Back to Journals » Journal of Inflammation Research » Volume 19
Platelet-Derived LGALS9 Suppresses NLRP3 Inflammasome-Dependent Pyroptosis in Pancreatic Acinar Cells During Severe Acute Pancreatitis
Authors Xiao W ![]() , Xu X, Yang T, Zou X
, Xu X, Yang T, Zou X ![]() , Xu H, Zhang H, Qian J, Wu F, Wang L, Zhou L, Gan Z, Zhang G, Li C
, Xu H, Zhang H, Qian J, Wu F, Wang L, Zhou L, Gan Z, Zhang G, Li C
Received 5 February 2026
Accepted for publication 17 June 2026
Published 3 July 2026 Volume 2026:19 596409
DOI https://doi.org/10.2147/JIR.S596409
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Nadia Andrea Andreani
Wang Xiao,1 Xingda Xu,1 Tingkang Yang,1 Xiangsen Zou,1 Hao Xu,1 Hanya Zhang,2 Jianping Qian,3 Feng Wu,4 Liping Wang,3 Lei Zhou,5 Zhe Gan,5 Guowei Zhang,5 Chuanjiang Li,3
1The First School of Clinical Medicine, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 2The First School of Clinical Medicine, Guangdong Medical University, Zhanjiang, Guangdong, People’s Republic of China; 3Department of Hepatobiliary Surgery, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 4Department of Critical Care Medicine, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China; 5Department of Hepatobiliary and Pancreatic Surgery, Peking University Shenzhen Hospital, Shenzhen, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guowei Zhang, Department of Hepatobiliary and Pancreatic Surgery, Peking University Shenzhen Hospital, Shenzhen, Guangdong, People’s Republic of China, Email [email protected];
Chuanjiang Li, Department of Hepatobiliary Surgery, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, People’s Republic of China, Email [email protected]
Background: Pyroptosis is a significant contributor to the development of severe acute pancreatitis (SAP), and the presence of SAP together with a reduced platelet count often indicates a poor prognosis. In this study, we screened targets from platelets and explored the role of lectin galactoside-binding soluble 9 (LGALS9) in acinar cell pyroptosis and inflammation in SAP.
Methods: SAP cell models, alongside mouse models showing diminished platelet counts, were established using caerulein and lipopolysaccharide. We conducted transcriptomic sequencing on platelets from mice with and without SAP to identify possible targets, and we determined LGALS9 levels in platelet releasates and serum from both patients and mice with and without SAP. Bioinformatic analysis was then applied to predict the relationship between LGALS9 and pyroptosis-related proteins. Subsequently, recombinant LGALS9, α-lactose, and RG9-35 were used to treat SAP mice and acinar cells to evaluate the effect of LGALS9 on inflammation and its regulatory effects on acinar cell pyroptosis in SAP.
Results: LGALS9 expression was upregulated in SAP platelets, and LGALS9 levels were found to be significantly elevated in platelet releasates and serum. Bioinformatic analysis illustrated the correlations and interactions between LGALS9 and the pyroptosis-related proteins ASC, NLRP3, caspase-1, and gasdermin D. Within the SAP context, LGALS9 may be potentially administered to diminish acinar cell pyroptosis-related protein levels, inhibit acinar cell pyroptosis, mitigate inflammatory factor levels, and alleviate pancreatic tissue inflammatory damage.
Conclusion: Our data indicated that LGALS9 expression was elevated in platelets of patients and mice with SAP. LGALS9 also inhibited NLRP3 inflammasome-dependent pyroptosis and suppressed inflammation in SAP.
Keywords: LGALS9, severe acute pancreatitis, pyroptosis, platelet, NLRP3 inflammasome
Introduction
Acute pancreatitis (AP) is a common acute digestive disorder that is principally caused by autodigestion of pancreatic tissue and severe local inflammation due to abnormal trypsinogen activation after damage to acinar cells.1 AP is classified by severity into mild acute pancreatitis (MAP), moderately severe acute pancreatitis (MSAP), and severe acute pancreatitis (SAP).2 Approximately 20% of patients with AP develop SAP, which may readily result in the dysfunction of numerous organs and even sepsis, with mortality rates attaining 20–40%.3,4 The pathogenesis of SAP is presently unclear, and there is a paucity of pharmacologic agents that specifically target the disease. Moreover, the clinical treatments for SAP have limited efficacy and result in poor patient prognosis.5 Therefore, an in-depth exploration of the underlying pathological mechanisms and their potential therapeutic value is crucial for developing objective management strategies for SAP.
Pyroptosis is a programmed lytic cell death that is characterized by cellular swelling, rupture, and the release of pro-inflammatory mediators, and it was first reported in 1998.6,7 The canonical and non-canonical inflammasome pathways play a primary role in triggering pyroptosis. Inflammasome complexes form the canonical inflammasome pathway, and four major prototypes are now known: NLRP3, NLRP1, NLRC4, and AIM2. These inflammasome complexes activate caspase-1, which cleaves gasdermin D (GSDMD) to release its N-terminal domain. Pores form in the plasma membrane that result in cellular swelling, lysis, and the release of the proinflammatory cytokines IL-1β and IL-18, which then amplify the inflammatory response. The non-canonical inflammasome pathway is primarily based upon the activation of caspase-4/5/11, which cleaves GSDMD to facilitate pyroptosis.8 Studies have confirmed that pyroptosis is involved in the key pathogenic mechanisms of multiple inflammatory diseases—including sepsis,9 Crohn’s disease,10 and pneumonia.11 NLRP3-dependent pyroptosis represents one of the critical factors that drive the pathological progression of AP to SAP.12 Clarification of the regulatory mechanisms governing pyroptosis in SAP would be critical to the development of therapeutic targets.
In addition to their functions in hemostasis and thrombosis, activated platelets are immune cells that initiate and accelerate the inflammatory process and contribute to various pathological inflammatory conditions.13 Current research indicates that activated platelets participate in the onset of AP by releasing inflammatory factors and forming microthrombi.14,15 Notably, a decrease in platelet count is associated with the severity of AP and can serve as one of the indicators for assessing disease severity and prognosis. A reduction in platelet count suggests a poor prognosis16 in SAP, indicating the involvement of activated platelets in the progression of the disease. Nevertheless, the mechanisms are not well understood.
Activated platelets produce and secrete soluble factors that mediate their inflammatory and immune functions.17 Lectin galactoside-binding soluble 9 (LGALS9), also known as galectin-9, is a secreted lectin and a member of the galectin family that selectively binds to β-galactosides.18 In recent years, LGALS9 has drawn significant attention due to its role in several physiological processes, including cellular growth, differentiation, communication, adhesion, and cell death.19 LGALS9 manifests diverse functions in various inflammatory states. For example, LGALS9 promotes inflammatory progression in osteoarthritis and rheumatoid arthritis20,21 while alleviating inflammatory damage in acute lung injury and colitis.22,23 The role of LGALS9 in SAP, however, remains unclear. Although previous studies have revealed that LGALS9 promotes the degradation of NLRP3,24 there is no direct evidence indicating that LGALS9 is involved in the regulation of pyroptosis.
In this study, we hypothesized that platelet-derived LGALS9 functions as an anti-inflammatory compensatory mechanism to suppress NLRP3-mediated pyroptosis in SAP. We investigated the expression of LGALS9 in platelets during SAP and demonstrated its effects on acinar cell pyroptosis in SAP. Our findings revealed that LGALS9 is upregulated in platelets in SAP and reduces inflammatory damage by blocking acinar cell pyroptosis, implying its future possible therapeutic use in the treatment of SAP.
Material and Methods
Antibodies and Reagents
Caerulein (CAE, S62702) and modified Tyrode’s buffer (R20269) were purchased from Yuanye Biotechnology (Shanghai, China). Lipopolysaccharide (LPS; ST1470), thrombin (ST1701), calcium chloride solution (ST365), RIPA buffer (P0013B), and PMSF (ST506) were purchased from Beyotime (Shanghai, China). α-Lactose (L2643) was purchased from Sigma-Aldrich (St. Louis, MO, USA); anti-LGALS9 (RG9-35, Ab01088-2.0-VXS) was purchased from Absolute Antibody (Redcar, UK). Recombinant LGALS9 protein (HY-P70535) and isotype control protein (mouse IgG2a kappa, HY-P99978) were purchased from MedChemExpress (Princeton, NJ, USA); PGE1 (Prostaglandin-E1, V2425) was purchased from InvivoChem (Libertyville, IL, USA). VAHTS® Universal V8 RNA-seq Library Prep Kit for Illumina (NR605) was purchased from Vazyme (Nanjing, China); a TUNEL In Situ Apoptosis Kit (E-CK-A320) was purchased from Elabscience (Wuhan, China); and calcein/PI cell viability/cytotoxicity assay kit (C2015) was purchased from Beyotime (Shanghai, China).
