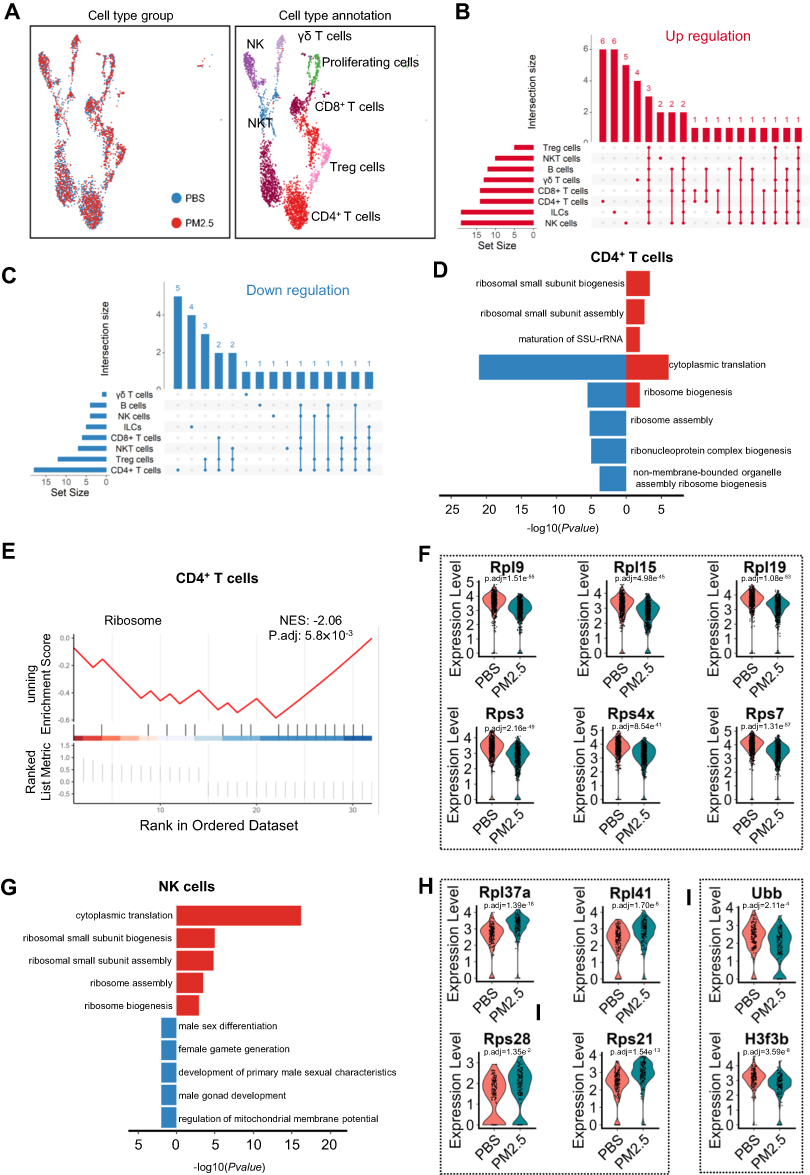 |
Figure 5 Ribosome-related transcriptional changes in lymphocytes following PM2.5 exposure. (A) NK and T lymphocytes were further annotated into seven subtypes; panels show their distribution and final classification. (B and C) UpSet plots highlighting DEGs across NK cells and CD4+ T cells, the most affected subtypes. (D and E) GOBP enrichment analysis of downregulated genes in CD4+ T cells. (D) shows bar plots; (E) presents the corresponding GSEA result. (F) Violin plots illustrate the expression levels of six genes (Rpl9, Rpl15, Rpl19, Rps3, Rps4x, and Rps7) in CD4+ T cells from control and PM2.5-treated groups, suggesting impaired protein synthesis machinery in response to particulate matter exposure. (G) Bar plot of GOBP enrichment analysis showing ribosome-related upregulation in NK cells post PM2.5 exposure. (H) Violin plots display the expression levels of four representative ribosomal protein genes—Rpl37a, Rpl41, Rps28, and Rps21—in NK cells from control and PM2.5-exposed mice. (I) Violin plots illustrate the expression levels of two representative differentially expressed genes—Ubb and H3f3b—which are associated with chromatin remodeling and gene regulation in the context of sex-linked biological processes. |
PM2.5 Exposure Promotes Interferon Response in Ciliated Cells and Enhances Ribosomal Functions in Adventitial Fibroblasts
Epithelial cells, endothelial cells, and stromal cells are critical components of the lung, but they exhibited relatively fewer differential changes following PM2.5 exposure in juvenile mice (Figure 2C). Dimensionality reduction and clustering of these cell types based on characteristic markers identified five subtypes of epithelial cells, five subtypes of endothelial cells, and six subtypes of stromal cells (Figure S5, Figure 6A). Differential gene expression analysis across these 16 cell subtypes revealed that six subtypes exhibited significant changes in gene expression, with adventitial fibroblasts and ciliated cells showing the highest number of differentially expressed genes (DEGs) (Figure 6B and C). GO enrichment analysis of DEGs revealed that ciliated cells were significantly enriched in the regulation of interferon-related genes and viral defense pathways (Figure 6D). Two key upregulated DEGs associated with these processes, Ifitm1 and Ifitm3, were highlighted (Figure 6E). These results were further supported by quantitative RT-PCR analysis, which confirmed the dysregulation of Ifitm1 mRNA expression in lung tissue (Figure 6F). DEGs in adventitial fibroblasts showed predominant enrichment in biological processes associated with ribosome assembly and calcium ion channel regulation (Figure 6G). Notably, five significant DEGs linked to ribosome assembly, including