Back to Journals » Journal of Inflammation Research » Volume 14
Platelet-Derived Growth Factor Regulates the Biological Behavior of Oral Mucosal Fibroblasts by Inducing Cell Autophagy and Its Mechanism
Authors Wang J, Yang L, You J, Wen D, Yang B, Jiang C
Received 1 April 2021
Accepted for publication 24 June 2021
Published 17 July 2021 Volume 2021:14 Pages 3405—3417
DOI https://doi.org/10.2147/JIR.S313910
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Jie Wang,1,2 Lina Yang,2 Jialing You,2 Dada Wen,2 Bo Yang,2 Canhua Jiang1
1Department of Oral and Maxillofacial Surgery, Xiangya Hospital, Central South University, Changsha, 410078, People’s Republic of China; 2Department of Immunology, Xiangya School of Medicine, Central South University, Changsha, 410078, People’s Republic of China
Correspondence: Canhua Jiang
Department of Oral and Maxillofacial Surgery, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, 410078, Hunan, People’s Republic of China
Tel/Fax +86-731-89753046
Email [email protected]
Objective: To explore the effect of platelet-derived growth factor (PDGF) on oral mucosal fibroblast autophagy and further elucidate the molecular mechanism by which PDGF-BB regulates the biological behavior of oral mucosal fibroblasts by inducing autophagy.
Methods: Primary oral mucosal fibroblasts were isolated and cultured by the tissue block and trypsin methods and identified by indirect immunofluorescence vimentin detection. We detected the autophagy marker Beclin-1 and fibrosis marker Col-I of the primary oral mucosal fibroblasts at different time points after stimulating the fibroblasts with different PDGF-BB concentrations by Western blotting and determined the best experimental concentration and stimulation time of PDGF-BB. Then, indirect immunofluorescence, Western blotting, and quantitative real-time polymerase chain reaction (PCR) were used to detect the effect of PDGF-BB on the expression of autophagy-related and fibrotic proteins before and after 3-methyladenine (3-MA) intervention. Additionally, the effect of 3-MA on the proliferation and migration of primary oral mucosal fibroblasts stimulated by PDGF-BB was detected by the MTT method and a scratch experiment. The effect of PDGF-BB on Beclin-1 and phosphatidylinositol-3 kinase class 3 (PI3KC3) interaction was detected by co-immunoprecipitation.
Results: The results demonstrated that PDGF-BB could induce autophagy of the oral mucosal fibroblasts, showing a certain time and dose correlation. It induced cell autophagy through Beclin-1 and PI3KC3 interaction to promote the proliferation, migration, conversion, and collagen synthesis of the fibroblasts. However, 3-MA inhibited the combination of Beclin-1 and PI3KC3 and weakened the fibroblasts’ proliferation, migration, conversion, and collagen synthesis activities.
Conclusion: Overall, PDGF-BB induces autophagy through the Beclin-1 pathway to regulate the biological behavior of oral mucosal fibroblasts.
Keywords: PDGF-BB, primary oral mucosal fibroblasts, autophagy, 3-MA, biological behavior, signaling pathway
Introduction
Oral submucous fibrosis (OSF) is an insidious and chronic oral mucosal disease, which has been recognized as a precancerous lesion of the oral cavity. Its incidence is increasing year by year and its biological behavior is highly malignant, which play a huge physical and mental burden on patients. It is prevalent primarily in East and Southeast Asia and has a high incidence in Taiwan and Hunan1,2 among those chewing betel nuts, which can increase the risk of OSF by 109~287 times.3 Studies have found that the arecoline in betel nuts is the primary factor inducing OSF, which is mainly caused by continuous chemical and mechanical stimulation that causes chronic persistent inflammation and induces cells to release inflammatory mediators. Simultaneously, collagen synthesis increases and degradation decreases, leading to collagen fiber deposition in oral tissues, an important cause of fibrosis.4,5 OSF is a process of multi-cellular multi-molecule participation, but the precise pathogenesis of OSF is not fully clear.
Various fibrotic diseases have one thing in common at the cellular level: Phenotypic transformation of fibroblasts (FBs) in quiescent state into myofibroblasts (MFBs) occurs under the action of certain stimulating factors. Compared with FBs, MFBs has an increased contractibility of smooth muscle cells (expression of α-smooth muscle actin (α-SMA)) and can synthesize a large amount of ECM;6,7 simultaneously, ECM degradation is reduced, leading to the occurrence of fibrosis. Studies have found that the process of OSF is related to a variety of inflammatory cytokines, such as platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β). The expression is upregulated in tissues and is mainly distributed in the membrane or cytoplasm of FBs and vascular endothelial cells.8,9 PDGF is a strong stimulator of the proliferation and differentiation of FBs in the oral mucosal tissue. It activates PDGF-receptor tyrosine phosphorylation by binding to the corresponding receptor, promotes cell mitosis, and increases ECM synthesis and secretion.10,11 There are five subtypes of PDGF: PDGF-AA, PDGF-BB, PDGF-CC, PDGF-DD, and PDGF-AB. Among them, PDGF-BB plays an important role in fibrotic diseases. Studies have shown that in tissue injury and inflammation, PDGF-BB can stimulate the proliferation and migration of hepatic stellate cells (HSC)12 or renal tubular mesenchymal cells,13 and induce it to be transformed into MFBs, thereby synthesize a large amount of collagen, leading to organ structural dysfunction and promote the occurrence of fibrosis disease. According to related literature reports, PDGF-BB can induce autophagy in vascular smooth muscle cells,14 and Li et al15 found that TGF-β induces an increase in the autophagy level of oral mucosal FBs and that inhibiting autophagy can reduce the level of type I collagen expression. As such, we speculate that PDGF-BB may participate in the occurrence and development of OSF by inducing autophagy in oral mucosal FBs.