Primary antibodies anti-NLRP3 (WL02635) and anti-ASC (WL02462) were procured from Wanleibio (Shenyang, China); anti-GSDMD (TA4012) and anti-caspase-1 (P79884R2) were from Abmart (Shanghai, China); and anti-MPO (22225-1-AP) and anti-GAPDH (81640-5-RR) were from Proteintech (Wuhan, China). Anti-CD44 (DF6392), anti-caspase1 (AF4005), anti-GSDMD (AF4012), and second antibodies (goat anti-rabbit IgG H+L, S0001) were purchased from Affinity (Cincinnati, OH, USA).
Mouse TNF-α (E-EL-M3063), mouse IL-6 (E-EL-M0044), mouse IL-1β (E-EL-M0037), mouse IL-18 (E-EL-M0730), and human LGALS9 (E-EL-H1059) ELISA kits were purchased from Elabscience (Wuhan, China). Mouse LGALS9 (CSB-EL012895MO) ELISA kits were purchased from CUSABIO (Wuhan, China).
SAP and Healthy Controls
Whole blood samples were collected from five normal controls and five SAP patients aged 18–80 years at Nanfang Hospital of Southern Medical University with approval from the Ethics Committee (Approval No. NFEC-2021-433). SAP patients met the corresponding diagnostic criteria,4 and samples were collected when their platelet counts were below 100 × 109/L.
SAP Mouse Model
All animal experiments were conducted in accordance with the ARRIVE guidelines.25 Preclinical male PRSS1 (GenBank Accession No. NM_002769.4) transgenic (PRSS1Tg) mice that were 6–8 weeks of age and that weighed 20–25 g each were grouped by simple randomization with a random number method. Compared with wild-type mice, PRSS1Tg mice more accurately recapitulates the pathological progression of human pancreatitis, exhibiting more pronounced phenotypic manifestations and disease severity. These mice were then used to establish the SAP model by 10 intraperitoneal (i.p.) injections of CAE (20 μg/mL) at 100 μg/kg administered at hourly intervals and one intraperitoneal injection of LPS (1 mg/mL) at 5 mg/kg with the final CAE injection; normal control animals received an equivalent volume of saline (Figure 1A). An equal volume of recombinant LGALS9 (rLGALS9) protein (30 μg/mouse) or PBS was intraperitoneally injected 30 minutes before the first CAE treatment in the SAP treatment groups, while α-lactose (30 mg/mouse), RG9-35 (60 μg/mouse), IgG2a isotype control antibody (60 μg/mouse), or PBS was administered intraperitoneally 30 minutes after LPS treatment. All mice were housed in randomly positioned static cages (with five mice per cage) under a 12/12 hour dark/light cycle and at a temperature of 22–24°C in the specific pathogen free–grade animal care facility, with free access to water and food. At the designated time after the first CAE injections, mice were anesthetized via intraperitoneal injections of pentobarbital sodium (150 mg/kg), after which they were sacrificed to harvest their blood and pancreas for subsequent experiments.
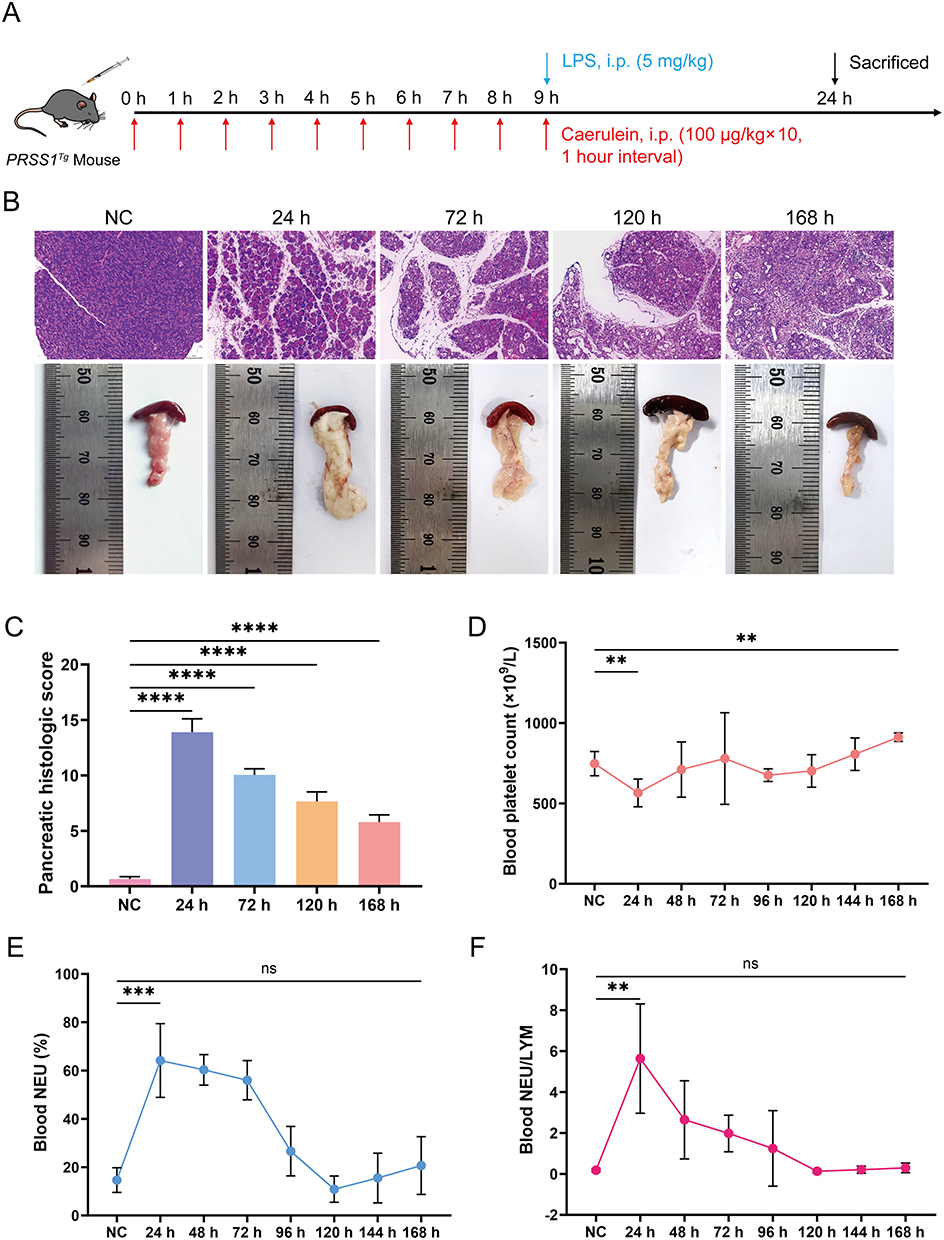 |
Figure 1 Establishment of SAP mouse models with diminished platelet counts. (A) SAP mouse models were established by intraperitoneal injection of CAE and LPS. (B and C) H&E staining, pancreatic images, and pancreatic histology scores of normal control (NC) and SAP at various time points (Scale bar: 100 μm). (D–F) Platelet counts, neutrophil percentages, and neutrophil/lymphocyte ratios in peripheral blood of NC and SAP from 24 h to 168 h. Data are presented as means ± standard error of the mean (SEM). n=5. NS, not significant, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-sided Student’s t-test. |
SAP Cell Model
Mouse pancreatic acinar MPC-83 cells were obtained from Jennio Biotech (Guangzhou, China) and cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, to prevent bacterial contamination, at 37°C in an incubator with 5% CO2 in compressed air with high humidity. The cell model was established by treating cells with CAE (100 nM) and LPS (10 μg/mL) or rLGALS9 (500 ng/mL) for 24 h. In the SAP treatment groups, cells were pretreated with rLGALS9 (500 ng/mL) or α-lactose (30mM) for 30 min before CAE and LPS treatment. The culture medium and cells were subsequently collected for further analysis.
Bioinformatic Analysis
The GSE194331 dataset containing RNA-sequencing data from 32 healthy controls, 57 MAP, 20 MSAP, and 10 SAP blood samples was obtained from The Gene Expression Synthesis Database (https://www.ncbi.nlm.nih.gov/geo/). Based on this dataset, we analyzed the differences in serum expression levels and the relationships among LGALS9, PYCARD (ASC), NLRP3, CASP1 (caspase-1), GSDMD, IL-1β, and IL-18 between patients with AP and healthy controls. STRING (https://cn.string-db.org) was adopted to detect the interaction network between LGALS9 and these genes.