Rpl31, Rps21, Rpl41, Rps28, and Rpl37a, were prominently identified (Figure 6H). Concurrently, three downregulated DEGs related to calcium ion channel regulation, namely Fos, Jun, and Jund, were detected (Figure 6I). Subsequent quantitative RT-PCR analysis further validated these findings by confirming the altered expression patterns of Jun and Jund mRNA in lung tissue (Figure 6J). Collectively, these results suggest that PM2.5 exposure affects distinct cellular processes in both ciliated cells and adventitial fibroblasts in juvenile mice.
 |
Figure 6 Ribosome-related transcriptional changes in lymphocytes following PM2.5 exposure. (A) Further classification of parenchymal cells into 16 subtypes across epithelial, endothelial, and interstitial compartments. (B and C) UpSet plots indicating a higher DEG burden in ciliated cells and adventitial fibroblasts following PM2.5 exposure. (D) GOBP enrichment analysis of DEGs in ciliated cells, with magenta/blue indicating up-/downregulation. (E) Violin plots of Ifitm1 and Ifitm3 expression in ciliated cells of PM2.5 compared to control mice. (F) qPCR analysis of Ifitm1 mRNA in lung tissues; significance tested via two-tailed Student’s t-test (*p < 0.05). (G) GOBP enrichment analysis of DEGs in adventitial fibroblasts. (H and I) Violin plots display the expression levels of five key differentially expressed genes (Rpl31, Rps21, Rpl41, Rps28, and Rpl37a) associated with ribosome assembly, and the expression of three representative DEGs—Fos, Jun, and Jund—involved in calcium channel regulation. (J) qPCR validation of Jun and Jund expression in lung tissues; *p < 0.05 (Student’s t-test). |
PM2.5 Exposure Alters the Interactions Between Alveolar Macrophages and Other Inflammatory Cells
We next investigated the impact of PM2.5 exposure on cellular interactions within the lung. By analyzing changes in the number of receptor-ligand pairs and their interaction strength between various cell types following PM2.5 exposure, we intersected these pairs with differentially expressed genes (DEGs). This analysis revealed that alveolar macrophages (AMs) exhibited significant alterations in receptor-ligand expression after PM2.5 exposure (Figure 7A). Based on the probability values of receptor-ligand pairs in AMs, four prominent cell interaction patterns were identified (Figure 7B), with AMs primarily exerting regulatory effects on neutrophils and B cells. Notably, following PM2.5 exposure, the expression of seven ligands was found to influence neutrophils and B cells, while monocytes and neutrophils could modulate AMs via three distinct receptors on AMs (Figure 7C). These findings suggest that PM2.5 exposure activates AMs, which then engage in interactions with other immune cells, particularly neutrophils and B cells, in the lungs of juvenile mice.
