")
Back to Journals » OncoTargets and Therapy » Volume 11
Platelets enhance the ability of bone-marrow mesenchymal stem cells to promote cancer metastasis
Authors Wang Q, Li Z, Sun L, Chen B, Zhao Y, Shen B, Zhu M, Zhao X , Xu C, Wang M , Xu W, Zhu W
Received 27 July 2018
Accepted for publication 15 October 2018
Published 21 November 2018 Volume 2018:11 Pages 8251—8263
DOI https://doi.org/10.2147/OTT.S181673
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Qianqian Wang,1 Zhuqian Li,2 Li Sun,1 Bin Chen,1 Yuanyuan Zhao,1 Bo Shen,3 Miaolin Zhu,3 Xiangdong Zhao,2 Changgen Xu,2 Mei Wang,1 Wenrong Xu,1 Wei Zhu1
1School of Medicine, Jiangsu University, Zhenjiang, Jiangsu, China; 2Department of Clinical Laboratory, Zhenjiang Provincial Blood Center, Zhenjiang, Jiangsu, China; 3Department of Oncology, Jiangsu Cancer Hospital Affiliated to Nanjing Medical University, Nanjing, Jiangsu, China
Background: Bone marrow-derived mesenchymal stem cells (BM-MSCs) have been identified to be closely associated with cancer progression. Our previous experimental results showed that BM-MSCs promote tumor growth and metastasis of gastric cancer through paracrine-soluble cytokines or exosomes. However, the elements that affect the role of BM-MSCs in promoting tumor metastasis are not clear. It is known that thrombocytosis in cancer patients is very common. Recently, platelets are recognized to play a critical role in tumor progression.
Purpose: This study aims to observe the effect of BM-MSCs which were co-cultured with platelets on tumor cell metastasis.
Methods: Platelet aggregation rate and the expression of P-selectin of platelets co-incubated with conditioned medium of SGC-7901 cells and BM-MSCs were detected by flow cytometry and platelet aggregometer. We also analyzed the change of BM-MSCs after co-incubation with platelets or platelets which were treated with SGC-7901 cells using transwell assay and Western blot analysis. The proliferation and migration ability and expression of VEGF, c-Myc, and sall-4 in SGC-7901 cells treated with medium of BM-MSCs which were co-cultured with platelets were detected. SGC-7901 cells were injected into Balb/c nude mice and the extent of lung metastasis was observed. Both in vitro and in vivo assays were used to analyze the effect of platelets on enhancing the ability of BM-MSCs to promote cancer metastasis.
Results: Results suggested that BM-MSCs and tumor cells can promote platelet aggregation rate and the expression of P-selectin. The protein levels of α-smooth muscle actin, vimentin, and fibroblast activation protein in BM-MSCs were higher after co-incubation with platelets, and SB431542 was used to confirm the effect of TGF-β on transdifferentiation of BM-MSCs into cancer-associated fibroblasts. Medium of BM-MSCs treated with platelets enhanced the proliferation and migration ability of SGC-7901 cells. More lung metastases were found in mice which were injected with SGC-7901 cells treated with conditioned medium from BM-MSCs co-incubated with platelets.
Conclusion: Tumor cells and BM-MSCs activate platelets which can change the characteristics of BM-MSCs through secretion of TGF-β. Moreover, we found that platelets enhanced the effect of BM-MSCs on tumor metastasis, which suggested a potential target and approach for gastric cancer therapy.
Keywords: mesenchymal stem cells, platelets, gastric cancer, metastasis, bone marrow
Introduction
It is reported that tumor involves complex multicellular interactions between cancer cells and stromal cells, especially mesenchymal stem cells (MSCs).1,2 MSCs were originally isolated from bone marrow and have been defined as a plastic-adherent fibroblastic cell population, expressing a defined immunophenotype such as CD105, CD73, and CD90.3 Multiple evidence indicates that bone marrow-derived MSCs (BM-MSCs) promote tumor growth and metastasis.4–6 Our previous findings confirmed that MSCs can enhance cancer proliferation, migration, and stemness and impair the anti-tumor immune response of peripheral blood mononuclear cells through disruption of Treg/Th17 balance.7,8 The potential of MSCs to inhibit anti-tumor immunity is widely recognized.9 Besides the immunosuppressive effect, BM-MSCs can promote colorectal cancer progression through paracrine neuregulin 1/HER3 signaling.10 We also demonstrated that BM-MSCs play an important role in promoting tumor growth in gastric cancer, which may be through paracrine-soluble cytokines or exosomes.11–13 BM-MSCs can be attracted to inflammatory sites and then they can differentiate into several cell types.14 Due to BM-MSCs tropism to cancer cells, BM-MSCs were found to have the ability to transdifferentiate into cancer-associated fibroblasts (CAFs) and perivascular-like cells.15–17 Our previous results have shown that BM-MSCs can be transdifferentiated into Gastric cancer-MSCs (GC-MSCs).18 In addition, TGF-β1 was identified as the key molecule regulating BM-MSCs tropism to prostate carcinoma cells, and TGF-β1 secreted by prostate carcinoma cells and tumor microenvironment cells sustained the transdifferentiation of MSCs into pro-tumor supporting CAFs.19 However, the exact mechanism of transformation is not clear now. We made a hypothesis that platelets may have an important role in the process.
Recently, the relationship between platelets and cancer has received much attention. It has been widely recognized that as a component of the tumor microenvironment, platelets can influence tumor growth, tumor metastasis, tumor angiogenesis, inflammation in tumor, and chemotherapy efficiency.20 Blood platelets can interact with different types of cells, including inflammatory cells, tumor cells, and MSCs.21 Platelets can be activated by tumor cells and the expression of P-selectin, glycoprotein IIb/IIIa, glycoprotein VI and glycoprotein Ib can also be upregulated.22 It is demonstrated that P-selectin-mediated adhesion between cancer cells and platelets can promote tumor progression.23 More and more studies focused on the role played by platelets in cancer metastasis. Studies show that platelets can enhance cancer cell survival and metastasis in vitro and in vivo by interaction between TLR4 and tumor cell-released high-mobility group box1 protein, shielding cancer cells from immune defense, inducing anoikis resistance, and so on.24–26 In addition, the interaction between platelets and cancer cells can contribute to metastasis via preventing natural killer cell recognition and the epithelial–mesenchymal transition (EMT) of tumor cells through the TGF-β and nuclear factor-kB pathway.24
In present study, we demonstrated that tumor cells and BM-MSCs induced platelet activation. Then, we investigated the influences of platelets on BM-MSCs. Our results showed that platelets enhanced the effect of BM-MSCs on tumor proliferation and metastasis. It will be of great significance in the diagnosis and treatment of tumor.