Platelet Isolation and RNA Sequencing and Analysis
Peripheral blood collected in 3.2% sodium citrate tubes was used to isolate platelets, and the samples were placed in an ice bath for 30 min immediately after collection. Following the addition of PBS and Ficoll separation medium, the sample was centrifuged at 200 g for 4 min at 4°C. The platelet-rich plasma was then extracted and washed with PBS. Following this, the platelet-rich plasma was centrifuged at 500 g for 20 minutes and then washed with PBS again. After centrifugation, TRIzol and chloroform were added to the platelets, and the mixture was centrifuged at 12,000 g for 10 min at 4°C. Anhydrous ethanol and glycogen were then added, and the mixture was mixed thoroughly and incubated at −80°C for 1 hour. A further centrifugation was then performed, and the supernatant was removed. The pellet was dissolved in RNase-free water, and the concentration was determined using a Qubit fluorometer (Thermo Fisher). We performed RNA extraction using a VAHTS® Universal V8 RNA-seq Library Prep Kit according to the manufacturer’s instructions. The obtained libraries were ultimately quantified, normalized, pooled, and sequenced using Illumina.
The quality of the FASTQ files from RNA sequencing was checked using FASTQC v0.11.7. Clean reads were then aligned to the reference genome quickly and accurately using HISAT2, followed by count quantification using the feature Counts tool in the Subread package. RNA sequencing data were analyzed using DESeq2 for batch correction and normalization. The sequencing depth was ≥10 million reads per sample. Differentially expressed genes (DEGs) were selected using the criteria |log2FC|>1 and an adjusted p-value (padj) < 0.05. We conducted pairwise statistical tests with the Wilcoxon rank-sum test and executed correlations using the non-parametric Spearman correlation.
Platelet Releasates
Sodium citrate (3.8%) was mixed with peripheral blood from patients or mice at a 9:1 ratio (v/v) and placed on ice immediately. Platelet-rich plasma (PRP) was isolated by centrifugation at 100 g for 5 min at room temperature and then diluted with Tyrode’s buffer at a 1:1 (v/v) ratio. After mixing with PGE1 to prevent platelet activation, the platelets were pelleted by centrifugation at 1000 g for 10 min at 4°C and washed again with Tyrode’s buffer containing 1 μM PGE1; following centrifugation, the platelet pellets were resuspended in Tyrode’s buffer without PGE1. To initiate the activation process, we adjusted the platelet concentration to 300 × 109/L and stimulated them with 10 U/mL thrombin and 2 mM calcium chloride solution for 30 minutes at 37°C. We then centrifuged the solution at 3000 g for 30 min at 4°C to remove platelet fragments. The supernatant was collected as platelet releasates and stored at −80°C for further analysis.
Western-Blot Assay
The pancreatic tissues or pancreatic acinar cells were lysed in RIPA buffer containing PMSF using an ultrasonic processor. The lysates were then centrifuged at 14,000 g for 10 min at 4°C to remove cellular debris, and denatured proteins were separated on SDS-PAGE gels by electrophoresis and transferred to PVDF membranes. Skimmed milk (5%) was used to block membranes for 1 h, and the membrane was then incubated with primary antibody overnight at 4°C and further incubated with secondary antibody at room temperature for 1 h. Finally, hypersensitive chemiluminescence reagents were used to visualize immunostained bands. ImageJ (NIH, Bethesda, MD, USA) was used to quantify the relative levels of protein expression.
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentrations of LGALS9, TNF-α, IL-6, IL-1β, and IL-18 in the MPC-83 cell culture medium and in the mouse serum were determined using ELISA kits according to the manufacturer’s protocols and analyzed using a microplate reader (TECAN, Männedorf, Switzerland).
Histopathological and Immunohistochemical Staining
For histopathological staining, fresh pancreatic tissues were fixed with 4.0% paraformaldehyde solution, treated with a concentration gradient of ethanol and xylene, and embedded in paraffin. Sections (4 μm thick) were then prepared, deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E). Histopathological scoring of the pancreas was performed blindly by two pathologists following the criteria reported by Schmidt et al26 The final pathological score was then the average of both values.
For immunohistochemical staining, the paraffin sections were dewaxed and rehydrated, and antigen retrieval took place in 0.01 μm citrate buffer. After blocking with 5% goat serum, the sections were incubated with an MPO antibody at 4°C overnight. The secondary antibody was added for incubation the following day, and color development was commenced with 200 µL of diaminobenzidine (DAB) solution.
Calcein/PI Staining and TUNEL Assay
Cells were incubated with calcein/PI for 30 min, and cell death was determined with PI (for dying/dead cells) and calcein (for living cells). Paraffin sections of pancreatic tissue were stained using a TUNEL In Situ Apoptosis Kit according to the instructions. We captured images using a fluorescence microscope (BX63, Olympus), and the percentage area of the fluorescence signal was calculated with ImageJ.
Statistical Analysis
We statistically analyzed all data collected throughout the study with GraphPad Prism 10 and IBM SPSS Statistics 26. The data are presented as a mean ± standard deviation (SD), with a minimum of three independent replicates. Correlation analyses were conducted using Spearman’s rank correlation test. For two-group comparisons of normally distributed data, we applied the Student’s unpaired t test, while the Mann–Whitney nonparametric test was used when normality was not satisfied. For paired-sample comparisons, the paired t test or the Wilcoxon signed-rank test was implemented. The outcome assessors and data analysts were blinded to group allocation. A P value ˂ 0.05 was considered to be statistically significant.
Results
Establishment of Our SAP Model and Target Gene
To establish a mouse model suitable for this investigation, we successfully constructed SAP mouse models for 1–7 days (Figure 1B). Hematological analysis showed that inflammatory damage to pancreatic tissue was most severe and pancreatic tissue edema was most pronounced 24 hours after model construction, with the platelet count significantly reduced and the neutrophil percentage (NEU%) and neutrophil/lymphocyte (NEU/LYM) ratio significantly increased compared with normal levels. However, inflammation was alleviated by 168 hours, and the platelet count recovered (Figure 1C–F). Consequently, the animal model that we constructed 24 hours after the first intraperitoneal injection of CAE in mice was the optimal choice for this study.
To investigate the contribution of platelets to SAP pathogenesis, RNA sequencing was performed on platelets isolated from five mice individually 24 h and 168 h after construction of the SAP mouse model and normal controls. To minimize interference, the upregulated DEGs in platelets from the SAP mice at 24 h and 168 h were analyzed separately using the edgeR, DESeq2, and limma packages, and the intersections yielded 1769 and 1373 genes, respectively (Figure 2A and B). The intersection of these lists resulted in 414 commonly upregulated genes (Figure 2C), and we then performed functional enrichment analysis with Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Given that the occurrence of AP is attributed to the activation of trypsinogen under various stimuli,27 we further intersected the genes enriched in three pathways: GO:0050777, GO:0002832, and GO:0031342. After retaining genes that encoded secretory proteins, we isolated three putative candidate genes: LGALS9, TGFB1, and CEACAM1. An extensive literature review ultimately resulted in the choice of LGALS9 as the most suitable target for further study (Figure 2D). The RNA expression levels of platelet LGALS9 were also significantly elevated in SAP mice compared with normal control mice (Figure 2E).