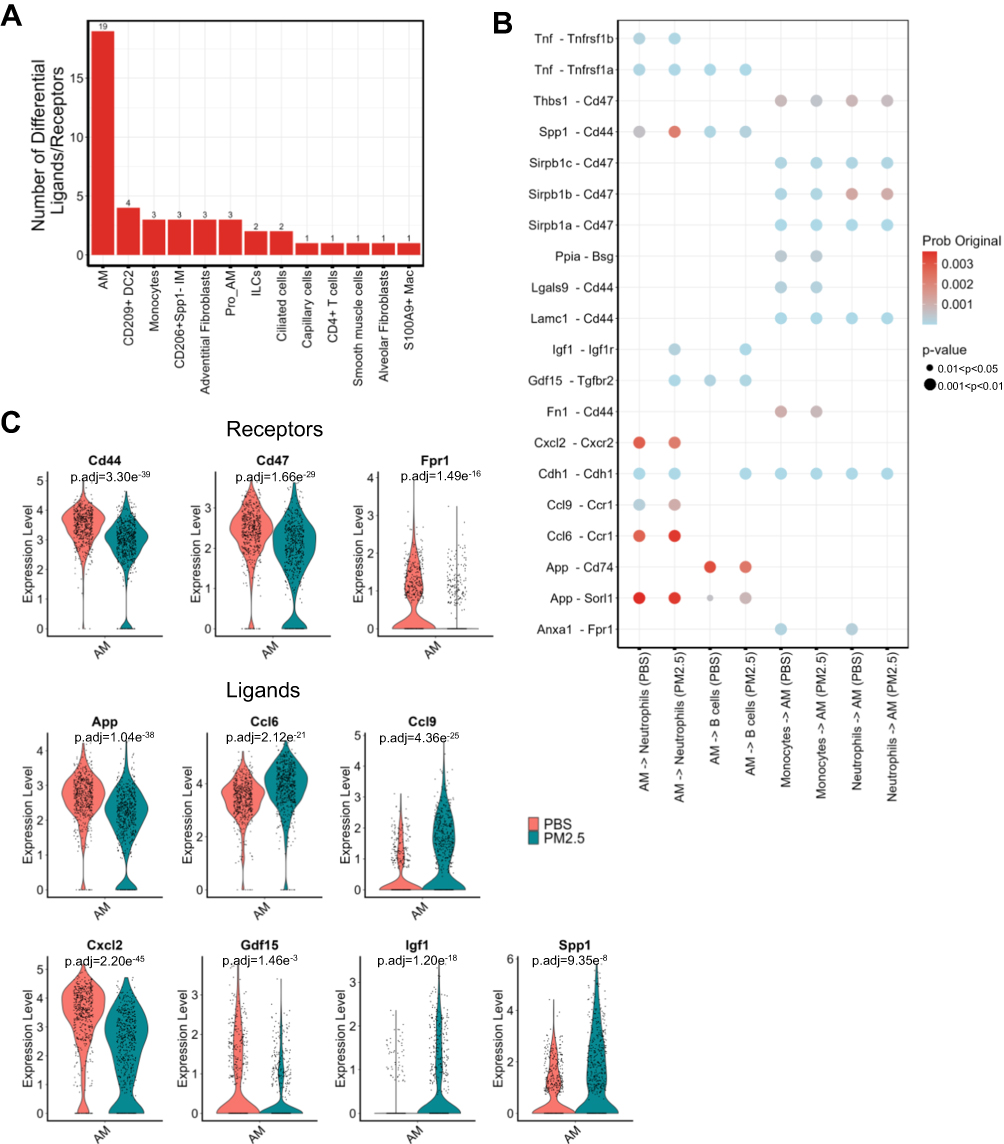 |
Figure 7 Alveolar macrophage-mediated intercellular signaling is altered by PM2.5 exposure. (A) Bar plot indicating the number of differentially expressed ligand-receptor pairs across cell subtypes. (B) CellChat-predicted interaction networks, highlighting the top four signaling patterns mediated by AMs. (C) The violin graph shows the expression of receptors and ligands in the control group and the PM2.5 exposure group. |
Discussion
This study investigates the effects of PM2.5 exposure on both immune and parenchymal cells in the lungs of juvenile mice. We found that myeloid immune cells, particularly alveolar macrophages (AMs) and CD209+ dendritic cells (DCs), were the most significantly affected populations. Specifically, PM2.5 exposure led to notable alterations in the energy metabolism pathways of AMs and CD209+ DCs. Furthermore, PM2.5 exposure impaired the antibacterial functions of AMs and the antigen presentation capabilities of DCs in juvenile mice. In contrast, the changes observed in parenchymal cells were less pronounced, though specific responses were still evident. Among the parenchymal cells, vascular endothelial cells were minimally affected, whereas ciliated epithelial cells and adventitial fibroblasts in the stromal layer exhibited the most significant alterations. These findings underscore the heightened sensitivity of the lung immune environment to PM2.5 exposure.
It is not unexpected that macrophages, as key phagocytic cells, exhibit the most profound alterations following PM2.5 exposure. We observed that PM2.5 exposure activated energy metabolism in AMs while downregulating transcripts associated with antibacterial defense. This suggests that while AMs are involved in clearing PM2.5, they concurrently lose their ability to defend against bacterial infections. This impairment of innate immunity in the lungs of children may increase the susceptibility to respiratory infections and hospitalizations. Previous