Materials and methods
Isolation of human platelets and BM-MSCs
As described previously, human BM-MSCs were isolated from bone marrow aspirates.27 Primary BM-MSCs were obtained from normal healthy subjects from Affiliated Hospital of Jiangsu University (Jiangsu, China). Human platelets were obtained from healthy volunteers from Jiangsu University. All the procedures were approved by the Ethics Committee of Affiliated Hospital of Jiangsu University and the written informed consents were obtained from all subjects. Whole blood was drawn from healthy subjects via median cubital vein and collected in EDTA-K2 anticoagulant tubes. Blood was centrifuged at 800× g for 5 minutes at room temperature to obtain platelet-rich plasma (PRP), and this fraction was further centrifuged at 2,000× g for 5 minutes. Then we obtained platelet-poor plasma (PPP) and platelet pellet. Platelet pellet was resuspended in 1 mL PBS and was centrifuged at 2,000× g for 5 minutes twice. Washed platelets were resuspended in 1 mL PBS.
Cell culture and preparation of conditioned medium (CM)
SGC-7901 gastric cancer cell lines were obtained from Shanghai Cell Bank (Shanghai, China) and cultured in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10% FBS within a humidified incubator containing 5% CO2 at 37°C. BM-MSCs were cultured in low-glucose DMEM medium (L-DMEM) (Thermo Fisher Scientific) supplemented with 10% FBS within a humidified incubator containing 5% CO2 at 37°C. When the confluence of passage 3 BM-MSCs reached 80%, we substituted L-DMEM supplemented with 2% FBS for L-DMEM supplemented with 10% FBS. Then, the conditioned medium of BM-MSCs (BM-MSCs-CM) was collected after 48 hours. Similarly, when the confluence of SGC-7901 reached 70%, we substituted RPMI-1640 supplemented with 2% FBS for RPMI-1640 supplemented with 10% FBS. Then, the conditioned medium of SGC-7901 (SGC-7901-CM) was collected after 48 hours.
Flow cytometry
We detected the expression of platelet P-selectin by flow cytometry. Platelets (1×105) were co-incubated with 2 mL SGC-7901-CM or BM-MSCs-CM in six-well plate at 37°C for 2 hours, and then platelets were centrifuged at 2,000× g for 5 minutes. Platelet pellet was resuspended in 1 mL PBS and co-incubated with monoclonal antibodies against P-selectin (fluorescein isothiocyanate [FITC]-conjugated) (BD Biosciences, San Jose, CA, USA) for 30 minutes at 4°C in the dark. After washing twice, platelets were analyzed with a FACSCanto (BD Biosciences). Results were analyzed using CFlow PLUS. To identify the isolated BM-MSCs, 1×105 cells were incubated with the monoclonal antibodies against CD19, CD34, and CD45 (FITC-conjugated); and CD29, CD105, and CD90 (phycoerythrin-conjugated) (BD Biosciences). Steps are the same as P-selectin.
Determination of platelet aggregation
PRP and PPP were prepared as described earlier.28 PRP was co-incubated with PBS, BM-MSCs-CM, and SGC-7901-CM, respectively, for 30, 60, 120, and 180 minutes at 37°C. Then PRP was centrifuged at 2,000× g for 5 minutes at room temperature and resuspended in 0.2 mL PPP. About 0.2 mL of PPP was placed in a cuvette, and the platelet aggregation rate of PPP was set as zero. The level of platelet aggregation was determined using SC-2000 Platelet Aggregometer (Succeeder, Beijing, China). Results were recorded as percent of maximal aggregation.
Western blot analysis
Proteins were extracted by using total protein extraction kit (Vazyme, Nanjing, China) and qualified by NanoDrop-2000 spectrophotometer (Thermo Fisher Scientific). Total protein lysates were separated in 12% SDS-PAGE and transferred to polyvinylidene fluoride membranes. Then the PVDF membranes were blocked with 5% nonfat milk. After 60 minutes, membranes were washed three times in Tris-Buffered Saline and Tween 20 (TBS-T) and incubated with primary antibodies overnight at 4°C. Subsequently, the membranes were incubated with secondary antibody for 1 hour at room temperature on the following day. All primary antibodies used were purchased from Cell Signaling Technology (Danvers, MA, USA). The gray densitometric was detected with Image Quant LAS 4000. GAPDH served as the loading control.
RNA isolation and qRT-PCR
Total mRNA of cells in each group was extracted with TRIzol reagent (Thermo Fisher Scientific) and the yield and purity of mRNA were determined by NanoDrop-2000 spectrophotometer (Thermo Fisher Scientific). The cDNA synthesis was carried out using reverse transcription kit (Vazyme), and the synthesized cDNA was stored at −20°C. TGF-β mRNA expression was calculated by duplicate amplification and a comparative cycle threshold method (ΔCt) as previously described.28 Primers of human TGF-β and β-actin are shown in Table 1.
 | Table 1 Primer sequences for the amplification of target genes |
Transwell migration assay and cell count
Cells were loaded into the upper compartment (5×104, final volume, 200 μL) in serum-deprived L-DMEM or RPMI-1640 medium. The transwell chambers (diameter: 8 μm; Corning Inc., Corning, NY, USA) were placed into 24-well plates with 600 μL conditioned medium and incubated at 37°C for 12 hours. After cell migration, upper side of the filter was swabbed to remove the nonmigratory cells. The filters were then fixed in 4% paraformaldehyde and stained with 0.5% crystal violet for 15 minutes. Results were analyzed by microscope. All the experiments in each group were performed in triplicate. About 1×105 BM-MSCs or SGC-7901 cells were seeded into six-well cell culture plates with different treatment in a humidified incubator containing 5% CO2 at 37°C for 24, 48, and 72 hours. The cell proliferation was detected by Neubauer-improved counting chamber.
Tube formation assay
Tube formation assay was performed to analyze the regulating effect of platelets and BM-MSCs on tumor angiogenesis. The procedure was carried out in accordance with a previously published method.29 First, Matrigel (50 μL/well; BD Pharmingen, San Diego, CA, USA) was added to each well of a 96-well plate and then incubated for 30 minutes at 37°C for matrix formation. Human umbilical vein endothelial cells (Shanghai Institutes for Biological Sciences, Shanghai, China) were added onto the gel at a density of 2×104 cells/well in L-DMEM with 10% FBS or in different conditioned medium. After incubation for 6 hours at 37°C, the cells were visualized and photographed by an inverted microscope to evaluate the tube-like structure formation. Results were quantified by measuring node numbers in the field.