In the cells of higher vertebrates, autophagy is ubiquitous,16 mainly dealing with damaged, degenerated, senescent, and out-of-function organelles, denatured proteins, nucleic acids, and other biological macromolecules through lysosomal degradation pathways.17,18 It includes micro-autophagy, macro-autophagy, and molecular chaperone-mediated autophagy. Among these, macro-autophagy has been studied the most extensively and is what we usually refer to as autophagy.19 The formation and regulation of autophagy is a complex process involving multiple molecules. Among the many autophagy pathways, mammalian target of rapamycin (mTOR), Beclin-1, and p53 are the three most common. Under physiological conditions, cells maintain a low level of background autophagy intensity. When the body is infected, mechanically damaged, or nutritionally deficient, autophagy is activated that provides essential nutrients for cell reconstruction, regeneration, and repair, thereby maintaining a stable cell environment. However, the abnormal activation of autophagy is widely involved in the pathophysiological processes of many diseases such as infection, tumor, and organ fibrosis. Studies have found that the regulation of miR-200b by DNMT3A can control cardiac FB autophagy in the process of cardiac fibrosis20 and slow down the occurrence and development of the disease. When HSCs is impaired in the mouse hepatic fibrosis model, the lack of autophagy in the mice reduces fibrogenesis and matrix accumulation. Therefore, selective reduction of the autophagy activity of fibrotic cells in the liver and other tissues can be used to treat fibrotic diseases.21 The role of autophagy in renal fibrosis is bidirectional. The unilateral ureteral obstruction model exhibits a time-dependent induction of autophagy with renal tubular atrophy, tubular cell death, and interstitial fibrosis, while autophagy inhibitor 3-methyladenine (3-MA) enhances obstructive renal tubular cell apoptosis and interstitial fibrosis. On the contrary, however, several studies have shown that the activation of autophagy leads to renal fibrosis.22,23 It has been reported that in pulmonary fibrosis ATG4B protease and autophagy protect epithelial cells from bleomycin-induced stress and apoptosis and also play a vital role in regulating inflammation and fibrosis.24 A number of studies have shown that autophagy is related to fibrosis of the heart, liver, kidneys, lungs, and other organs, and more and more evidence is demonstrating that the mechanism of autophagy is closely related to the occurrence, development, and outcome of fibrotic diseases.25
In essence, PDGF-BB works by binding to PDGF receptor-β (PDGFR-β). Han et al8 found that in the FB, epithelial, and tissue vascular endothelial cell membranes of OSF patients, the expression level of PDGFR-β is upregulated, and the interaction of PDGF and PDGFR can induce phosphorylation of PDGFR and promote the activation of phosphatidylinositol-3 kinase (PI3K).26 PI3K has three subtypes: PI3K class 1 (PI3KC1), PI3KC2, and PI3KC3 (a homolog of Vps34). After phosphorylation, it forms a complex with Beclin-1 to promote the occurrence of autophagy.27,28 Therefore, this study intended to explore the effect of PDGF-BB on the autophagy of oral mucosal FBs and further clarify the molecular mechanism of how it induces autophagy to regulate the biological behavior of oral mucosal FBs through the interaction of Beclin-1 and PI3KC3. In conclusion, these findings suggest that the interaction of autophagy with PDGF-BB is a key mechanism for the development of OSF and that inhibition of the autophagy pathway may provide a new way to prevent fiber formation.
Materials and Methods
Primary FB Isolation and Culture
The specimens were the gum tissues obtained from the extraction of molar teeth in a healthy 22-year-old Chinese female periodontal patient on October 23, 2019. The patient was in good health without systemic disease and periodontal inflammation. This study was conducted in accordance with the declaration of Helsinki. This study was conducted with approval from the Ethics Committee of Xiangya Hospital of Central South University. Written informed consent was obtained from this participant.
The normal human oral mucosal tissue block was packed in dulbecco’s modified eagle medium (DMEM) (Gibco) containing 15% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. The tissue block was washed three times with phosphate buffer saline (PBS) containing double antibodies, minced, and inoculated into a culture bottle before being cultured in a 37°C incubator in a humid environment containing 5% CO2 for 15–20 days. Then, it was digested with 0.25% trypsin; the digestion was terminated with DMEM containing 15% fetal bovine serum (FBS). After centrifugation and resuspension, the cells were incubated in a 37°C incubator in a humid environment containing 5% CO2. The primary oral mucosal FBS was automatically purified after two passages to obtain uniform oral mucosal FBS. Oral mucosal FBS after two passages were used for subsequent experiments.
Ribonucleic Acid (RNA) Extraction and Quantitative Real-Time Polymerase Chain Reaction (PCR) Analysis
Total RNA extractor lysate was used to extract the total Messenger RNA (mRNA) of the cells, which was reverse transcribed into complementary Deoxyribonucleic acid (cDNA) according to the instructions of the cDNA kit used. The mRNA level was quantified by Hieff qPCR SYBR Green Master Mix (Yeasen, Shanghai, China) and detected by RT-qPCR (ABI7500, U.S.A.). The PCR reaction conditions were as follows: pre-denaturation at 95°C for five minutes, denaturation at 95°C for 10 s, annealing/extension at 60°C for 30 s, and a total of 40 cycles of denaturation to extension. The relative expression levels of LC3 mRNA, Beclin-1 mRNA, COl-I mRNA, and α-SMA mRNA were calculated by the 2−ΔΔCt method relative to GAPDH mRNA expression. The primer list is given in Table 1.
 |
Table 1 The Primers List |
Western Blot Analysis
Proteins were separated from the cell lysates using Radio Immunoprecipitation Assay (RIPA) lysate solution, and the protein concentration was determined by a Bicinchoninic acid (BCA) kit. Then, 30 μg protein was separated by 12% SDS-PAGE and transferred to a PVDF membrane. The membrane was incubated for an hour in blocking buffer containing 5% skimmed milk, followed by incubation overnight at 4°C with the primary antibodies, which included GAPDH, LC3, Beclin-1, and COl-I, and then incubation with the secondary antibodies in the dark for two hours at room temperature. The membranes were washed using 0.1% Tween 20/TBS solution three times. The blots were visualized by an enhanced chemiluminescence system (Amersham Pharmacia Biotech, Arlington Heights, IL). Each blot was repeated three times.
Immunofluorescence Assay
The cells were seeded at 1×104 in a pre-selected 24-well plate with spreading slides and incubated with 10 mM 3-MA for 2 h before the corresponding conditions were added to culture for 24 h. Then, the cell slides were washed with pre-chilled PBS and fixed with 4% paraformaldehyde at room temperature for 30 min, after which the membrane was penetrated with 0.2% Triton-100 at 4°C for 10 min and blocked with 1% BSA for one hour at 37°C. The cells were incubated with primary antibodies vimentin (1:100, CST), Beclin-1 (1:200, Bioworld), and LC3 (1:500, CST) at 4°C overnight before being incubated with a fluorescently labeled secondary antibody (Cwbiotech, China) and Hoechst (1:500) simultaneously. The images were analyzed under a fluorescent microscope (Ficol, LEICA DMi8).