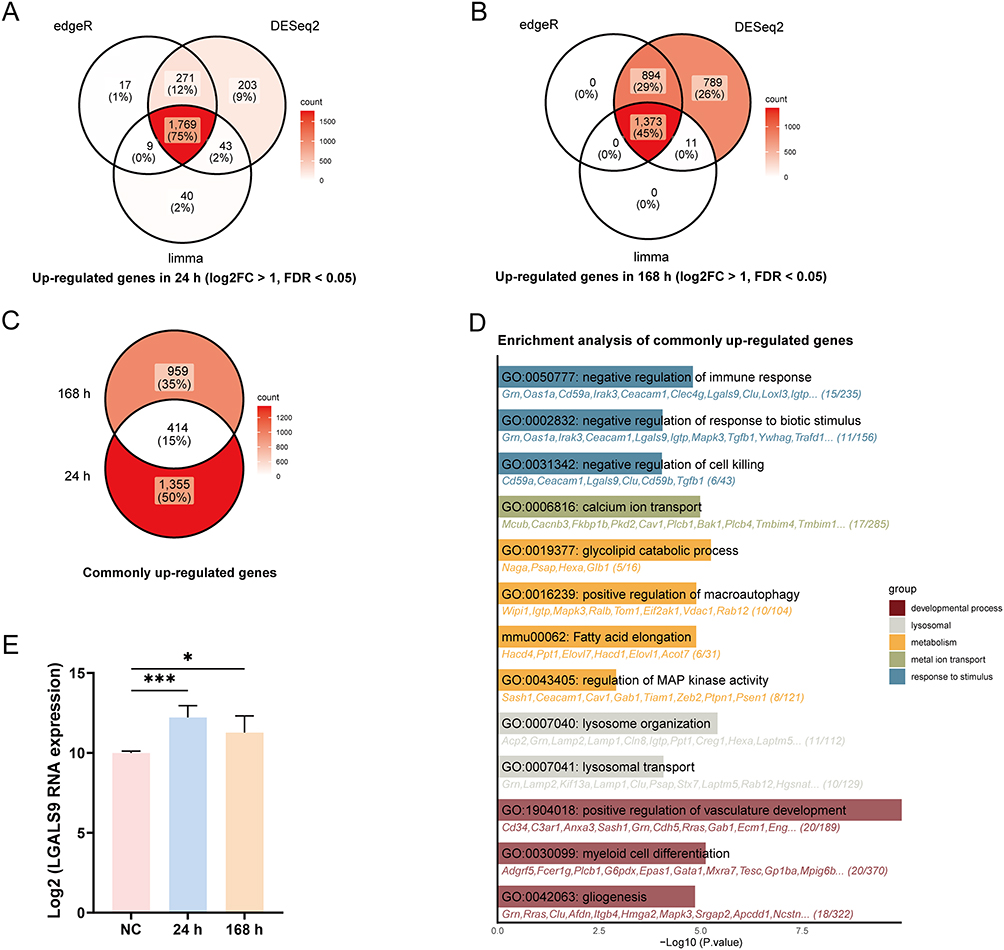 |
Figure 2 LGALS9 RNA expression was elevated in platelets from SAP mice. (A and B) Upregulated genes in platelets from SAP mouse models at 24 h and 168 h. (C) 414 commonly upregulated genes identified in both 24 h and 168 h groups. (D) Functional enrichment analysis of the common upregulated genes was performed using GO and KEGG. (E) LGALS9 RNA expression in platelets from the NC, 24 h, and 168 h groups. All data represented the means ± SEM. n=5. *P < 0.05, ***P < 0.001, two-sided Student’s t-test. |
LGALS9 is Upregulated in SAP Platelets and Serum and Correlates with Pyroptosis-Related Proteins
We subsequently confirmed by ELISA that LGALS9 protein concentrations were significantly augmented in both platelet releasates and serum from SAP mice and humans compared with normal controls (Figure 3A–D), which supported our transcriptomic results. Additionally, platelet-derived LGALS9 was discerned to be discharged to the extracellular space after platelet activation.
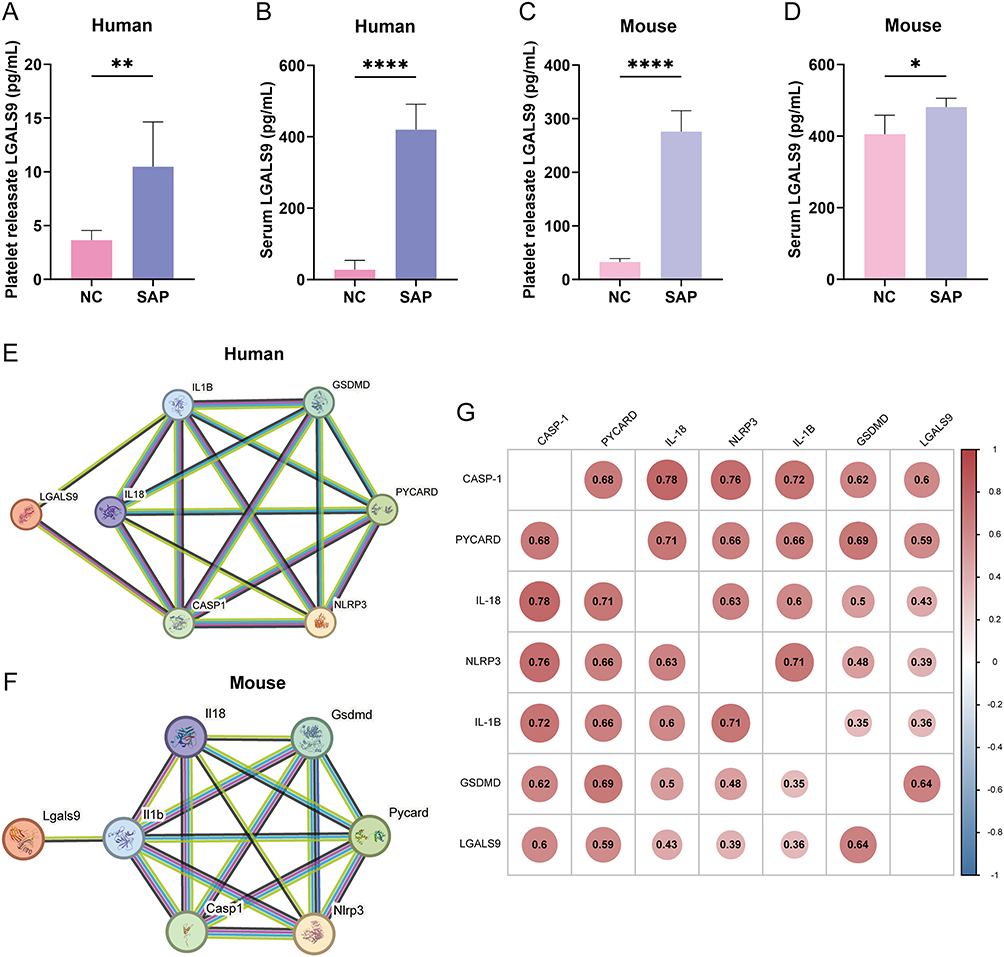 |
Figure 3 LGALS9 protein expression was elevated in platelets and serum and correlated with pyroptosis-related proteins. (A and B) Expression levels of LGALS9 in platelet releasates and serum from SAP patients compared with NC, n=6. (C and D) Expression levels of LGALS9 in platelet releasates and serum from SAP mice compared with NC, n=5. (E and F) Correlations between LGALS9 and pyroptosis-related proteins, including NLRP3, PYCARD (ASC), CASP1 (caspase-1), GSDMD, IL1B (IL-1β), and IL-18 in humans and mice. (G) Correlation analysis of LGALS9 with pyroptosis-related proteins using the mRNA expression profile dataset GSE194331. Means ± SEM; *P < 0.05, **P < 0.01, ****P < 0.0001 as determined by two-sided Student’s t-test. |
We next investigated the relationships between LGALS9 and pyroptosis-related proteins, and STRING analysis (https://cn.string-db.org) confirmed direct or indirect interaction relationships between LGALS9 and pyroptosis-related proteins in both humans and mice (Figure 3E and F). We then applied the mRNA expression profile dataset GSE194331 for analysis, and correlation analysis revealed that circulating LGALS9 expression was significantly correlated with the levels of pyroptosis-related proteins, particularly those of CASP1 and GSDMD (Figure 3G).
LGALS9 Alleviates Pyroptosis in Acinar Cells During SAP in vitro
Since LGALS9 is a secretory protein, we subsequently exploited pancreatic acinar cells (MPC83) for the induction of SAP in vitro to investigate the role of circulating LGALS9 in regulating acinar cell pyroptosis during SAP, and we conducted a series of functional experiments using exogenous rLGALS9. Calcein/PI staining revealed that CAE and LPS induction caused substantial death of SAP acinar cells, whereas the addition of rLGALS9 significantly attenuated acinar cell death. However, the effect of rLGALS9 was reversed in the presence of α-lactose (an inhibitor of LGALS9 activity) (Figure 4A and B).