studies have shown that PM2.5 exposure induces nuclear factor-κB (NF-κB) activation and nitric oxide production in macrophages.20,21 Moreover, PM2.5 alters macrophage functions by inducing intracellular Ca2+ dysregulation and excessive endoplasmic reticulum stress.22 The toxic effects of PM2.5 on AMs extend beyond alterations in intracellular metabolism to include modulation of metabolic pathways and intercellular interactions. PM2.5 has been shown to promote inflammation through the regulation of macrophage-epithelial interactions in the lung.23 In this study, we also observed that PM2.5 exposure led to AMs modulating their interactions with neutrophils via various ligand-receptor pairs, such as Spp1-Cd44, Igf1-Igf1r, Gdf15-Tgfbr2, and Cxcl2-Cxcr2. These interactions influence neutrophil chemotaxis and recruitment. Notably, Cxcl2 is transcriptionally regulated by the NLR signaling pathway. Although no significant changes were observed in the proportion or function of neutrophils, we hypothesize that PM2.5 exposure may downregulate the NLR pathway in AMs, leading to reduced Cxcl2 release and limiting excessive neutrophil recruitment. This modulation could be essential in regulating lung repair processes. The observed alterations in the population of IM and other immune cell subsets following exposure to PM2.5 suggest a potential modulation of immune responses at the cellular level. Transcriptomic analysis of these cell subpopulations revealed a significant downregulation of genes involved in immune activation, cytoskeletal remodeling, and lipid metabolism.24–26 These findings are indicative of a shift in macrophage phenotypes toward a reparative or alternatively activated state, which may reflect an adaptive response to chronic inflammation or tissue damage induced by PM2.5 exposure.27 This transition could have important implications for the resolution of inflammation and the restoration of tissue homeostasis in the context of environmental stressors.