Oral Mucosal FB Proliferation Assay
The MTT method was used to evaluate cell proliferation. Cells were seeded in a 96-well plate with 200 μL/well (1 × 103cells/well). When conditioned medium was added for 0, 24, 48, 72, and 96 h, The cells were incubated with 20 μL of 5-mg/mL MTT solution for 4 H and added into the test hole on the same day of measurement. After the old culture medium was discarded, 150 μL DMSO solution was transferred into a new 96-well plate, which was shaken at a low speed for 10 min to examine the absorbance at 490 nm using a microplate reader. The experiment was repeated three times.
Wound Healing Assay
Scratch experiments were used to assess cell migration. The cells were evenly seeded in a six-well culture plate with conditioned medium for 24 h. When the cell density reached over 90%, the cells were scraped with a P200 pipette tip to create a scratch. The cells were washed with PBS three times to remove the suspended cells. The adherent cells were incubated with DMEM 0.1% FBS medium and photographed at 0, 12, 18, and 24 h post-scratch under a 10× objective.
Co-Immunoprecipitation
The protein interaction was evaluated by the immunoprecipitation method. RIPA was used to lyse the cells to obtain total protein, and the protein concentration was determined by the BCA method. The concentration of each group was 1 μg/μL. Agarose beads were added for adsorption and shaken at 4°C for two hours. The total protein was centrifuged at 2,500 rpm × 30 s at 4°C before the supernatant was leaved and incubated with the primary antibodies (Beclin-1 = 1:200, PI3KC3 = 1:50) at 4°C overnight. Then, beads were added again and shaken at 4°C for two hours. The supernatant was discarded by centrifugation at 4°C, the remaining cells were added to an EP tube with a loading buffer, given a boiling water bath, and denatured for five minutes.
Statistical Analysis
The GraphPad Prism software was used for the data plotting, and all statistical analyses were performed using SPSS 18.0 (SPSS, Chicago, IL, U.S.A.). The results were expressed as the mean ± standard deviation (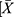 ± S). Comparisons between two groups were evaluated by the Student’s t-test. The inspection level was α = 0.05, and a P-value <0.05 indicated statistical significance.
± S). Comparisons between two groups were evaluated by the Student’s t-test. The inspection level was α = 0.05, and a P-value <0.05 indicated statistical significance.
Results
Isolation and Culture of High-Purity Primary Oral Mucosal FBs
To obtain primary oral mucosal FBs, oral mucosal FBs were separated by the tissue block and trypsin methods and observed under an inverted microscope after 15 days. Figure 1Aa and b represent the morphology of the cells swimming out of the tissue block under different magnifications. The cells isolated after digestion were bright and spherical with a clear outline and single or aggregated into a mass. Figure 1Ac and d display the growth states of the FBs at different magnifications after purification. The cell morphology was uniform, long, and fusiform, with clear boundaries between cells (Figure 1A). The expression of vimentin in the primary cultured cells was further detected by Immunological Fluorescence Assay (IFA). As shown in Figure 1B, vimentin was expressed in the cytoplasm of almost all cells (>99%). The cells had obvious synapses and typical FB characteristics (Figure 1B). The results showed that primary oral mucosal FBs were successfully isolated and cultured.
 |
Figure 1 Isolation and culture of high-purity primary oral mucosal FBs. (A) Morphological observation of primary cells under inverted microscope. (a and b) The cells swam out from the tissue blocks and expanded into the surrounding area. (a) ×100; (b) ×200. (c and d) Morphological observation of cell subculture. (c) ×100; (d) ×200. (B) The expression of vimentin in the primary cells under a fluorescent microscope. When the cell density reached 60–80%, the cells were taken out, and rabbit anti-human vimentin antibody was used as the primary antibody; goat anti-rabbit IgG-Cy2 was used as the secondary antibody to detect vimentin expression. Blue represents the nucleus, and green represents the vimentin protein. The results showed that the cells expressed vimentin positively and that the rate was over 99%. |
Best Experimental Concentration of 3-MA
To screen out the best experimental concentration of 3-MA, the concentration was divided into seven groups: 0, 2.5, 5, 7.5, 10, 15, 20, and 25 mmol/L. After stimulating oral mucosal FBs with these 3-MA concentrations for 24 h, the cells’ proliferation was detected by the MTT method, and the OD490nm value was detected by a microplate reader. The results showed that the half-maximal inhibitory concentration rate of the 3-MA was about 10 mmol/L (Figure 2), which was thus used as the subsequent experimental concentration.
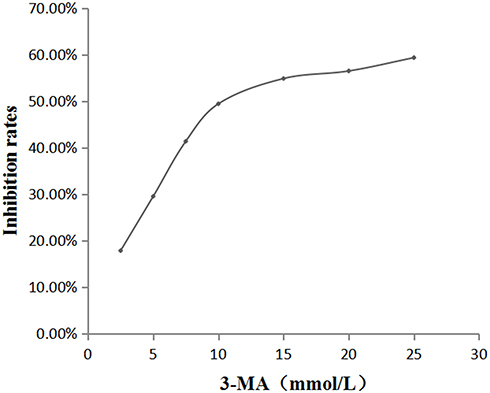 |
Figure 2 The effect of different concentrations of 3-MA on the amount of oral mucosal FBs. |
Effect of Different Concentrations of PDGF-BB on the Expression of Col-I and Beclin-1
After stimulating oral mucosal FBs with different concentrations of PDGF-BB (0, 10, 20, 30, 40, 60 ng/mL) for 24 h and extracting the total cellular protein, the expressions of fibrosis-related protein Col-I and autophagy-related protein Beclin-1 were detected by Western blot analysis. The results showed that the expression of COl-I changed with the concentration, showing a trend of increasing initially (reaching a peak at 40 ng/mL) and then decreasing. The expression of Beclin-1 also changed with the concentration and was highest at 40 ng/mL as well (Figure 3).