Figure 4 LGALS9 inhibited inflammation and NLRP3-dependent pyroptosis in vitro. (A and B) Representative images and results of calcein (green), PI (red), and DAPI (blue) staining in mouse pancreatic acinar cells from each group (Scale bar: 100 μm). (C) ELISA detection of IL-1β, IL-18, IL-6, and TNF-α levels in cell culture supernatants from each group. (D–G) Western Blot analysis of the expression levels of pyroptosis-related proteins, including ASC, NLRP3, caspase1-FL, caspase1-cleaved, GSDMD-FL, and GSDMD-N expression in acinar cells from each group. Means ± SEM. n=3. NS, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-sided Student’s t-test. Figure 4 continued.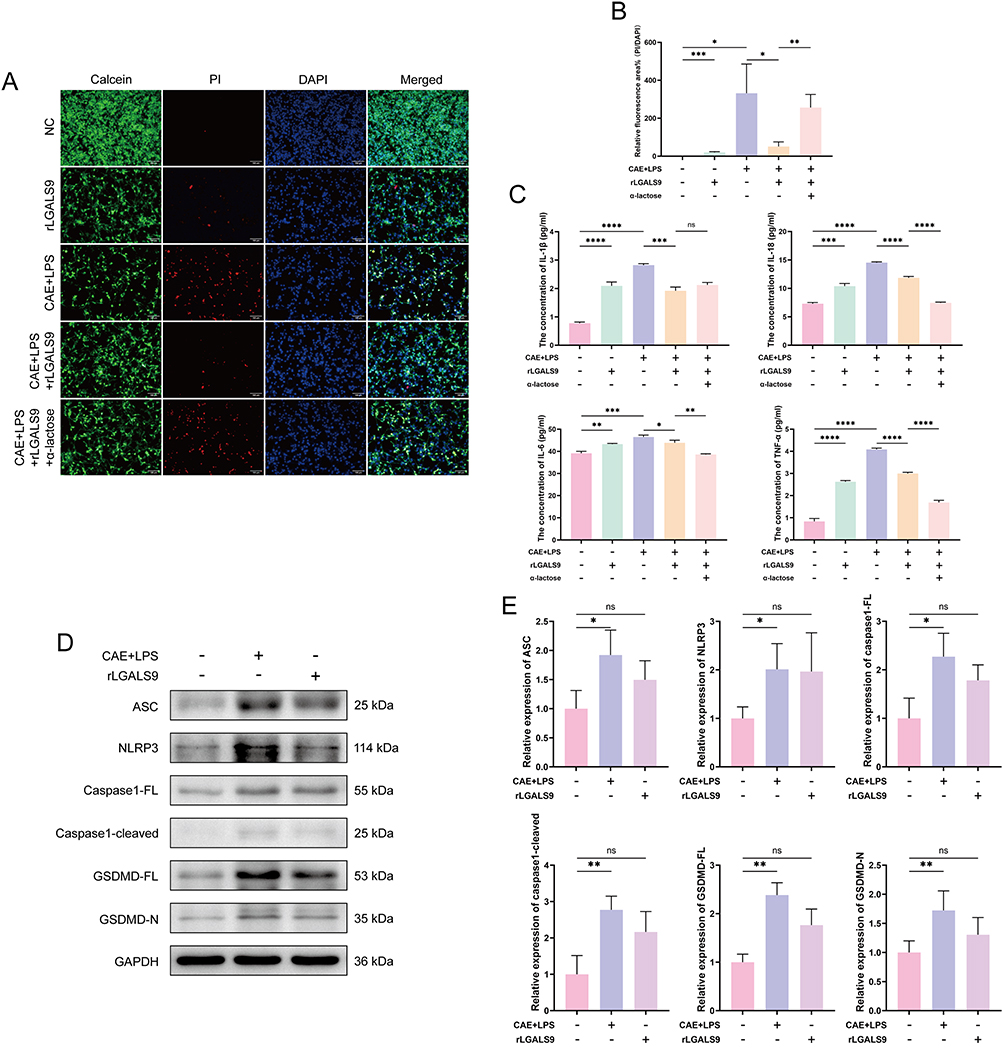
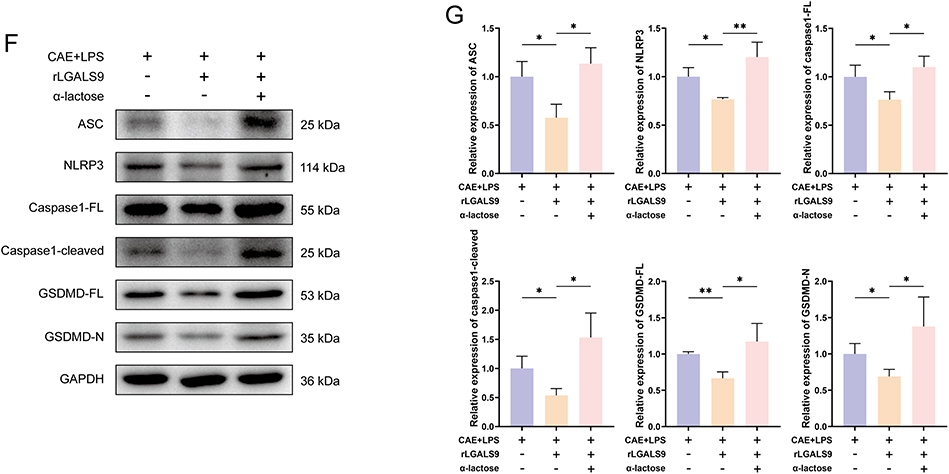
We adopted ELISA to measure the levels of inflammatory cytokines in the cell culture supernatants. As shown in Figure 4C, the inflammatory cytokines IL-1β, IL-18, IL-6, and TNF-α in the supernatants from cells from individuals with SAP were significantly elevated compared with the normal group, whereas the addition of rLGALS9 markedly reduced these cytokine levels. Notably, in the presence of α-lactose, the levels of IL-18, IL-6, and TNF-α diminished further, without blocking rLGALS9 function.
Western-blot analysis was then used to evaluate the expression levels of the pyroptosis-related proteins NLRP3, ASC, caspase1-full length (caspase1-FL), caspase1-cleaved, GSDMD-full length (GSDMD-FL), and GSDMD-N in pancreatic acinar cells subjected to different treatments, in order to observe the effects of rLGALS9. Compared with the normal group, the expression of pyroptosis-related proteins in SAP acinar cells was significantly increased (Figure 4D and E), while rLGALS9 inhibited the expression of pyroptosis-related proteins, with such suppressive activity almost completely abrogated by α-lactose (Figure 4F and G).
LGALS9 Inhibits Inflammation and Acinar Cell Pyroptosis in SAP in vivo
We wished to verify whether LGALS9 inhibited inflammation and pancreatic acinar cell pyroptosis in SAP mice in vivo. PRSS1Tg mice were exploited to establish our SAP model, histopathological staining was employed to assess inflammatory injury in their pancreatic tissues, and MPO immunohistochemical staining was applied to evaluate NEU infiltration in pancreatic tissue. Our results revealed that pancreatic tissue-injury scores in mice with SAP and NEU infiltration were both significantly increased, while IgG2a (isotype control antibody) had no effect on SAP. rLGALS9 reduced pancreatic tissue inflammation and NEU infiltration, while α-lactose and RG9-35 (a specific antibody to murine LGALS9) blocked the effects of rLGALS9 (Figure 5A–C).
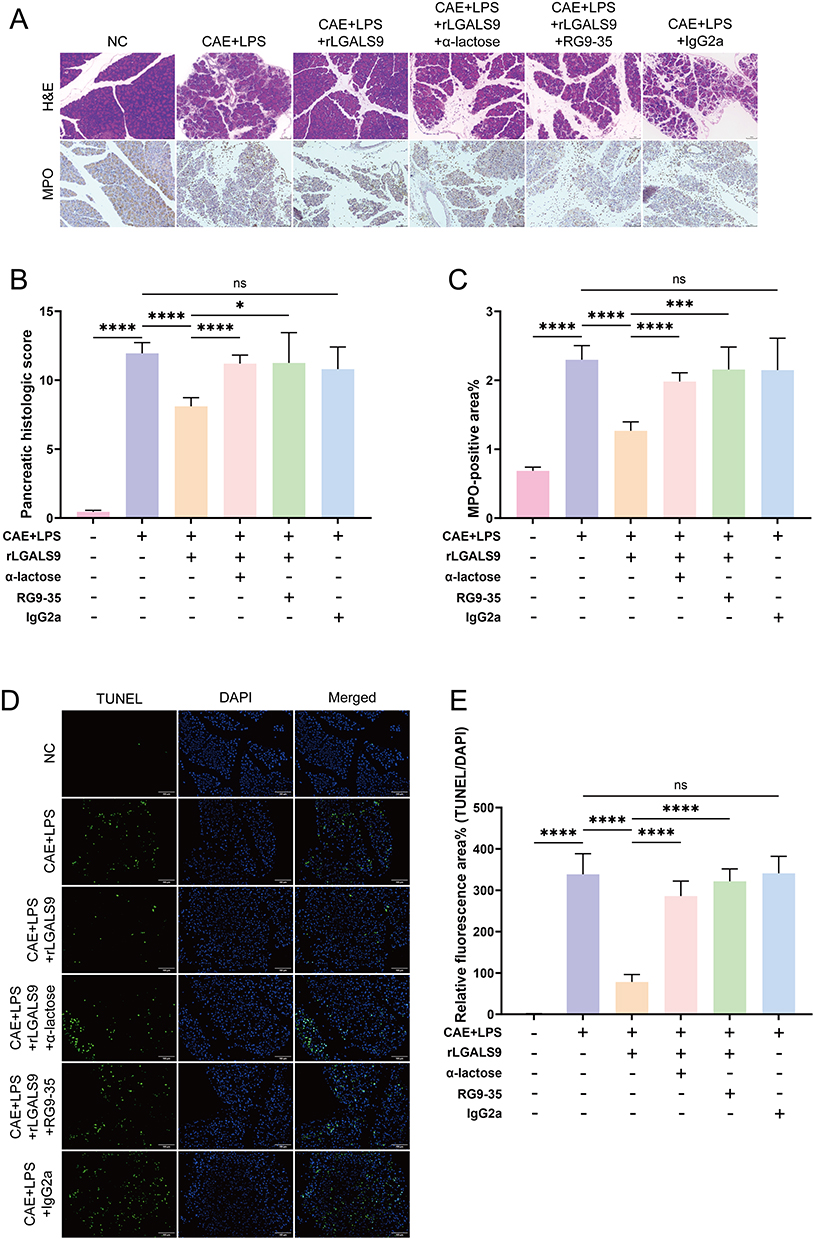 |
Figure 5 LGALS9 alleviated pancreatic tissue inflammation and injury in SAP in vivo. (A) Representative images of H&E staining and MPO immunohistochemical staining in each group (Scale bar: 100 μm). (B) Pancreatic histology scores in each group. (C) Percentage of MPO-positive area in each group. (D and E) Representative images and results of TUNEL staining in pancreatic tissue from each group (Scale bar: 100 μm). Means ± SEM. n=5. NS, not significant, *P < 0.05, ***P < 0.001, ****P < 0.0001 as determined by two-sided Student’s t-test. |
We implemented the TUNEL assay to assess acinar cell death, and we achieved results consistent with our calcein/PI staining in vitro. SAP mice exhibited increased acinar apoptosis that was reduced by treatment with rLGALS9 and exacerbated by blockade of LGALS9 protein (Figure 5D and E).