In comparison to our previous study using a chronic PM exposure model in adult mice,27 both juvenile and adult mice exhibited alterations in myeloid cells following PM2.5 exposure. However, the absence of significant epithelial cell proliferation or repair-related gene expression in juvenile mice following PM2.5 exposure, in contrast to the marked epithelial remodeling observed in our earlier work on adult mice,12 may be attributed to both the exposure route and developmental stage. The current experimental design employed intratracheal instillation, whereas the prior study utilized chronic inhalation, which may result in more gradual and sustained deposition of PM2.5 particles and thus a greater likelihood of eliciting long-term epithelial responses. Additionally, the developing lungs of juvenile mice exhibit distinct physiological and regenerative capacities compared to mature lungs, potentially dampening the extent of epithelial injury and compensating for inflammatory stimuli through more robust or flexible repair mechanisms.28 This developmental resilience could explain why overt epithelial damage was not observed in the current study, despite the detectable alterations in immune cell populations, including an expansion of interstitial macrophages and increased heterogeneity among dendritic cells. Some studies have indicated that long-term exposure to PM2.5 can result in pulmonary fibrosis.15,17 Previous research identified adventitial fibroblasts with high Pi16 expression as a source of pro-fibrotic fibroblasts,29 while more recent studies have suggested that alveolar fibroblasts are the primary source of pro-fibrotic cells.30 In our study, adventitial fibroblasts were the most significantly altered fibroblast population. Identifying the precise source of pro-fibrotic fibroblasts is crucial for understanding whether short-term PM2.5 exposure in juvenile mice induces early changes that could contribute to lung fibrosis. Although we observed differential changes in CD4+ and γδ T cells, the current evidence is insufficient to conclude that short-term PM2.5 exposure in juvenile mice induces a strong, specific immune response. In this model, short-term PM2.5 exposure primarily triggers innate immune responses in the lungs with minimal effects on alveolar epithelial cells. Whether this exposure leads to long-term conditions such as lung fibrosis, asthma, or other diseases requires further investigation and the development of appropriate disease models.
Overall, we are the first to utilize a juvenile mouse PM2.5 airway instillation model to assess overall lung changes at the single-cell level. Our findings highlight myeloid immune cells, particularly AMs and DCs, as central to modulating the immune response to short-term PM2.5-induced lung injury. This approach provides a single-cell perspective on how PM2.5 influences the development of lung inflammation through immune pathways. Despite revealing significant effects of PM2.5 on the lung immune environment, our study has several limitations. First, the study was conducted exclusively in a mouse model, and the short observation period warrants further validation to determine whether these findings are translatable to humans. Second, the composition of PM2.5 is highly complex, and the specific effects of certain components (eg, metal particles or organic compounds) on immune cells remain unclear. Third, the pulmonary toxicities of PM2.5 are dose-dependent, and since only one dosage was used in this study, some interpretations may be biased. Future studies could employ single-cell transcriptomics to explore the impact of PM2.5 exposure on juvenile disease models, simulating the effects of PM2.5 on populations with specific disease susceptibilities. Epidemiological studies have shown that PM2.5 impacts children during both developmental and post-developmental stages. Our study provides insight into the possible mechanisms underlying PM2.5 effects on children in the post-developmental stage. Further research is needed to elucidate the mechanisms by which PM2.5 affects children during their developmental stage.
Acknowledgments
This work was sponsored by the Tianjin Health Research Project (Grant No. TJWJ2022QN077), the National Natural Science Foundation of China (Grant No. 81773394), and the Key Laboratory of Medical Rescue Key Technology and Equipment, Ministry of Emergency Management (Open Fund Project No. YJBKFKT202410). The schematic figures were created with BioRender.com.
Disclosure
Authors declared no conflict of interest.