 |
Figure 3 Western blotting was used to detect the Col-I and Beclin-1 protein expression of oral mucosal FBs stimulated by different PDGF-BB concentrations. (A) Different concentrations of PDGF-BB (0, 10, 30, 40, 60 ng/mL) were used to stimulate the FBs for 24 h. (B) The effect of the different PDGF-BB concentrations on the Col-I and Beclin-1 protein expression in the FBs. When the Col-I in the 40-ng/mL group is compared with that in the 0-, 10-, 30-ng/mL groups, ***P < 0.001; when the Beclin-1 in the 40-ng/mL group is compared with that in the 0-, 10-, 30-, and 60-ng/mL groups, #P < 0.05. |
The Effect of PDGF-BB Stimulation Time on the Expression of Col-I and Beclin-1
Primary oral mucosal FBs were stimulated with PDGF-BB (40 ng/mL) at different times (6, 12, 24, 48, 72 h); a negative control group without stimulation was created as well. The expression of Col-I and Beclin-1 in the cells at the different time points was detected by Western blotting. Compared with the control group, fibrosis-related protein Col-I increased as the stimulation time increases. Moreover, autophagy-related protein Beclin-1 first increased and then decreased as the stimulation time increased, reaching a peak at 24 h (Figure 4). These findings suggested that the longer PDGF-BB stimulates primary oral mucosal FBs, the higher the incidence of fibrosis. Additionally, PDGF-BB could induce the occurrence of autophagy, which is time-dependent, increasing initially and then decreasing.
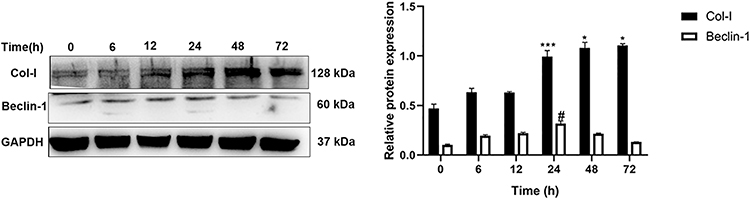 |
Figure 4 Western blotting was used to detect the Col-I and Beclin-1 protein expression in oral mucosal FBs after different PDGF-BB stimulation times. ***Indicates the comparison of Col-I in the 24-h group with that in the 6- and 12-h groups; ***P < 0.001. *Indicates the comparison of Col-I in the 48- and 72-h groups with that in the 24-h group; *P < 0.05. #Indicates the comparison of Beclin-1 in the 24-h group with that in the rest of the time groups; #P < 0.05. |
PDGF-BB Induces Autophagy of Primary Oral FBs and the Expression of Col-I
Regarding the effect of PDGF-BB on primary oral mucosal FBs, RT-qPCR showed that the expressions of Col-I, Beclin-1, and LC3 in the PDGF-BB group were significantly higher than those in the control group and that the difference was statistically significant (P < 0.05) (Figure 5A). The expression levels of Col-I, Beclin-1, and LC3 were also detected by Western blotting. Compared with the control group, the expressions of these proteins in the PDGF-BB group were significantly increased, and the difference was statistically significant (P < 0.05) (Figure 5B). The results of the indirect immunofluorescence showed that there was no obvious red fluorescence in the control group; however, the PDGF-BB group showed red fluorescence. The intensity of the red fluorescence in the PDGF-BB group was significantly higher than that in the control group (Figure 5C). Overall, these findings revealed that PDGF-BB can promote the autophagy of primary oral mucosal FBs while stimulating primary oral mucosal FB fibrosis.
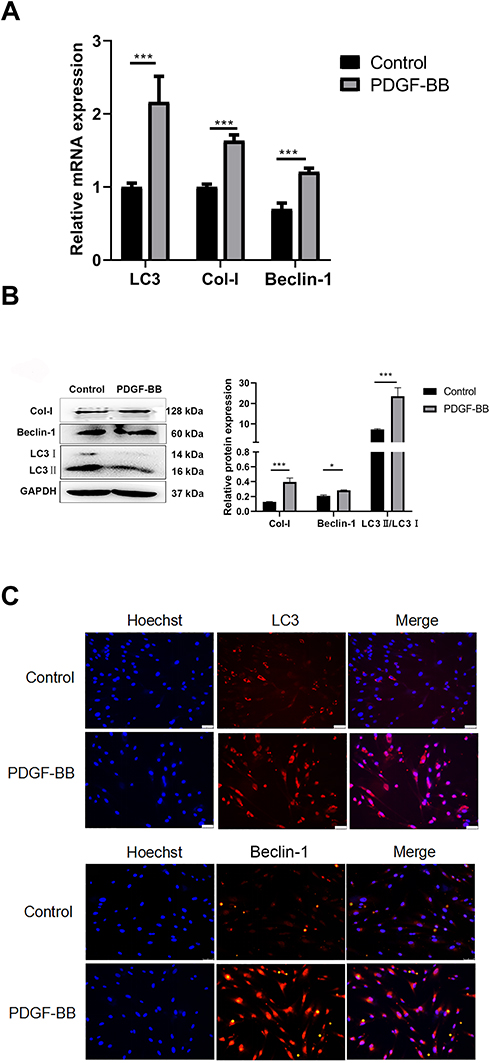 |
Figure 5 PDGF-BB promoted the fibrosis and activated the autophagy of the oral mucosal FBs. (A) Detection of the expression of Col-I, Beclin-1, and LC3 at the mRNA level by RT-qPCR. Compared with the control group, the PDGF-BB group’s data are represented as the means ± SD of at least three independent experiments. P < 0.05; (***P < 0.01). (B) Western blot detection of autophagy and fibrosis in primary oral mucosal FBs. The expression of fibrosis-related protein Col-I and autophagy-related proteins Beclin-1 and LC3 in primary oral mucosal FBs were detected after PDGF-BB stimulation for 24 h. PDGF-BB group vs control group, P < 0.05. (*P < 0.05, ***P < 0.01). (C) Indirect immunofluorescence detection of cell autophagy (×200). Indirect immunofluorescence was used to detect the expression and localization of autophagy-related proteins LC3 and Beclin-1 in primary oral mucosal FBs after PDGF-BB stimulation for 24 h. Hoechst 33342 was used to stain the nucleus; rabbit anti-LC3 and rabbit anti-Beclin-1 were the primary antibodies, and Cy3-goat anti-rabbit IgG was the secondary antibodies. LC3 and Beclin-1 showed red fluorescence. |
3-MA Can Downregulate the Expression of Col-I, α-SMA, Beclin-1, and LC3 in Oral Mucosal FBs Induced by PDGF-BB
After autophagy inhibitor 3-MA was pre-incubated with primary oral mucosal FBs for 2 h, PDGF-BB (40 ng/mL) condition culture medium was used to continue the culturing for 24 h. The expressions of Col-I, α-SMA, Beclin-1, and LC3 at the mRNA level were detected by RT-qPCR. The results showed that the expression levels in the PDGF-BB group were significantly higher than those in the control group, 3-MA group, and 3-MA/PDGF-BB group; the difference was statistically significant (P < 0.05) (Figure 6A).