Furthermore, when the levels of the pyroptosis-related proteins NLRP3, ASC, caspase-1-FL, caspase1-cleaved, GSDMD-FL, and GSDMD-N in each group of pancreatic tissues were investigated, we ascertained according to western-blot results that the expression of pyroptosis-related proteins was elevated in SAP. Although GSDMD-FL did not show significant differences, its levels in the pancreatic tissues of the SAP group remained higher than those in the normal group. Although LGALS9 inhibited the expression of pyroptosis-related proteins, this downregulation was counteracted by α-lactose and RG9-35 (Figure 6A and B).
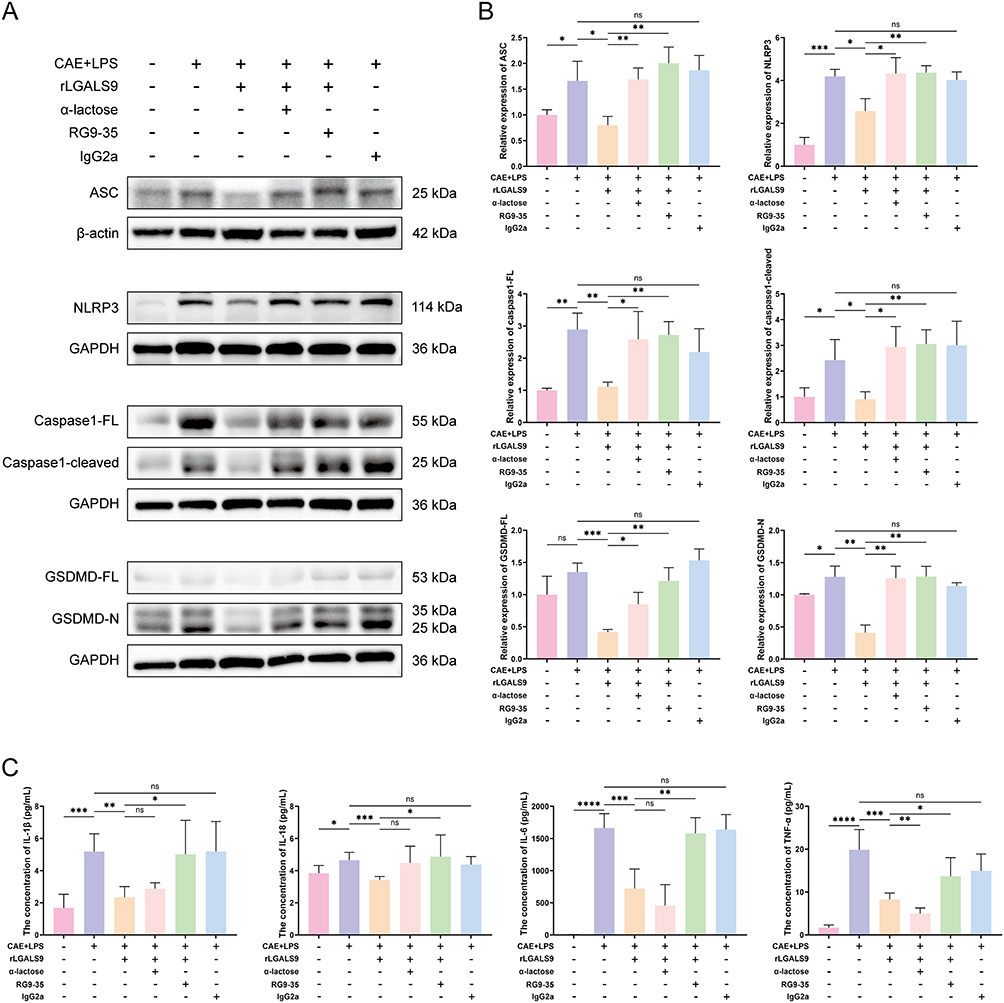 |
Figure 6 LGALS9 suppressed SAP inflammation and NLRP3-dependent pyroptosis in vivo. (A and B) Western Blot analysis of the expression levels of pyroptosis-related proteins in pancreatic tissues from each group, n=3. (C) ELISA detection of IL-1β, IL-18, IL-6, and TNF-α levels in serum from each group, n=5. Means ± SEM; NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as determined by two-sided Student’s t-test. |
Finally, we measured the levels of IL-1β, IL-18, IL-6, and TNF-α in the serum of mice from each group and found that rLGALS9 significantly reduced the levels of inflammatory factors in the serum of SAP mice, while RG9-35 reversed this reduction. Notably, α-lactose did not completely reverse the diminution in IL-1β and IL-18, and the levels of IL-6 and TNF-α fell further after the addition of α-lactose (Figure 6C). These results further confirmed that LGALS9 ameliorated inflammatory injury and pancreatic acinar cellular pyroptosis, demonstrating a specific protective effect against SAP.
Discussion
SAP is a disease that is difficult to treat, exhibiting a high mortality rate caused by systemic injury and multiple-organ failure, and activated platelets appear to play an important role in its development. In this study, we screened the alterations in gene expression in platelets of normal mice and those in mice with SAP by transcriptomics and found that LGALS9 RNA was upregulated in SAP platelets. The protein levels of LGALS9 were also significantly augmented in SAP platelet releasates and serum. In vitro and in vivo experiments confirmed the onset of pancreatic acinar cell pyroptosis in SAP, while LGALS9 was able to alleviate the inflammation and tissue damage in SAP by inhibiting pyroptosis-related proteins in acinar cells and by reducing the levels of serum inflammatory factors.
Numerous studies have substantiated that NLRP3 inflammasome-dependent pyroptosis comprises a critical mechanism in the development of AP. The NLRP3 inflammasome is composed of NLRP3, ASC, and caspase-1,28 and blocking its related proteins can inhibit acinar cell pyroptosis and mitigate AP inflammation.29–31 The intraperitoneal injection of CAE with LPS is frequently used to create SAP models, as it simulates the common clinical scenario of SAP complicated by infection; moreover, LPS is a key activator of the NLRP3 inflammasome.32 Based on these studies, we successfully established an SAP model and observed the onset of pyroptosis in pancreatic acinar cells. By inhibiting the protein expression of NLRP3, ASC, caspase1, and GSDMD in acinar cells and the secretion of IL-1β and IL-18, we ameliorated acinar cell pyroptosis and pancreatic tissue inflammation. These findings further demonstrated the significant therapeutic value of inhibiting acinar cell pyroptosis in SAP treatment.
Although platelets have been traditionally shown to play a key role in hemostasis and thrombosis, they also serve as important modulators of inflammation and immunity.33 Platelets store three types of granules: α-granules, dense granules, and lysosomes, with α-granules being the most plentiful storing molecules. In addition to containing proteins that are critical to hemostasis, platelets also store abundant chemokines, growth factors, and immune mediators; when platelets are activated, these inflammatory factors are discharged into the blood and contribute to the pathogenesis of various inflammatory conditions.34 Platelets might therefore be important in the pathogenesis of AP. Activated platelets are able to mediate NEU infiltration and tissue injury in the pancreatic tissues of individuals with AP through the release of the chemokine CXCL4.14 Platelet microparticles also enhance pancreatic tissue damage by modulating the development of NEU extracellular traps (NETs).35 Excessive platelet activation and aggregation appear to contribute to the formation of thromboses in the pancreatic microcirculation, further leading to the exacerbation of pancreatic ischemia, necrosis, and edema.15 Studies have indicated that AP patients with decreased platelet counts also exhibit significantly higher incidence and mortality rates of infected pancreatic necrosis.16 On the basis of these findings, we established an SAP model with decreased platelet counts and conducted a series of experiments. We demonstrated that the expression of LGALS9 was elevated in SAP platelets and LGALS9 could be discharged into the immediate environment during activation. Contrary to the previously acknowledged concept of platelets as pro-inflammatory, LGALS9 was shown to suppress inflammatory damage and acinar cell pyroptosis in SAP. LGALS9 as a protective factor implies a dual identity of platelets in the development of SAP.