In vivo experiments
Eighteen 4–5-weeks old male Balb/c nude mice (Model Animal Research Center of Nanjing University, Nanjing, China) were divided into three groups randomly and each group had six mice. All animal experiments were conducted under the approval of Animal Experiments Ethics Committee, Jiangsu University, China, and carried out according to the guidelines of the National Institutes of Health Guides for the Care and Use of Laboratory Animals. BM-MSCs were co-cultured with platelets or T-platelets for 48 hours, and then the medium of BM-MSCs was collected. SGC-7901 cells were co-cultured with the condition medium from BM-MSCs for 48 hours. Then SGC-7901 cells (2×106) with different treatment were injected into the tail vein of mice. SGC-7901 cells co-cultured with medium of individual BM-MSCs were used as the control group. We observe these mice every day and the extent of lung metastasis was determined 3 months later. Tumors from lung tissue of three groups were immunostained for VEGF, Ki-67, c-Myc, and Sall-4. The procedure was carried out according to a previously described method.30
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM) of three independent experiments. Results were analyzed using GraphPad Prism software 5.0 (GraphPad Software, La Jolla, CA, USA). Student’s t-test was used to evaluate statistical significance when two data sets were compared. When more groups were analyzed, we used ANOVA. P<0.05 was considered statistically significant.
Results
Tumor cells and BM-MSCs induce platelet activation
Based on the criteria of the International Society for Cellular Therapy,31 we detected characteristic surface markers of BM-MSCs. Flow cytometry analysis conveyed that BM-MSCs were positive for CD90, CD29, and CD105, but negative for hematopoietic markers (CD19, CD45, and CD34), which displayed the characteristic surface markers of BM-MSCs (Figure S1). To investigate the interaction between tumor cells, BM-MSCs, and platelets, we first examined the effects of tumor cells and BM-MSCs on platelets. The platelet aggregation rate and P-selectin were widely considered as indicators of platelet activation. We detected the platelet aggregation after treatment of platelets with SGC-7901-CM or BM-MSCs-CM for 30, 60, 120, and 180 minutes. As is shown in Figure 1A, at 120 minutes, compared with the PBS control group (8.00±2.31), SGC-7901-CM and BM-MSCs-CM increased the platelet aggregation obviously (P<0.01). Since the highest platelet aggregation was observed at 120 minutes, we decided to use 120 minutes as the major end point for platelet co-incubation with SGC-7901 in vitro. In keeping with this observation, we detected the expression of platelet P-selectin using flow cytometry. The expression of P-selectin in SGC-7901-CM group was 31.4%±1.71% and the BM-MSCs-CM group was 21.37%±1.00%. Both of them were higher than control group which was 3.17%±0.40% (Figure 1B). The results showed that after treatment with SGC-7901-CM or BM-MSCs-CM for 120 minutes, there was a significant increase in the expression of platelet P-selectin (P<0.01). In Figure S2, platelets were co-cultured with SGC-7901 cells and BM-MSCs for 2 hours at 37°C and the platelets aggregation was photographed by microscope. Compared with image at 0 hours, the image at 2 hours showed that platelets were activated and have a tendency to move toward cancer cells and BM-MSCs.
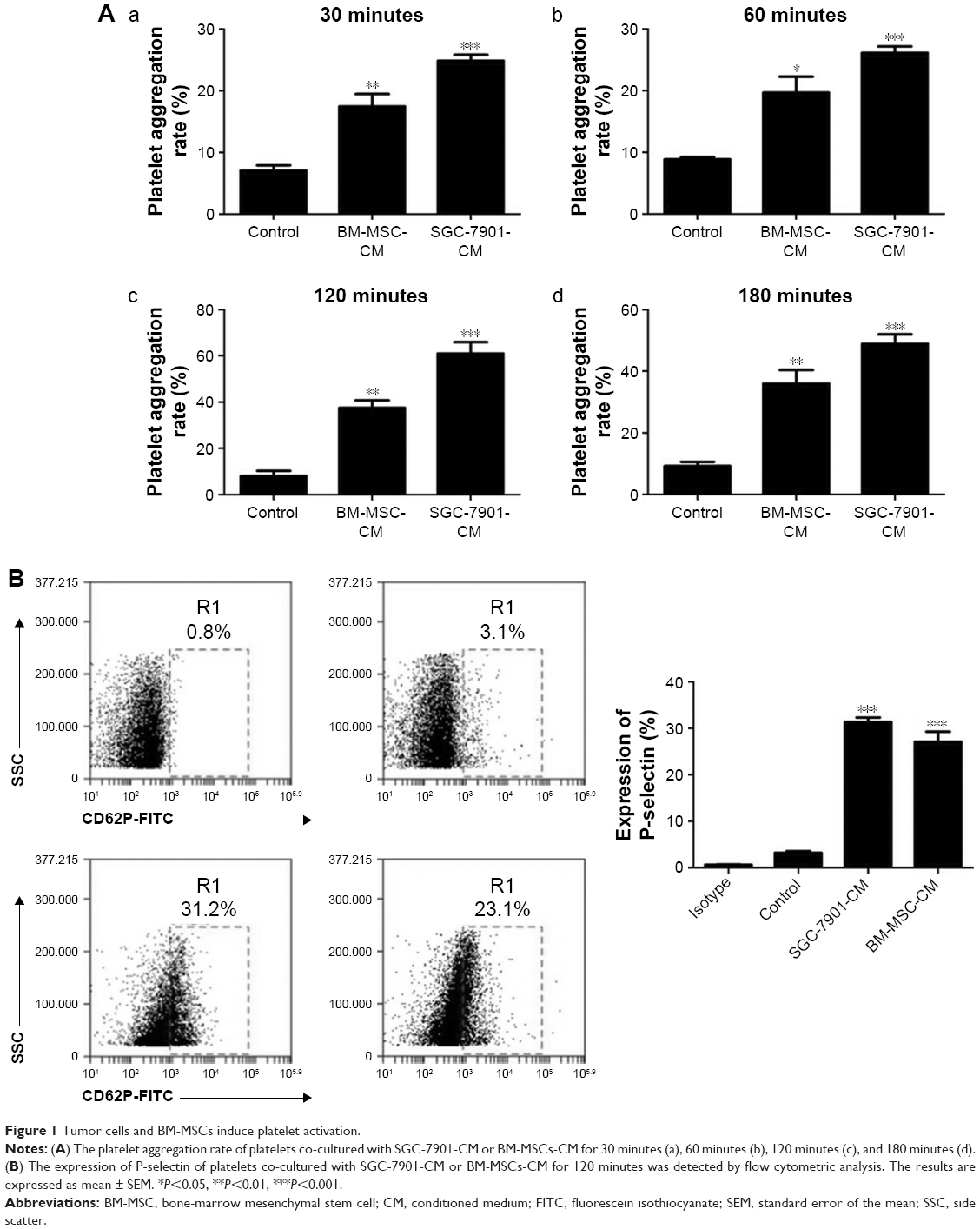 | Figure 1 Tumor cells and BM-MSCs induce platelet activation. |
Platelets induce BM-MSCs transdifferentiation into CAF-like cells by increasing TGF-β
Tumor-educated blood platelet (TEP) is found in cancer patients and is demonstrated to contribute to tumor progression. In our study, we collected the medium of SGC-7901 cells which were cultured in RPMI-1640 supplemented with 2% FBS (SGC-7901-CM). Then 2×108 platelets were cultured with 2 mL SGC-7901-CM in order to mimic platelets in cancer patients (TEP). After 2 hours, platelets were centrifuged at 2,000× g for 5 minutes and resuspended with 1 mL PBS. We called the platelets as T-platelets in this study. The BM-MSCs were randomly divided into the MSCs group (1×105), platelets + MSCs group (2×108+1×105), and T-platelets+ MSCs group (2×108+1×105). The expression of α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), and vimentin was analyzed by Western blotting. We found that co-incubation of BM-MSCs with platelets, especially with T-platelets, induced a significant increase of markers associated with CAF-like phenotype (Figure 2A). BM-MSCs and CAFs have similar cell morphology. They were both plastic-adherent and exhibited spindle-shaped morphology. BM-MSCs were evaluated by microscopy for homogenous, elongated, spindle-shaped morphology. CAFs have uneven sizes and more branched synapses and were larger in size than BM-MSCs. We observed the morphology of BM-MSCs before and after T-platelet treatment by microscope (Figure S3). As shown in Figure S3, we found that BM-MSCs became larger in size with the appearance of some branched synapses. However, the change in BM-MSCs morphology was not very obvious because of the similar cell morphology of BM-MSCs and CAFs.