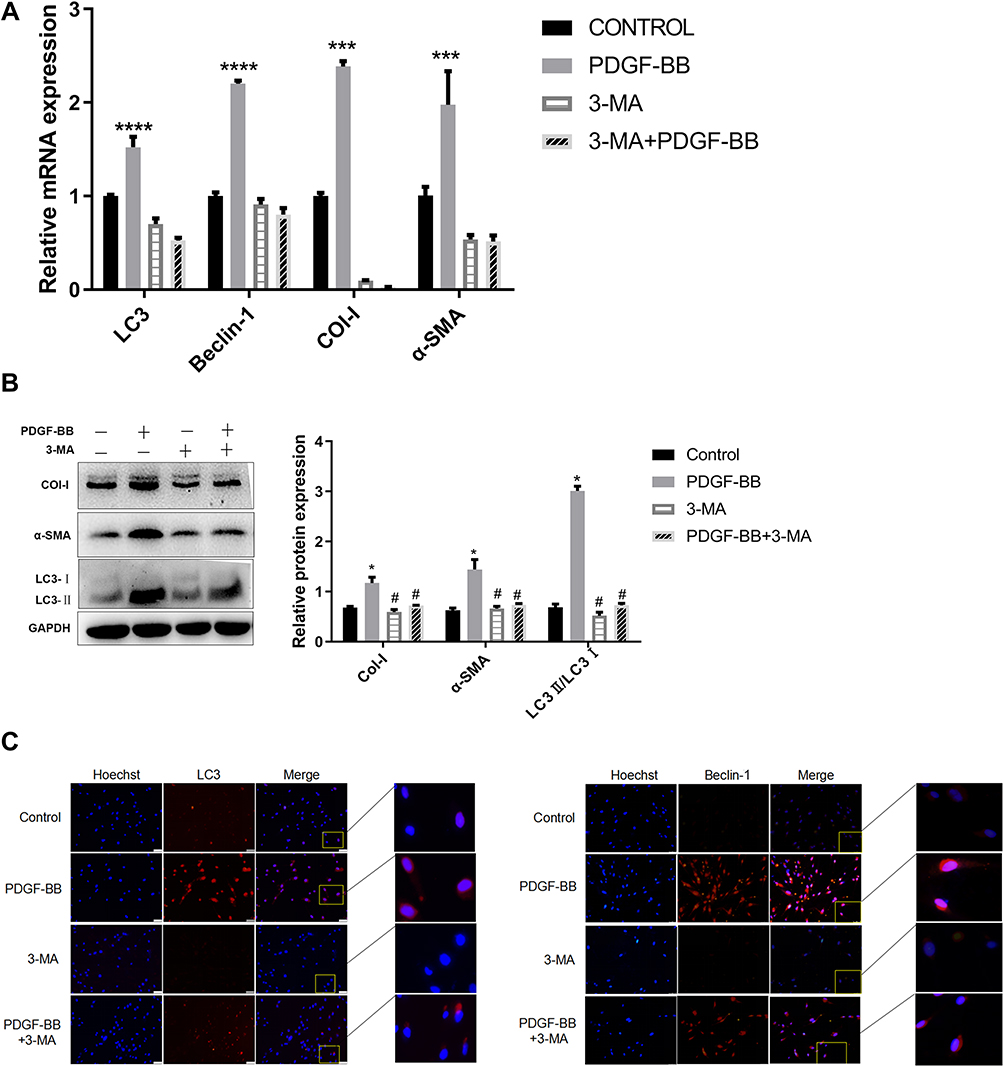 |
Figure 6 Effect of autophagy inhibitor 3-MA on PDGF-BB-induced oral mucosal FB fibrosis and autophagy. (A) Detection of the expression of Col-I, α-SMA, Beclin-1, and LC3 at the mRNA level by RT-qPCR. The effects of PDGF-BB and 3-MA on the autophagy and fibrosis of primary oral mucosal FBs. The data are represented as the means ± SD of at least three independent experiments. *Indicates comparison with the control group, P < 0.05; #Indicates comparison with the PDGF-BB group, P < 0.05. (B) Detection of autophagy and fibrosis in primary oral mucosal FBs by Western blotting; effects of PDGF-BB and 3-MA on the autophagy and fibrosis of the FBs. *Indicates comparison with the control group, P < 0.05; #Indicates comparison with the PDGF-BB group, P < 0.05. (C) Indirect immunofluorescence was used to detect the effect of 3-MA on the PDGF-BB-stimulated autophagy and the expression of autophagy-related proteins LC3 and Beclin-1 of the FBs. Hoechst 33342 was used to stain the nucleus; rabbit anti-LC3 and rabbit anti-Beclin-1 were the primary antibodies, and Cy3-goat anti-rabbit IgG was the secondary antibodies. LC3 and Beclin-1 showed red fluorescence. |
Upon extracting the total protein of each group for Western blot analysis, the protein expressions of Col-I, Beclin-1, and LC3 in the PDGF-BB group were significantly higher than those in the control group, and the difference was statistically significant (P < 0.05). Moreover, the protein expressions of Col-I, Beclin-1, and LC3 in the 3-MA and 3-MA/PDGF-BB groups were lower than those in the PDGF-BB group, and the difference was statistically significant (P < 0.05) (Figure 6B).