References
1. Bu X, Xie Z, Liu J, et al. Global PM2.5-attributable health burden from 1990 to 2017: estimates from the global burden of disease study 2017. Environ Res. 2021;197:111123. doi:10.1016/j.envres.2021.111123
2. Ye Z, Li X, Lang H, Fang Y. Long-term PM2.5 exposure, lung function, and cognitive function among middle-aged and older adults in China. J Gerontol a Biol Sci Med Sci. 2023;78(12):2333–2341. doi:10.1093/gerona/glad180
3. McCarron A, Semple S, Braban CF, Gillespie C, Swanson V, Price HD. Personal exposure to fine particulate matter (PM2.5) and self-reported asthma-related health. Soc Sci Med. 2023;337:116293. doi:10.1016/j.socscimed.2023.116293
4. Lin S, Xue Y, Thandra S, et al. PM2.5 and its components and respiratory disease healthcare encounters - unanticipated increased exposure-response relationships in recent years after environmental policies. Environl Pollut. 2024;360:124585. doi:10.1016/j.envpol.2024.124585
5. Walker ES, Semmens EO, Belcourt A, et al. Efficacy of air filtration and education interventions on indoor fine particulate matter and child lower respiratory tract infections among rural US homes heated with wood stoves: results from the KidsAIR randomized trial. Environ Health Perspect. 2022;130(4):047002. doi:10.1289/EHP9932
6. George PE, Thakkar N, Yasobant S, Saxena D, Shah J. Impact of ambient air pollution and socio-environmental factors on the health of children younger than 5 years in India: a population-based analysis. Lancet Regional Health Southeast Asia. 2024;20:100328. doi:10.1016/j.lansea.2023.100328
7. Lei J, Chen R, Liu C, et al. Fine and coarse particulate air pollution and hospital admissions for a wide range of respiratory diseases: a nationwide case-crossover study. Int J Epidemiol. 2023;52(3):715–726. doi:10.1093/ije/dyad056
8. Lei J, Sun Q, Chen R, et al. Respiratory benefits of multisetting air purification in children: a cluster randomized crossover trial. JAMA Pediatrics. 2025;179(2):122–128. doi:10.1001/jamapediatrics.2024.5049
9. Zhang Y, Guo Z, Zhang W, et al. Effect of acute PM2.5 exposure on lung function in children: a systematic review and meta-analysis. J Asthma Allergy. 2023;16:529–540. doi:10.2147/JAA.S405929
10. Liu Q, Weng J, Li C, et al. Attenuation of PM2.5-induced alveolar epithelial cells and lung injury through regulation of mitochondrial fission and fusion. Particle Fibre Toxicol. 2023;20(1):28. doi:10.1186/s12989-023-00534-w
11. Zhang J, Cheng H, Wang D, et al. Revealing consensus gene pathways associated with respiratory functions and disrupted by PM2.5 nitrate exposure at bulk tissue and single cell resolution. Environl Pollut. 2021;280:116951. doi:10.1016/j.envpol.2021.116951
12. Li Y, Lin B, Hao D, et al. Short-term PM2.5 exposure induces transient lung injury and repair. J Hazard Mater. 2023;459:132227. doi:10.1016/j.jhazmat.2023.132227
13. Luo L, Jiang M, Xiong Y, et al. Fine particulate matter 2.5 induces susceptibility to Pseudomonas aeruginosa infection via expansion of PD-L1high neutrophils in mice. Respir Res. 2024;25(1):90. doi:10.1186/s12931-023-02640-x
14. Harding JN, Gross M, Patel V, Potter S, Cormier SA. Association between particulate matter containing EPFRs and neutrophilic asthma through AhR and Th17. Respir Res. 2021;22(1):275. doi:10.1186/s12931-021-01867-w
15. Zhang R, Chen S, Chen L, et al. Single-cell transcriptomics reveals immune dysregulation mediated by IL-17A in initiation of chronic lung injuries upon real-ambient particulate matter exposure. Particle Fibre Toxicol. 2022;19(1):42. doi:10.1186/s12989-022-00483-w