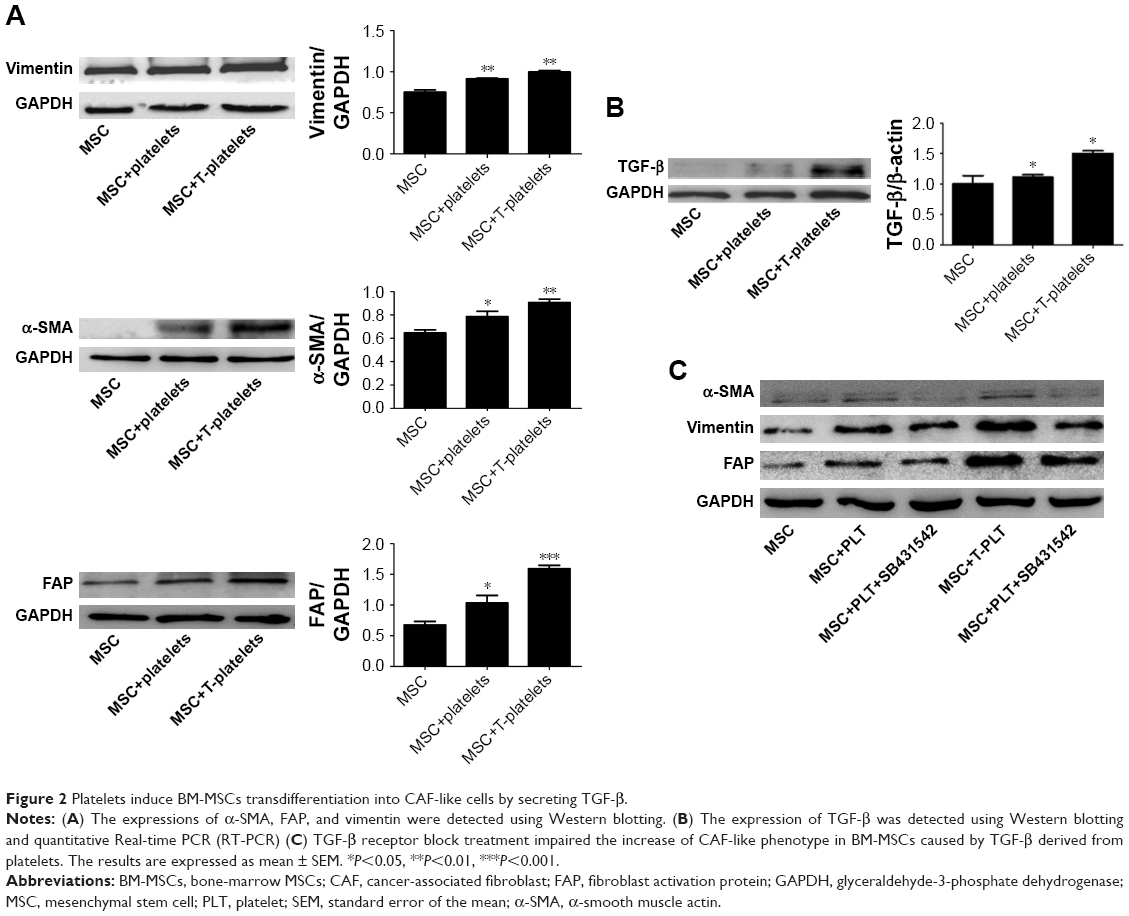 | Figure 2 Platelets induce BM-MSCs transdifferentiation into CAF-like cells by secreting TGF-β. |
To investigate whether TGF-β plays a critical role in BM-MSCs transdifferentiation into CAF-like cells, the expression of TGF-β in BM-MSCs was detected using Western blotting and quantitative Real-time PCR (RT-PCR). We found that the expression of TGF-β in both platelet+MSCs group and T-platelet+MSCs group increased obviously compared with that in control group. Western blotting for TGF-β protein showed the same trends as observed for mRNA expression (Figure 2B). We supposed that the increased secretion of TGF-β in platelets and BM-MSCs may be one of the possible reasons for the activation of BM-MSCs transdifferentiation into CAF-like cells. In tumor microenvironment, platelets are affected by tumor cells which result in higher TGF-β concentrations in platelets. It is also possible that TEPs stimulate BM-MSCs to secret more TGF-β. That is why we found that MSC+T-platelets group expressed the highest levels of α-SMA, vimentin, and FAP. To confirm our investigation that TGF-β has an important role in BM-MSCs transdifferentiation into CAF-like cells induced by platelets, we added MSC + PLT+SB431542 group and MSC+T-PLT+SB431542 group (Figure 2C). SB431542 (MedChem Express, Monmouth Junction, NJ, USA) is a potent and selective inhibitor of ALK5 and also an inhibitor of TGF-β receptor. BM-MSCs were cultured in L-DMEM supplemented with 10% FBS. When the confluence of BM-MSCs reached 80%, platelets or T-platelets were added in BM-MSCs and 10% FBS was replaced by 2% FBS. Eight hours before adding platelets, SB431542 was added to MSC+PLT+SB431542 group and MSC+T-PLT+SB431542 group in order to inhibit TGF-β receptor in BM-MSCs (the final concentration of SB431542 was 10 μM). Total proteins were extracted from BM-MSCs 48 hours after adding platelets and the expression of CAF-like phenotype (α-SMA, vimentin, and FAP) was analyzed by Western blotting. Figure 2C conveys that SB431542 reduced the expression of α-SMA, vimentin, and FAP in BM-MSCs. In other words, TGF-β receptor block treatment impaired the increase of CAF-like phenotype in BM-MSCs caused by TGF-β.