The IFA results showed that regarding LC3 and Beclin-1, the PDGF-BB group had the strongest red fluorescence, the 3-MA group had almost no fluorescence, and the 3-MA/PDGF-BB group had stronger fluorescence than the control and 3-MA groups (Figure 6C). These results further indicated that PDGF-BB can induce autophagy in oral mucosal FBs and that 3-MA can inhibit this autophagy. Therefore, PDGF-BB may regulate the biological behavior of oral mucosal FBs by inducing autophagy.
3-MA Attenuates PDGF-BB’s Effect on the Proliferation and Migration of Oral Mucosal FBs
The MTT method was used to examine the effect of 3-MA on the PDGF-BB stimulation of primary oral mucosal FB proliferation. The results showed that the OD490nm value of the PDGF-BB group was higher than that of the control and 3-MA groups at 48 h (P < 0.05) and that the difference between the groups increased with time. The difference was highly significant (P < 0.01). At 72 and 96 h, the OD490nm value of the 3-MA/PDGF-BB group was significantly lower than that of the PDGF-BB group (Figure 7A). These data demonstrated that 3-MA can reduce the ability of PDGF-BB to promote the proliferation of oral mucosal FBs.
 |
Figure 7 3-MA attenuated the effect of PDGF-BB on the proliferation and migration of the oral mucosal FBs. (A) The effect of PDGF-BB on the proliferation of the FBs at different time points was detected by the MTT method. *Indicates vs control group, P < 0.05; #Indicates vs PDGF-BB group, P < 0.05. (B) The migration of the oral mucosal FBs was detected by cell scratch assay. Real-time photographing was performed to detect the relative migration width of each group of cells (×50), comparing the average relative mobility of the cells at 6, 12, and 24 h. The data are represented as the means ± SD of at least three independent experiments. *P < 0.05 vs control group; #P < 0.05 vs PDGF-BB group. |
The cell scratch test is a common method for detecting cell migration ability. The stronger the cell migration ability, the faster peripheral cells grow toward the central scratch area and the smaller the average scratch width. In this study, the scratch width in the PDGF-BB group gradually reduced as the culture time increased. Moreover, the scratch width in the 3-MA and 3-MA/PDGF-BB groups also gradually reduced as the culture time increased but was significantly smaller compared with that of the PDGF-BB group (Figure 7B). The average relative mobility of the cells in the PDGF-BB group was significantly greater than that in the control, 3-MA, and 3-MA/PDGF-BB groups (P < 0.05 in each group) (Figure 7B). These findings confirmed that 3-MA can weaken the ability of PDGF-BB to promote the migration of oral mucosal FBs.
PDGF-BB Induces Autophagy Through Beclin-1 and PI3KC3 Interaction
The cell lysates of each group were extracted and subjected to mutual immunoprecipitation analysis with Beclin-1 and PI3KC3 antibodies. The results showed that there was no protein band in the negative control mixed with IgG, indicating that there was no non-specific protein interference. However, Beclin-1 protein was detected in the PI3KC3 precipitated protein, indicating that the two interacted. The interaction between Beclin-1 and PI3KC3 in the PDGF-BB group was significantly stronger than that in the control, 3-MA, and 3-MA/PDGF-BB groups (Figure 8). The combination of the two decreased with the addition of 3-MA. These results demonstrated that PDGF-BB can induce interaction between Beclin-1 and PI3KC3, promoting oral mucosal FB autophagy, and that 3-MA can reduce the autophagy intensity by inhibiting the proteins’ combination.
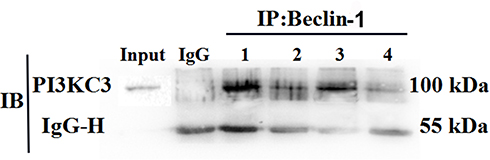 |
Figure 8 Co-immunoprecipitation to detect the expression levels and interactions of Beclin-1 and PI3KC3 in the different groups. This figure shows the result of detecting PI3KC3 with anti-PI3KC3 antibody as the primary antibody; inorganic phosphorus precipitated the Beclin-1 protein. Here, 1, 2, 3, and 4 represent the PDGF-BB group, control group, PDGF-BB + 3-MA group, and 3-MA group, respectively. |
Discussion
Platelet-derived growth factor is a cytokine that promotes cell activation, division, and proliferation, and PDGF-BB is a common pro-fibrotic factor whose expression increases in fibrotic diseases and is closely related to OSF formation.8,9 Various physical, chemical, genetic, immune defense, and nutritional factors affect the occurrence and development of OSF. However, the body is immune to the fibrosis caused by PDGF-BB through a series of mechanisms, among which autophagy plays an important role.
In this study, we provide strong evidence that PDGF-BB can induce autophagy in primary oral mucosal FBs. To start, we separated and purified primary oral mucosal FBs by the tissue block method combined with trypsin digestion. To identify primary oral mucosal FBs, we used vimentin antibody. Vimentin is mainly expressed in interstitial cells.29 Oral mucosal FBs can express vimentin in cytoplasm, but epithelial cells cannot. Thus, the results proved that high-purity primary oral mucosal FBs were obtained.
To study the effect of PDGF-BB on the autophagy of oral mucosal FBs and their collagen synthesis and transformation, we used RT-qPCR, Western blotting, and IFA experiments to verify that PDGF-BB stimulates cells to trigger an obvious autophagy response. After PDGF-BB stimulation for 24 h, a significant increase in Beclin-1 and LC3 expression was detected by RT-qPCR and Western blotting, accompanied by an increase in Col-I and α-SMA expression. The protein expressions in PDGF-BB group were significantly stronger than those in the control group (P < 0.05). Moreover, the fluorescence of Beclin-1 and LC3 in the PDGF-BB group was significantly enhanced by IFA. The increase in the expression of the autophagy-related markers also indicated that the autophagy level was enhanced. Thus, these results revealed that PDGF-BB promoted the occurrence of primary oral mucosal FB fibrosis and enhanced the FBs’ autophagy level.