16. Zhou W, Yuan W, Chen Y, et al. Single-cell transcriptomics reveals the pulmonary inflammation induced by inhalation of subway fine particles. J Hazard Mater. 2024;463:132896. doi:10.1016/j.jhazmat.2023.132896
17. Zhao C, Pu W, Wazir J, et al. Long-term exposure to PM2.5 aggravates pulmonary fibrosis and acute lung injury by disrupting Nrf2-mediated antioxidant function. Environl Pollut. 2022;313:120017. doi:10.1016/j.envpol.2022.120017
18. Cheng P, Chen Y, Wang J, et al. PM2.5 induces a senescent state in mouse AT2 cells. Environl Pollut. 2024;347:123686. doi:10.1016/j.envpol.2024.123686
19. Liu T, De Los Santos FG, Phan SH. The Bleomycin model of pulmonary fibrosis. Methods Mol Biol. 2017;1627:27–42. doi:10.1007/978-1-4939-7113-8_2
20. Huang Y-P, Huang Y-T, Wu H-Y, et al. Armillaria mellea mycelia alleviate PM2.5-induced pulmonary inflammation in murine models. Antioxidants. 2024;13(11):1381. doi:10.3390/antiox13111381
21. Zhang J, Zhang W-H, Morisseau C, et al. Genetic deletion or pharmacological inhibition of soluble epoxide hydrolase attenuated particulate matter 2.5 exposure mediated lung injury. J Hazard Mater. 2023;458:131890. doi:10.1016/j.jhazmat.2023.131890
22. Ma Z, Du X, Sun Y, Jia Y, Liang X, Gao Y. Attenuation of PM2.5-induced lung injury by 4-phenylbutyric acid: maintenance of [Ca2+]i stability between endoplasmic reticulum and mitochondria. Biomolecules. 2024;14(9):1135. doi:10.3390/biom14091135
23. Xu H, Li X, Liu K, Huang P, Liu X-J. PM2.5 promotes macrophage-mediated inflammatory response through airway epithelial cell-derived exosomal miR-155-5p. J Inflamm Res. 2024;17:8555–8567. doi:10.2147/JIR.S482509
24. Choubey D, Duan X, Dickerson E, et al. Interferon-inducible p200-family proteins as novel sensors of cytoplasmic DNA: role in inflammation and autoimmunity. J Interferon Cytokine Res. 2010;30(6):371–380. doi:10.1089/jir.2009.0096
25. Iizuka Y, Cichocki F, Sieben A, et al. UNC-45A is a nonmuscle myosin IIA chaperone required for nk cell cytotoxicity via control of lytic granule secretion. J Iimmunol. 2015;195(10):4760–4770. doi:10.4049/jimmunol.1500979
26. Gautier T, Deckert V, Nguyen M, Desrumaux C, Masson D, Lagrost L. New therapeutic horizons for plasma phospholipid transfer protein (PLTP): targeting endotoxemia, infection and sepsis. Pharmacol Ther. 2022;236:108105. doi:10.1016/j.pharmthera.2021.108105
27. Zhao T, Qi W, Yang P, et al. Mechanisms of cardiovascular toxicity induced by PM2.5: a review. Environ Sci Pollut Res Int. 2021;28(46):65033–65051. doi:10.1007/s11356-021-16735-9
28. Niethamer TK, Planer JD, Morley MP, et al. Longitudinal single-cell profiles of lung regeneration after viral infection reveal persistent injury-associated cell states. Cell Stem Cell. 2025;32(2):302–321.e6. doi:10.1016/j.stem.2024.12.002
29. Buechler MB, Pradhan RN, Krishnamurty AT, et al. Cross-tissue organization of the fibroblast lineage. Nature. 2021;593(7860):575–579. doi:10.1038/s41586-021-03549-5
30. Tsukui T, Wolters PJ, Sheppard D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature. 2024;631(8021):627–634. doi:10.1038/s41586-024-07660-1
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Altered Immune Cell Profiles in the Follicular Fluid of Patients with Poor Ovarian Response According to the POSEIDON Criteria
Zhou L, Zhao S, Luo J, Rao M, Yang S, Wang H, Tang L
Journal of Inflammation Research 2024, 17:10663-10679
Published Date: 8 December 2024