Platelets promote BM-MSCs proliferation and migration
The migratory ability of BM-MSCs co-incubated for 12 hours with platelets or with T-platelets was analyzed by transwell assay. The result indicated that platelets strongly increased the migratory ability of BM-MSCs (Figure 3A) and the BM-MSCs displayed more significant tropism to T-platelets than platelets. However, the specific mechanism needs further study. Besides the migratory ability of BM-MSCs, we also explored the effects of platelets on BM-MSCs proliferation. As expected, compared with MSCs group, the proliferation of platelet+MSCs group and T-platelet+MSCs group was enhanced (Figure 3B).
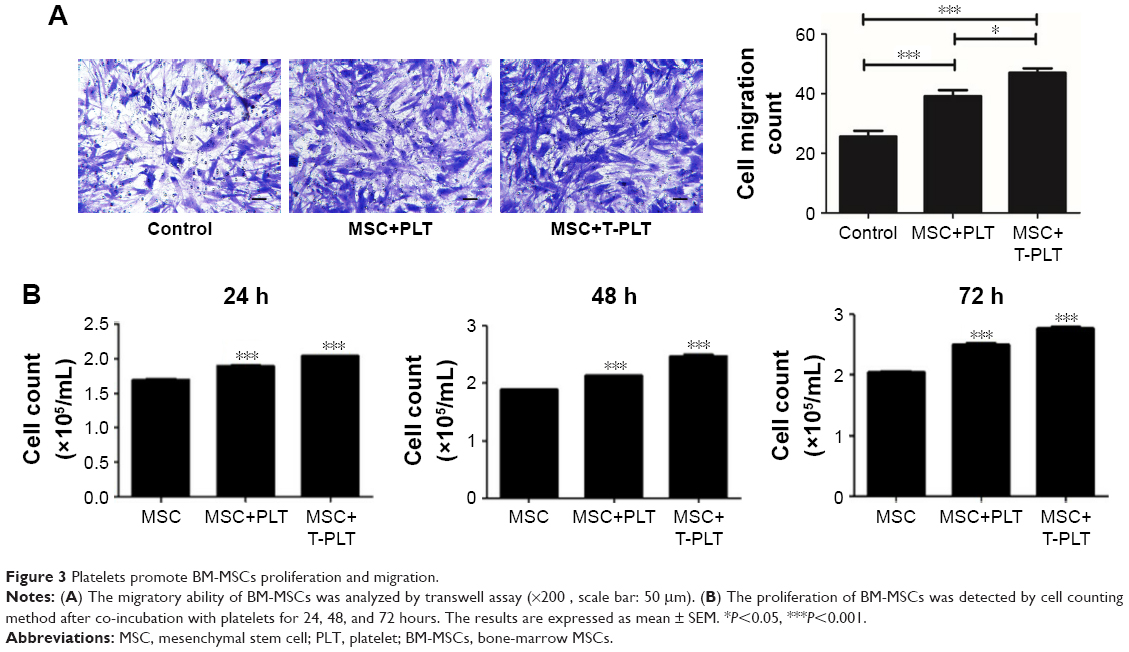 | Figure 3 Platelets promote BM-MSCs proliferation and migration. |
Platelets enhanced the effect of BM-MSCs on proliferation and metastasis of tumor cells
To investigate the role of platelets in the interaction between BM-MSCs and gastric cancer cells, we firstly detected the proliferation of SGC-7901 cells co-cultured with platelets and conditioned medium from BM-MSCs treated with platelets or T-platelets. The results suggested that compared with MSCs-CM group and platelets group, the proliferation of cells in (platelet+MSCs)-CM group and (T-platelet)+MSCs-CM group was enhanced (Figure 4A). Next, the angiogenesis ability of BM-MSCs was detected by tubule formation assay. The result of tubule formation assay in vitro showed that compared with the MSCs-CM group, the angiogenic ability was enhanced in the (platelet+MSCs)-CM group and (T-platelet)+MSCs-CM group (Figure 4B). Then we made a hypothesis that platelets enhanced the effects of BM-MSCs on tumor metastasis. We analyzed the migration ability of SGC-7901 cells co-cultured with or without (BM-MSCs+PLT)-CM in vitro by transwell assay (Figure 4C). Results indicated that the migration ability of SGC-7901 cells was upregulated by (BM-MSCs+PLT)-CM. Also, we detected the expression of VEGF, c-Myc, and sall-4 in SGC-7901 cells in vitro by Western blotting (Figure 4D). We also performed an experiment in vivo and the results showed that the (BM-MSCs+platelets)-CM+ SGC-7901 group and (BM-MSCs+T-platelets)-CM+SGC-7901 group had much more lung metastatic foci compared with the control group (Figure 5A). The round translucent nodules on lung are considered as the lung metastatic foci and they were calculated with naked eyes. Compared with the (BM-MSCs+platelets)-CM-SGC-7901 group, the number of lung metastatic foci in (BM-MSCs+T-platelets)-CM+SGC-7901 group increased greatly. The results of experiment in vivo confirmed that platelets enhanced the ability of BM-MSCs to promote gastric cancer metastasis. Tumors from lung tissue of three groups were immunostained for VEGF, Ki-67, c-Myc, and Sall-4. Then we analyzed the expression of VEGF, Ki-67, c-Myc, and Sall-4 by Pannoramic MIDI pathology section scanner (Figure 5B). Immunohistochemistry revealed that the anatomic organization of the lung was different in the presence of platelets and BM-MSCs. Those cells stained brownish yellow in nucleus or cytoplasm were regarded as the positive cells. The number of positive cells and depth of cell coloration determined the expression of VEGF, c-Myc, Ki67, and sall-4. Our results showed that the supernatants of the BM-MSCs when stimulated by platelets enhanced the expression of VEGF, Ki-67, c-Myc, and Sall-4.