To select the best experimental PDGF-BB concentration and best detection time point, we consulted the literature and found that PDGF-BB can induce autophagy in human umbilical vein endothelial cells and that its intensity is significantly higher than that of PDGF-BB stimulation in the middle stage of stimulation (24 h) Early stage (6 h).30 In this experiment, to study the difference in the expressions of Beclin-1 and Col-I in primary oral mucosal FBs stimulated by different concentrations of PDGF-BB at different times, Western blotting was performed. The expressions of Col-I and Beclin-1 in the 40 ng/mL group were significantly increased. The expression of Col-I increased over time, while Beclin-1 first increased and then decreased with a certain time–dose correlation. Therefore, we use 40 ng/mL PDGF - BB to stimulate oral mucosa FBs 24 h. The results showed that in the early stage of PDGF-BB stimulation, the FBs’ demand for nutrients increased, so PDGF-BB induced autophagy to maintain the cells’ survival. The autophagy level reached a peak at 24 h; hence, related proteins were degraded, and the autophagy intensity gradually weakened. After 24 h, the cells gradually adapted to the environment, and the autophagy weakened further. It has been reported in the literature that autophagy is related to the pathophysiological process of many diseases, such as cancer, metabolic and neurodegenerative diseases, and cardiovascular and pulmonary diseases.31,32 Therefore, it is vital to study the role of autophagy in OSF. Overexpression of autophagy marker LC3 has been observed in the tissue samples of OSF patients, and many studies have shown that pro-fibrotic cytokines (IL-1, IL-6, TGF-β, PDGF, bFGF, and IGF) promote collagen synthesis.33 According to relevant literature reports, TGF-β stimulation of primary oral mucosal FBs can induce Beclin-1 and LC3 overexpression.15 Our research showed that PDGF-BB can induce autophagy of FBs in the oral mucosa and promote the synthesis of Col-I. Therefore, PDGF-BB-mediated increase of Beclin-1 and LC3 may be a potential molecular mechanism that promotes the development of fibrosis.
According to relevant literature reports, pathologically activated autophagy is associated with a variety of fibrotic diseases34–36 as well as with collagen release in OSF.37 Therefore, we further studied the relationship between OSF and autophagy. After 3-MA intervention, the IFA results showed that the level of autophagy decreased, while the RT-qPCR and Western blotting results found that the expressions of Col-I and α-SMA were significantly reduced. When α-SMA increases, this means that FBs have transformed into MFBs, which can greatly enhance collagen synthesis, participate in wound reconstruction and aggravate organ fibrosis. These results indicated that 3-MA can inhibit the autophagy of primary oral mucosal FBs induced by PDGF-BB, reducing the ability of PDGF-BB-stimulated oral mucosal FBs to synthesize collagen and slow the occurrence of fibrosis. We explored the effect of 3-MA on PDGF-BB-induced oral mucosal FB proliferation and migration and found that inhibiting autophagy weakened these processes. The results indicated that PDGF-BB promotes the proliferation and migration of primary oral mucosal FBs by inducing autophagy. The key role of autophagy in cell survival was confirmed in a study on Atg gene knockout mice. Mice lacking Atg3, Atg5, Atg7, Atg9, or Atg16L1 failed to induce autophagy due to starvation after the interruption of the placental nutrient supply and died on the day of birth.38,39 In short, autophagy is a complex process, and its mechanism remains to be elucidated. Our current research showed that inhibiting autophagy can reduce the expression of Col-I and weaken the proliferation and migration effects of PDGF-BB on oral mucosal FBs. Therefore, autophagy may mediate a mechanism of fibrosis.
The activation of the autophagy pathway and its effect on promoting fibrosis suggest that autophagy may be a potential target of new anti-fibrotic methods. Beclin-1 plays an important role in autophagy regulation and is a BH3-only protein. Moreover, PI3K subtype PI3KC3 can phosphorylate the third-site protein of phosphatidylinositol in eukaryotes, thereby forming a complex with Beclin-1 (Beclin-1–PI3KC3) and promoting the occurrence of autophagy.27,28 Intracellular environment disorders, neurodegenerative diseases, aging, and tumors are related to loss of and abnormality in the structure and function of the Beclin-1–PI3KC3 complex.40 Our previous study found that PDGF-BB can activate the PI3K pathway and its phosphorylation, indicating that this PI3K subtype may be combined with Beclin-1 to induce autophagy. Therefore, this study tested the interaction between Beclin-1 and PI3KC3 in each group through a Co-immunoprecipitation (Co-ip) experiment and found that PDGF-BB can induce autophagy through the interaction between them. The Co-ip results indicated that PDGF-BB activates PI3KC3, which forms a complex with Beclin-1 to promote autophagy to regulate the proliferation, migration, transformation, and collagen synthesis of oral mucosal FBs. In summary, these findings suggest that the interaction between autophagy and PDGF-BB is a key mechanism of OSF occurrence and development and that inhibition of the autophagy pathway may provide a new method for preventing fibrosis.
In this study, we verified in vitro that PDGF-BB induces autophagy through the interaction of Beclin-1 and PI3KC3 to regulate the biological behavior of oral mucosal FBs. However, autophagy is a complicated process where different pathway activation can lead to different biological effect. This study lacks in vivo animal experiments to further clarify the pathogenesis of PDGF-BB/ PiskC3 /Beclin-1 axis in OSF. Therefore, our study results should be interpreted carefully. To clearly understand the mechanism of autophagy in fibrotic diseases, a more comprehensive study of the process’s signaling pathways is needed.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Funding
National Natural Science Foundation of China, No.: 81271154. College Students’ Innovation Project of Central South University, S2020105330686. Changsha Municipal Natural Science Foundation: kq2014140.
Disclosure
The authors declare that they have no competing interests.
References
1. Shih YH, Wang TH, Shieh TM, et al. Oral submucous fibrosis: a review on etiopathogenesis. Diagn Ther. 2019;20(12):2940.
2. Tang JG, Jian XF, Gao ML, et al. Epidemiological survey of oral submucous fibrosis in Xiangtan City, Hunan Province, China. Community Dent Oral Epidemiol. 1997;25(2):177–180. doi:10.1111/j.1600-0528.1997.tb00918.x
3. Jian X, Zheng L. Research advances of oral submucous fibrosis. Chin J Pract Stomatol. 2011;4(2):65–68.
4. Anand R, Dhingra C, Prasad S, et al. Betel nut chewing and its deleterious effects on oral cavity. J Cancer Res Ther. 2014;10(3):499–505. doi:10.4103/0973-1482.137958
5. Gao YJ, Peng HY, Yin XM, et al. [Epidemiological study of betel nut chewing among elementary and middle school students in Loudi city, Hunan province]. Chin J Stomatol. 2009;44(11):686–689. [Chinese].