LGALS9 was initially uncovered to be an auto-antigen in the lymphomatous tissue of Hodgkin lymphoma patients and as an eosinophil attractant in T-lymphocytes; investigators subsequently determined that LGALS9 is extensively distributed at variable expression levels in various organ systems and tissues.36 While LGALS9 has been localized to the cytoplasm, cell surface, and circulation, the source of LGALS9 in the serum has not been fully identified.37 We noted that serum LGALS9 concentrations were elevated following the drop in platelet count in SAP, suggesting that serum LGALS9 originates from diverse sources. LGALS9 manifests a variety of different biological activities that apparently depend upon disease type and localization. LGALS9 exacerbates inflammatory reactions in the articular cartilage of osteoarthritis by activating the JNK and ERK1/2 pathways20 but ameliorates inflammation in intestinal mucosa by inhibiting the TLR4/NF-κB pathway in ulcerative colitis.23 Recent research has shown that LGALS9 thus manifests the dichotomous effects of being pro-inflammatory or anti-inflammatory in inflammatory bowel disease depending upon context. Both a deficiency in endogenous LGALS9 and the treatment with exogenous rLGALS9 can attenuate intestinal inflammation.38 LGALS9 exhibits disparate regulatory effects on NLRP3 in various contexts. Although LGALS9 interacts with TLR4 on microglia and triggers the NLRP3 inflammasome in Alzheimer’s disease,39 it promotes NLRP3 autophagic degradation by mediating the binding of p62 to NLRP3 in LPS-stimulated mouse peritoneal macrophages, inhibiting NLRP3 inflammasome activation.24 With SAP, we herein demonstrated for the first time that treatment with exogenous rLGALS9 suppresses NLRP3 inflammasome-dependent pyroptosis in acinar cells, confirming a protective role for exogenous LGALS9 in SAP. The aforementioned results revealed the variety of LGALS9’s biological activities and mechanisms of action and its roles in a variety of diseases, and this should be explored further in the future.
The present study has several important implications. We demonstrated that LGALS9 expression was elevated in SAP platelets and that LGALS9 inhibited the pyroptosis of pancreatic acinar cells—alleviating inflammatory injury. These findings provide further supporting evidence linking platelets and inflammation in the pathogenesis of SAP, revealing the dual role of platelets in inflammation. The development and resolution of inflammation are generated by dynamic interactions between pro-inflammatory and anti-inflammatory factors. Although we found augmented LGALS9 in SAP serum, pro-inflammatory factors still dominated. We hypothesize that increasing serum LGALS9 would offer a novel avenue of therapy in SAP. This study thus presents a theoretical basis for mechanistic research focused on platelets, in particular LGALS9, and augurs for greater future understanding of the pathological development of SAP and for improvements in treatment strategies.
There were some limitations to the current study. First, α-lactose competitively inhibited the glycosidase activity of LGALS9 protein by binding to its carbohydrate recognition domains (CRDs). However, α-lactose can also inhibit other LGALS family members with CRDs in addition to LGALS9.40 The specific inhibitor of LGALS9 has yet to be identified. Second, we herein found reduced platelet counts in mice and humans with SAP and also elevations in LGALS9 concentrations in serum, without eliminating the possible involvement of other cells in circulating LGALS9. Finally, we were not able to confirm whether the regulation of acinar cell pyroptosis by LGALS9 was direct or indirect. The specific mechanisms by which LGALS9 regulates pyroptosis also remain unclear, including how extracellular LGALS9 protein transmits regulatory signals into cells to further affect the onset of acinar cell pyroptosis. These aspects require additional verification in future studies.
Conclusion
Our study showed that LGALS9 was a key suppressor of SAP and that LGALS9 expression was upregulated in the SAP platelets. Furthermore, LGALS9 inhibited NLRP3 inflammasome-dependent pyroptosis in acinar cells, alleviating inflammation. These findings highlight the promise of LGALS9 as a therapeutic target in the treatment of SAP.
Abbreviations
AP, acute pancreatitis; MAP, mild acute pancreatitis; MSAP, moderately severe acute pancreatitis; SAP, severe acute pancreatitis; GSDMD, gasdermin D; LGALS9, lectin, galactoside-binding, soluble, 9; RNA-seq, RNA sequencing; CAE, caerulein; LPS, lipopolysaccharide; PGE1, prostaglandin E1; Tg, transgenic; i.p. intraperitoneal; rLGALS9, recombinant LGALS9; DEGs, differentially expressed genes; ELISA, enzyme-linked immunosorbent assay; H&E, hematoxylin and eosin; DAB, diaminobenzidine; TUNEL, TdT-mediated dUTP Nick End Labeling; NEU, neutrophil; LYM, lymphocyte; GO, Gene Ontology.
KEGG, Kyoto Encyclopedia of Genes and Genomes; FL, full length; NETs, neutrophil extracellular traps; CRDs, carbohydrate recognition domains.
Data Sharing Statement
Data sets used or analyzed during the current study period are available from corresponding authors upon reasonable request.
Ethics Statement
Animal experiment protocols were approved by the Nanfang Hospital, Southern Medical University (License No. SYXK (Yue) 2023-0056), following ARRIVE guidelines. Patient blood samples were collected from Nanfang Hospital, Southern Medical University, and consents were obtained. All the programs complied with the ethical guidelines of the Declaration of Helsinki and approved by the Medical Ethics Committee of Nanfang Hospital, Southern Medical University (Approval No. NFEC-2021-433).
Acknowledgments
We thank LetPub (www.letpub.com.cn) for its linguistic assistance during the preparation of this article. We thank Shanghai Tengyun Biotechnology Co., Ltd. for developing Hiplot Pro platform (https://hiplot.com.cn/) and providing technical assistance and valuable tools for data analysis and visualization.
Author Contributions
Wang Xiao and Xingda Xu: Conceptualization, Formal analysis, Investigation, Writing – original draft, Writing – review and editing. Tingkang Yang and Hanya Zhang: Formal analysis, Investigation, Writing – review and editing. Xiangsen Zou and Hao Xu: Data curation, Methodology, Writing – review and editing. Jianping Qian and Lei Zhou: Project administration, Supervision, Data curation, Writing – review and editing. Feng Wu, Liping Wang and Zhe Gan: Resources, Investigation, Writing – review and editing. Guowei Zhang and Chuanjiang Li: Conceptualization, Funding acquisition, Supervision, Writing – review and editing. All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the Shenzhen Natural Science Foundation Program (JCYJ20250604183826036) and Guangdong Basic and Applied Basic Research Foundation (2023A1515012523).
Disclosure
The authors report no conflicts of interest in this article.