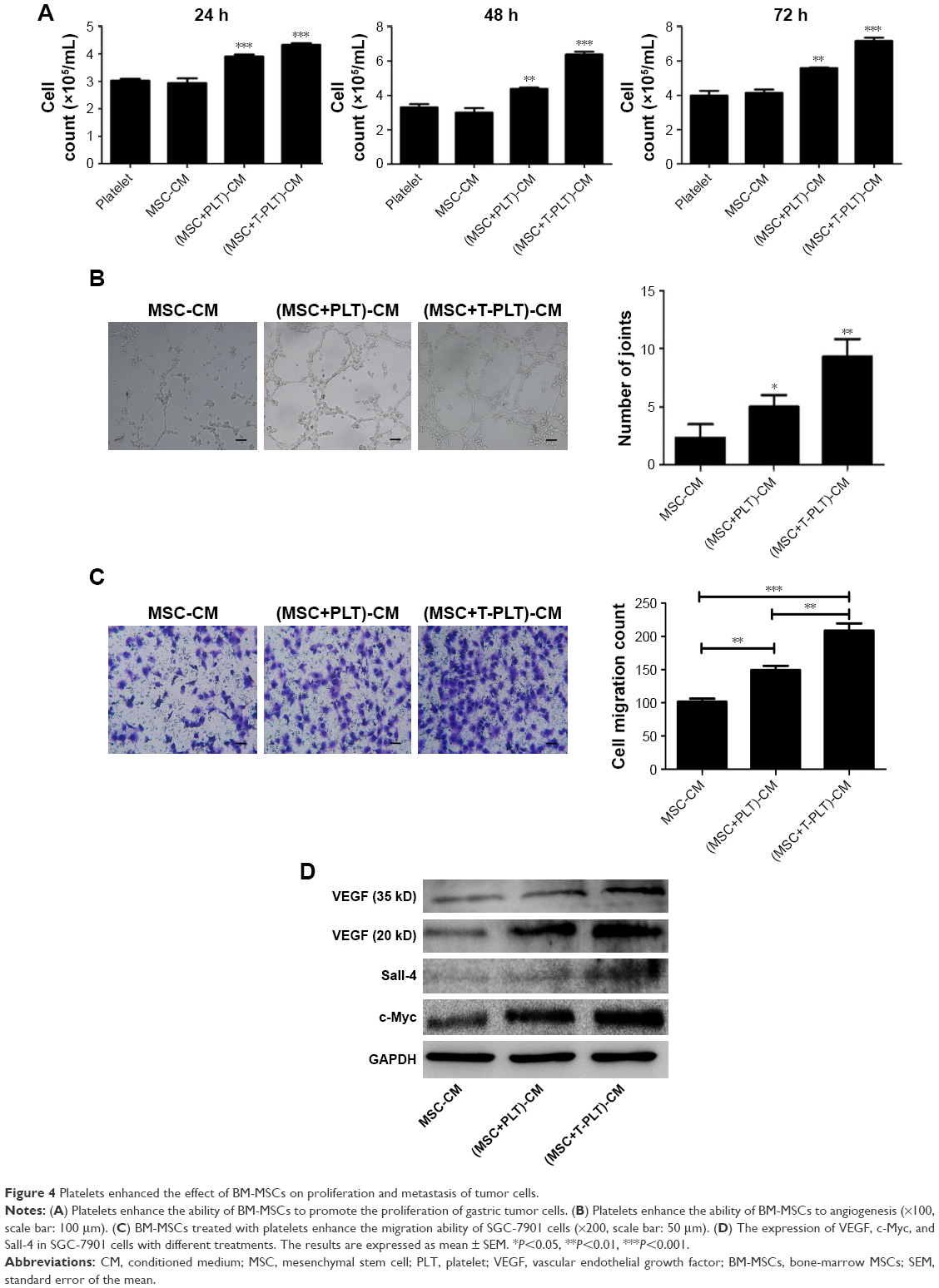 | Figure 4 Platelets enhanced the effect of BM-MSCs on proliferation and metastasis of tumor cells. |
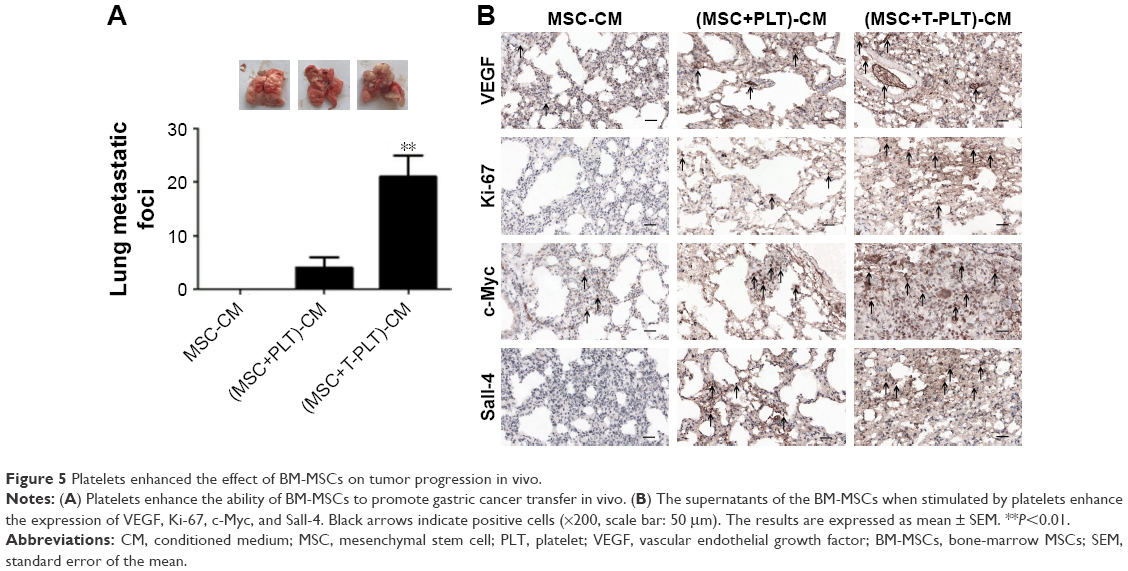 | Figure 5 Platelets enhanced the effect of BM-MSCs on tumor progression in vivo. |
Discussion
Platelets are recognized to halt hemorrhage after vascular injury and tissue trauma by releasing various bioactive factors.32 Other than coagulation function, contributions of platelets to tumor progression have received more and more attention in recent years. TEPs are involved in the progression and spread of tumors, and can be used for cancer detection and progression monitoring.33 Platelets are reported to contribute to cancer cell proliferation and metastasis by direct interactions and secretion of bioactive proteins.32 In this study, we found the tendency of platelets to move toward cancer cells and BM-MSCs and confirmed that platelets can be activated by BM-MSCs and cancer cells. Activated platelets secrete higher level of growth factors, proangiogenic regulatory proteins, chemokines, and proteolytic enzymes such as TGF-β, platelet-derived growth factor, platlet factor 4, and thrombospondin-1.33 It has been confirmed that the formation of platelet–tumor cell aggregates by integrin αIIbβ3 (glycoprotein IIb/IIIa) bridging in tumor microenvironment could promote immune evasion and cancer metastasis.34 In our research, we explored the role played by platelets in the process of homing MSCs to tumor sites. Our results revealed increased expression of CAF markers (α-SMA, vimentin, and FAP) and TGF-β in BM-MSCs. So we made a hypothesis that BM-MSCs were promoted to transdifferentiate to CAFs by TGF-β in tumor microenvironment, and this hypothesis was confirmed using SB431542. Weber et al demonstrated that TGF-β autocrine/paracrine signaling loop acted to initiate and maintain this CAF phenotype.35 In addition, it has been found that TGF-β from platelets, which was essential for EMT of tumor cells, and podoplanin promoted hematogenous metastasis in part by releasing TGF-β,36 which is consistent with our research. We subsequently focused on the potential effects of plateles on BM-MSCs in tumor progression. Previous research has revealed a tropism of BM-MSCs toward tumor cells.37 Our data showed that platelets enhanced BM-MSCs migration ability. In other words, the ability of BM-MSCs to migrate toward tumor cells was enhanced. In tumor microenvironment, tumors recruit MSCs, and MSCs convert into cancer-associated fibroblasts inducing an EMT, which ultimately promotes metastasis to secondary tumor sites.38 We next proceeded with analysis of proliferation and migration ability of cancer cells and expression of cancer-associated cytokines such as VEGF in cancer cells. As expected, platelets enhanced the ability of BM-MSCs to promote cancer progression. Angiogenesis is a prerequisite for primary tumor growth, cancer cell intravasation, extravasation, and growth of cancer foci at distant sites.39 In the present study, we found that BM-MSCs treated with platelets can enhance angiogenesis in vitro and in vivo. The expression of Ki67 is strongly associated with tumor cell proliferation and growth.40 The increased expression of Ki67 demonstrated that platelets enhanced the ability of BM-MSCs to promote cancer cell proliferation. In addition, the oncoprotein c-Myc is overexpressed in a large number of human cancers, including colon, gastric, breast, lung, and prostate cancers. c-Myc is frequently found to be constitutively activated and overexpressed in tumor cells at the metastatic niche compared with primary tumors through pathways triggered by microenvironmental signals.41 Sall-4 is a zinc-finger transcriptional factor for embryonic stem cell self-renewal and pluripotency. Previous research has demonstrated that sall-4 overexpression contributed to growth and metastasis of gastric cancer in vivo through the modulation of EMT.42 In our study, the fact that platelets enhance the ability of BM-MSCs to promote cancer cells metastasis is very clear but the underlying mechanism is still unknown. We detected the expression of sall-4 and c-Myc in order to explore whether sall-4 and c-Myc play a role during this process. According to our findings, sall-4 and c-Myc are worthy of concern for potential function in the interactions between platelets and MSCs in tumor microenvironment. However, the underlying mechanism by which platelets affect MSCs needs more exploration. Finally, it is worth noting that our research possibly devises novel treatment strategies for gastric cancer.
Conclusion
Our study confirmed that conditioned medium of tumor cells and BM-MSCs can active platelets, and that platelets promote BM-MSCs proliferation and migration. We found that platelets induce BM-MSCs transdifferentiation into CAF-like cells through TGF-β. Moreover, we demonstrated that platelets enhanced the effect of BM-MSCs on proliferation and metastasis of tumor cells.
Acknowledgments
This study was supported by the National Science Foundation of China (grant no.: 81472334), Jiangsu Province’s Project of Key Research and Development Plan (Social Development) (grant no.: BE2017694), and Social Development Plan of Zhenjiang City (grant no.: FZ2017065, SH2018039).
Disclosure
The authors report no conflicts of interest in this work.
References
de Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200(4):429–447. | ||
de Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123(10):2229–2238. | ||
Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. | ||
Shinagawa K, Kitadai Y, Tanaka M, et al. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer. 2010;127(10):2323–2333. | ||
Tsai KS, Yang SH, Lei YP, et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology. 2011;141(3):1046–1056. | ||
de Boeck A, Narine K, de Neve W, Mareel M, Bracke M, de Wever O. Resident and bone marrow-derived mesenchymal stem cells in head and neck squamous cell carcinoma. Oral Oncol. 2010;46(5):336–342. | ||
Xue J, Zhu Y, Sun Z, et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer. 2015;15:793. | ||
Wang M, Chen B, Sun XX, et al. Gastric cancer tissue-derived mesenchymal stem cells impact peripheral blood mononuclear cells via disruption of Treg/Th17 balance to promote gastric cancer progression. Exp Cell Res. 2017;361(1):19–29. | ||
Modiano JF, Lindborg BA, Mcelmurry RT, et al. Mesenchymal stromal cells inhibit murine syngeneic anti-tumor immune responses by attenuating inflammation and reorganizing the tumor microenvironment. Cancer Immunol Immunother. 2015;64(11):1449–1460. | ||
de Boeck A, Pauwels P, Hensen K, et al. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut. 2013;62(4):550–560. | ||
Zhu W, Xu W, Jiang R, et al. Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp Mol Pathol. 2006;80(3):267–274. | ||
Zhu W, Huang L, Li Y, et al. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012;315(1):28–37. | ||
Zhu W, Huang L, Li Y, et al. Mesenchymal stem cell-secreted soluble signaling molecules potentiate tumor growth. Cell Cycle. 2011;10(18):3198–3207. | ||
Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. | ||
Mishra PJ, Mishra PJ, Humeniuk R, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68(11):4331–4339. | ||
Au P, Tam J, Fukumura D, Jain RK. Bone marrow-derived mesenchymal stem cells facilitate engineering of long-lasting functional vasculature. Blood. 2008;111(9):4551–4558. | ||
Bexell D, Gunnarsson S, Tormin A, et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol Ther. 2009;17(1):183–190. | ||
Zhu M, Wang M, Yang F, et al. miR-155-5p inhibition promotes the transition of bone marrow mesenchymal stem cells to gastric cancer tissue derived MSC-like cells via NF-κB p65 activation. Oncotarget. 2016;7(13):16567–16580. | ||
Barcellos-de-Souza P, Comito G, Pons-Segura C, et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-Derived TGF-β1. Stem Cells. 2016;34(10):2536–2547. | ||