References
1. Mederos MA, Reber HA, Girgis MD. Acute pancreatitis: a review. JAMA. 2021;325(4):382–16. doi:10.1001/jama.2020.20317
2. Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62(1):102–111. doi:10.1136/gutjnl-2012-302779
3. Lou D, Shi K, Li H, et al. Quantitative metabolic analysis of plasma extracellular vesicles for the diagnosis of severe acute pancreatitis. J Nanobiotechnol. 2022;20(1):52. doi:10.1186/s12951-022-01239-6
4. Trikudanathan G, Yazici C, Evans Phillips A, Forsmark CE. Diagnosis and management of acute pancreatitis. Gastroenterology. 2024;167(4):673–688. doi:10.1053/j.gastro.2024.02.052
5. Chen X, Chen X, Yan D, et al. GV-971 prevents severe acute pancreatitis by remodeling the microbiota-metabolic-immune axis. Nat Commun. 2024;15(1):8278. doi:10.1038/s41467-024-52398-z
6. Bai Y, Pan Y, Liu X. Mechanistic insights into gasdermin-mediated pyroptosis. Nat Rev Mol Cell Biol. 2025;26(7):501–521. doi:10.1038/s41580-025-00837-0
7. Hilbi H, Moss JE, Hersh D, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem. 1998;273(49):32895–32900. doi:10.1074/jbc.273.49.32895
8. Rao Z, Zhu Y, Yang P, et al. Pyroptosis in inflammatory diseases and cancer. Theranostics. 2022;12(9):4310–4329. doi:10.7150/thno.71086
9. Liao Y, Zhang W, Zhou M, Zhu C, Zou Z. Ubiquitination in pyroptosis pathway: a potential therapeutic target for sepsis. Cytokine Growth Factor Rev. 2024;80:72–86. doi:10.1016/j.cytogfr.2024.09.001
10. Kirkik D, Kalkanli Tas S, Tanoglu A. Unraveling the blood microbiome: novel insights into inflammasome responses in crohn’s disease. Eur J Gastroenterol Hepatol. 2024;36(8):975–984. doi:10.1097/MEG.0000000000002695
11. Wei T, Zhang C, Song Y. Molecular mechanisms and roles of pyroptosis in acute lung injury. Chin Med J. 2022;135(20):2417–2426. doi:10.1097/CM9.0000000000002425
12. Wu Q, Ji Z. Pyroptosis in acute pancreatitis: from pathogenesis to treatment. Int J Surg. 2025;10–1097. doi:10.1097/JS9.0000000000003932
13. Parker WAE, Storey RF. The role of platelet p2y(12) receptors in inflammation. Br J Pharmacol. 2024;181(4):515–531. doi:10.1111/bph.16256
14. Wetterholm E, Linders J, Merza M, Regner S, Thorlacius H. Platelet-derived CXCL4 regulates neutrophil infiltration and tissue damage in severe acute pancreatitis. Translational Res. 2016;176:105–118. doi:10.1016/j.trsl.2016.04.006
15. Li L, Tan Q, Wu X, et al. Coagulopathy and acute pancreatitis: pathophysiology and clinical treatment. Front Immunol. 2024;15:1477160. doi:10.3389/fimmu.2024.1477160
16. Sun W, Zhao B, Wang Z, Mao E, Li Y, Che Z. Variations in platelet count associated with the occurrence of infected pancreatic necrosis, surgical intervention, and mortality in acute pancreatitis: a retrospective cohort study. J Inflamm Res. 2025;18:17421–17432. doi:10.2147/JIR.S552811
17. Scherlinger M, Richez C, Tsokos GC, Boilard E, Blanco P. The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol. 2023;23(8):495–510. doi:10.1038/s41577-023-00834-4
18. Wada J, Kanwar YS. Identification and characterization of galectin-9, a novel beta-galactoside-binding mammalian lectin. J Biol Chem. 1997;272(9):6078–6086. doi:10.1074/jbc.272.9.6078
19. Lv Y, Ma X, Ma Y, Du Y, Feng J. A new emerging target in cancer immunotherapy: galectin-9 (LGALS9). Genes Dis. 2023;10(6):2366–2382. doi:10.1016/j.gendis.2022.05.020
20. Ge L, Lin C, Qian J, Wu L, Zhu L. Study on pro-inflammatory effect and mechanism of galectin-9 (LGALS9) in osteoarthritis: exacerbating inflammatory response by activating JNK and ERK1/2 pathways. Int J Biol Macromol. 2024;280(Pt 1):135626. doi:10.1016/j.ijbiomac.2024.135626
21. Jia Q, Che Q, Zhang X, et al. Knockdown of galectin-9 alleviates rheumatoid arthritis through suppressing TNF-alpha-induced activation of fibroblast-like synoviocytes. Biochem Pharmacol. 2024;220:115994. doi:10.1016/j.bcp.2023.115994
22. Kojima K, Arikawa T, Saita N, et al. Galectin-9 attenuates acute lung injury by expanding CD14- plasmacytoid dendritic cell-like macrophages. Am J Respir Crit Care Med. 2011;184(3):328–339. doi:10.1164/rccm.201010-1566OC
23. Xiong H, Xue G, Zhang Y, et al. Effect of exogenous galectin-9, a natural TIM-3 ligand, on the severity of TNBS- and DSS-induced colitis in mice. Int Immunopharmacol. 2023;115:109645. doi:10.1016/j.intimp.2022.109645
24. Wang W, Qin Y, Song H, et al. Galectin-9 targets NLRP3 for autophagic degradation to limit inflammation. J Immunol. 2021;206(11):2692–2699. doi:10.4049/jimmunol.2001404
25. Percie Du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. doi:10.1371/journal.pbio.3000410
26. Schmidt J, Rattner DW, Lewandrowski K, et al. A better model of acute pancreatitis for evaluating therapy. Ann Surg. 1992;215(1):44–56. doi:10.1097/00000658-199201000-00007
27. Gukovskaya AS, Lerch MM, Mayerle J, et al. Trypsin in pancreatitis: the culprit, a mediator, or epiphenomenon? World J Gastroenterol. 2024;30(41):4417–4438. doi:10.3748/wjg.v30.i41.4417
28. Paik S, Kim JK, Shin HJ, Park E, Kim IS, Jo E. Updated insights into the molecular networks for NLRP3 inflammasome activation. Cell Mol Immunol. 2025;22(6):563–596. doi:10.1038/s41423-025-01284-9
29. Feng M, Qin B, Luo F, et al. Qingjie huagong decoction inhibits pancreatic acinar cell pyroptosis by regulating circhipk3/mir-193a-5p/NLRP3 pathway. Phytomedicine. 2024;126:155265. doi:10.1016/j.phymed.2023.155265
30. Lyu S, Liu S, Guo X, et al. Hp-MSCs attenuate severe acute pancreatitis in mice via inhibiting NLRP3 inflammasome-mediated acinar cell pyroptosis. Apoptosis. 2024;29(5–6):920–933. doi:10.1007/s10495-024-01946-5
31. Lu Y, Li B, Wei M, et al. HDL inhibits pancreatic acinar cell NLRP3 inflammasome activation and protect against acinar cell pyroptosis in acute pancreatitis. Int Immunopharmacol. 2023;125(Pt A):110950. doi:10.1016/j.intimp.2023.110950
32. Al Mamun A, Suchi SA, Aziz MA, et al. Pyroptosis in acute pancreatitis and its therapeutic regulation. Apoptosis. 2022;27(7–8):465–481. doi:10.1007/s10495-022-01729-w
33. Boncler M, Bartczak K, Rozalski M. Potential for modulation of platelet function via adenosine receptors during inflammation. Br J Pharmacol. 2024;181(4):547–563. doi:10.1111/bph.16146
34. Carestia A, Godin LC, Jenne CN. Step up to the platelet: role of platelets in inflammation and infection. Thromb Res. 2023;231:182–194. doi:10.1016/j.thromres.2022.10.001
35. Qi Q, Yang B, Li H, et al. Platelet microparticles regulate neutrophil extracellular traps in acute pancreatitis. Pancreas. 2020;49(8):1099–1103. doi:10.1097/MPA.0000000000001631
36. Moar P, Tandon R. Galectin-9 as a biomarker of disease severity. Cell Immunol. 2021;361:104287. doi:10.1016/j.cellimm.2021.104287
37. Cao Z, Leng P, Xu H, Li X. The regulating role of galectin-9 in immune cell populations. Front Pharmacol. 2024;15:1462061. doi:10.3389/fphar.2024.1462061
38. Tull S, Saviano A, Fatima A, et al. Dichotomous effects of galectin-9 in disease modulation in murine models of inflammatory bowel disease. Biomed Pharmacothe. 2025;184:117902. doi:10.1016/j.biopha.2025.117902
39. Guo X, Zhang G, Peng Q, et al. Galectin-9 activates the TLR4-NLRP3 inflammasome pathway and promotes tau pathology in Alzheimer’s disease. Brain Behav Immun. 2026;131:106158. doi:10.1016/j.bbi.2025.106158
40. Karkempetzaki AI, Schatton T, Barthel SR. Galectin-9-an emerging glyco-immune checkpoint target for cancer therapy. Int J Mol Sci. 2025;26(16):7998. doi:10.3390/ijms26167998
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Pyroptosis: A Novel Intervention Target in the Progression of Osteoarthritis
Chang X, Kang Y, Yang Y, Chen Y, Shen Y, Jiang C, Shen Y
Journal of Inflammation Research 2022, 15:3859-3871
Published Date: 11 July 2022
Characterization of Cathepsin B in Mediating Silica Nanoparticle-Induced Macrophage Pyroptosis via an NLRP3-Dependent Manner
Ma L, Han Z, Yin H, Tian J, Zhang J, Li N, Ding C, Zhang L
Journal of Inflammation Research 2022, 15:4537-4545
Published Date: 8 August 2022
Inhibition of Dectin-1 Alleviates Neuroinflammatory Injury by Attenuating NLRP3 Inflammasome-Mediated Pyroptosis After Intracerebral Hemorrhage in Mice: Preliminary Study Results
Ding Z, Zhong Z, Wang J, Zhang R, Shao J, Li Y, Wu G, Tu H, Yuan W, Sun H, Wang Q
Journal of Inflammation Research 2022, 15:5917-5933
Published Date: 19 October 2022
Lactobacillus Plantarum Promotes Wound Healing by Inhibiting the NLRP3 Inflammasome and Pyroptosis Activation in Diabetic Foot Wounds
Wang X, Li X, Liu J, Tao Y, Wang T, Li L
Journal of Inflammation Research 2024, 17:1707-1720
Published Date: 16 March 2024
The Impact of NLRP3 Inflammasome on Osteoblasts and Osteogenic Differentiation: A Literature Review
Yang Z, Xu J, Kang T, Chen X, Zhou C
Journal of Inflammation Research 2024, 17:2639-2653
Published Date: 29 April 2024