Zhang Q, Liu H, Zhu Q, et al. Patterns and functional implications of platelets upon tumor “education”. Int J Biochem Cell Biol. 2017;90:68–80. | ||
Contursi A, Sacco A, Grande R, Dovizio M, Patrignani P. Platelets as crucial partners for tumor metastasis: from mechanistic aspects to pharmacological targeting. Cell Mol Life Sci. 2017;74(19):3491–3507. | ||
Fuentes E, Palomo I, Rojas A. Cross-talk between platelet and tumor microenvironment: Role of multiligand/RAGE axis in platelet activation. Blood Rev. 2016;30(3):213–221. | ||
Qi C, Li B, Guo S, et al. P-selectin-mediated adhesion between platelets and tumor cells promotes intestinal tumorigenesis in Apc(Min/+) mice. Int J Biol Sci. 2015;11(6):679–687. | ||
Yu LX, Yan L, Yang W, et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat Commun. 2014;5:5256. | ||
Bambace NM, Holmes CE. The platelet contribution to cancer progression. J Thromb Haemost. 2011;9(2):237–249. | ||
Haemmerle M, Taylor ML, Gutschner T, et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. 2017;8(1):310. | ||
Koninckx R, Hensen K, Daniëls A, et al. Human bone marrow stem cells co-cultured with neonatal rat cardiomyocytes display limited cardiomyogenic plasticity. Cytotherapy. 2009;11(6):778–792. | ||
Zhang S, Li P, Yuan Z, Tan J. Effects of platelet-rich plasma on the activity of human menstrual blood-derived stromal cells in vitro. Stem Cell Res Ther. 2018;9(1):48. | ||
Zhang T, Lee YW, Rui YF, Cheng TY, Jiang XH, Li G. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res Ther. 2013;4(3):70. | ||
von der Heide EK, Neumann M, Vosberg S, et al. Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia. 2017;31(5):1069–1078. | ||
Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. | ||
Tesfamariam B. Involvement of platelets in tumor cell metastasis. Pharmacol Ther. 2016;157:112–119. | ||
Best MG, Sol N, Kooi I, et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 2015;28(5):666–676. | ||
Borsig L. The role of platelet activation in tumor metastasis. Expert Rev Anticancer Ther. 2008;8(8):1247–1255. | ||
Weber CE, Kothari AN, Wai PY, et al. Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene. 2015;34(37):4821–4833. | ||
Takemoto A, Okitaka M, Takagi S, et al. A critical role of platelet TGF-β release in podoplanin-mediated tumour invasion and metastasis. Sci Rep. 2017;7:42186. | ||
Pietrovito L, Leo A, Gori V, et al. Bone marrow-derived mesenchymal stem cells promote invasiveness and transendothelial migration of osteosarcoma cells via a mesenchymal to amoeboid transition. Mol Oncol. 2018;12(5):659–676. | ||
Jung Y, Kim JK, Shiozawa Y, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:4. | ||
Wojtukiewicz MZ, Sierko E, Hempel D, Tucker SC, Honn KV. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017;36(2):249–262. | ||
Lt L, Jiang G, Chen Q, Zheng JN. Ki67 is a promising molecular target in the diagnosis of cancer (Review). Mol Med Rep. 201;11(3):1566–1572. | ||
Mitrugno A, Sylman JL, Ngo AT, et al. Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: Implications for the oncoprotein c-MYC. Am J Physiol Cell Physiol. 2017;312(2):C176–C189. | ||
Zhang L, Xu Z, Xu X, et al. SALL4, a novel marker for human gastric carcinogenesis and metastasis. Oncogene. 2014;33(48):5491–5500. |
Supplementary materials
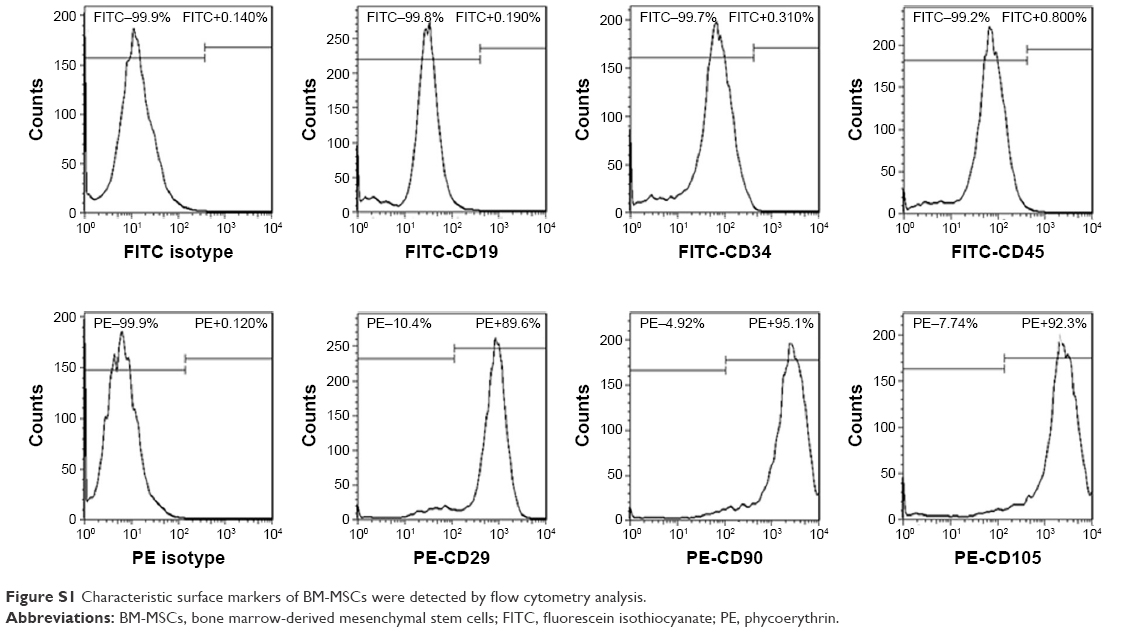 | Figure S1 Characteristic surface markers of BM-MSCs were detected by flow cytometry analysis. |
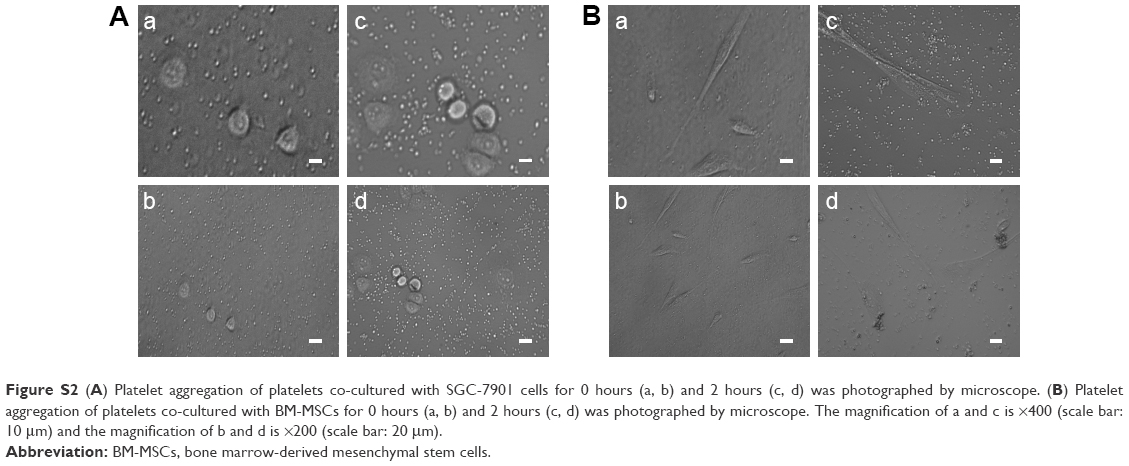 | Figure S2 (A) Platelet aggregation of platelets co-cultured with SGC-7901 cells for 0 hours (a, b) and 2 hours (c, d) was photographed by microscope. (B) Platelet aggregation of platelets co-cultured with BM-MSCs for 0 hours (a, b) and 2 hours (c, d) was photographed by microscope. The magnification of a and c is ×400 (scale bar: 10 μm) and the magnification of b and d is ×200 (scale bar: 20 μm). |
 | Figure S3 Cell morphology of BM-MSCs co-cultured with T-platelets for 0 hours (A) and 24 hours (B). |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.