6. Harris AK. Fibroblasts and myofibroblasts. Methods Enzymol. 1988;163:623–642.
7. Sun YB, Qu X, Caruana G, et al. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. 2016;92(3):102–107. doi:10.1016/j.diff.2016.05.008
8. Han WN, Peng JY, Liu SF. The expression and distribution of platelet-derived growth factor-BB in oral submucous fibrosis. Chin J Pathophysiol. 2002;112(2–3):112–130.
9. Yin-Fang W, Jie-Ying P, Wei-Nong H. Study on relationship of the expression of platelet-derived growthfactor BB receptor in oral mucous membrane with oral submucous fibrosis. J Clin Stomatol. 2004;1:48–51.
10. Peppel K, Zhang LS, Orman ES, et al. Activation of vascular smooth muscle cells by TNF and PDGF: overlapping and complementary signal transduction mechanisms. Cardiovasc Res. 2005;65(3):674–682. doi:10.1016/j.cardiores.2004.10.031
11. Wu JH, Goswami R, Kim LK, et al. The platelet-derived growth factor receptor-beta phosphorylates and activates g protein-coupled receptor kinase-2. J Biol Chem. 2005;280(35):31027–31035. doi:10.1074/jbc.M501473200
12. Si HF, Lv X, Guo A, et al. Suppressive effect of leflunomide on rat hepatic stellate cell proliferation involves on PDGF-BB-elicited activation of three mitogen- activated protein kinases. Cytokine. 2008;42(1):24–31. doi:10.1016/j.cyto.2008.01.017
13. Roeyen CRCV, Ostendorf T, Floege J. The platelet-derived growth factor system in renal disease: an emerging role of endogenous inhibitors. Eur J Cell Biol. 2012;91(6–7):542–551. doi:10.1016/j.ejcb.2011.07.003
14. Salabei J, Cummins T, Singh M, et al. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J. 2013;451(3):375–388. doi:10.1042/BJ20121344
15. Li J, Zhao T, Zhang P, et al. Autophagy mediates oral submucous fibrosis. Exp Ther Med. 2016;11(5):1859–1864. doi:10.3892/etm.2016.3145
16. Kourtis N, Tavernarakis N. Autophagy and cell death in model organisms. Cell Death Differ. 2009;16(1):21–30. doi:10.1038/cdd.2008.120
17. Hamaï A, Botti J, Codogno P. Autophagy and Inflammation. 2016.
18. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi:10.1038/nature09782
19. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
20. Zhao XD, Qin RH, Yang J, et al. DNMT3A controls miR-200b in cardiac fibroblast autophagy and cardiac fibrosis. Inflamm Res. 2018;67(8):681–690. doi:10.1007/s00011-018-1159-2
21. Hernandez-Gea V, Ghiassi-ne Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946. doi:10.1053/j.gastro.2011.12.044
22. Kimura T, Isaka Y, Yoshimori T. Autophagy and kidney inflammation. Autophagy. 2017;13(6):997–1003. doi:10.1080/15548627.2017.1309485
23. Livngston MJ, Ding HF, Huang S, et al. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy. 2016;12(6):976–998. doi:10.1080/15548627.2016.1166317
24. Cabrera S, Maciel M, Herrera I, et al. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy. 2015;11(4):670–684. doi:10.1080/15548627.2015.1034409
25. Ghavami S, Yeganeh B, Zeki AA, et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-β1 in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2018;314(3):L493–L504. doi:10.1152/ajplung.00372.2017
26. Heldin CH, Ostman A, Rönnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta. 1998;1378(1):F79–F113. doi:10.1016/s0304-419x(98)00015-8
27. Kong Q, Xu LH, Xu W, et al. HMGB1 translocation is involved in the transformation of autophagy complexes and promotes chemoresistance in leukaemia. Int J Oncol. 2015;47(1):161–170. doi:10.3892/ijo.2015.2985
28. Wang B, Yang H, Fan Y, et al. 3-Methyladenine ameliorates liver fibrosis through autophagy regulated by the NF-κB signaling pathways on hepatic stellate cell. Oncotarget. 2017;8(64):107603–107611. doi:10.18632/oncotarget.22539
29. Chang YC, Tsai CH, Tai KW, et al. Elevated vimentin expression in buccal mucosal fibroblasts by arecoline in vitro as a possible pathogenesis for oral submucous fbrosis. Oral Oncol. 2002;38(5):425–430. doi:10.1016/S1368-8375(01)00083-5
30. Dai Z, Zhu B, Yu H, et al. Role of autophagy induced by arecoline in angiogenesis of oral submucous fibrosis. Arch Oral Biol. 2019;102:7–15. doi:10.1016/j.archoralbio.2019.03.021
31. Wu YC, Wang WT, Lee SS, et al. Glucagon-like peptide-1 receptor agonist attenuates autophagy to ameliorate pulmonary arterial hypertension through Drp1/NOX and Atg-5/Atg-7/Beclin-1/LC3β pathways. Int J Mol Sci. 2019;20(14):3435–3450.
32. Giampieri F, Afrin S, Forbes-Hernandez TY, et al. Autophagy in human health and disease: novel therapeutic opportunities. Antioxid Redox Signal. 2019;30(4):577–634. doi:10.1089/ars.2017.7234
33. Pant I, Rao SG, Kondaiah P. Role of areca nut induced JNK/ATF2/Jun axis in the activation of TGF-β pathway in precancerous oral submucous fibrosis. Sci Rep. 2016;6(1):1–15. doi:10.1038/srep34314
34. He Y, Jin L, Wang J, et al. Mechanisms of fibrosis in acute liver failure. Liver Int. 2015;35(7):1877–1885. doi:10.1111/liv.12731
35. De Stefano D, Villella VR, Esposito S, et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy. 2014;10(11):2053–2074. doi:10.4161/15548627.2014.973737
36. He L, Livingston MJ, Dong Z. Autophagy in acute kidney injury and repair. Nephron Clin Pract. 2014;127(1–4):56–60. doi:10.1159/000363677
37. Junkins RD, Mccormick C, Lin TJ. The emerging potential of autophagy-based therapies in the treatment of cystic fibrosis lung infections. Autophagy. 2014;10(3):538–547. doi:10.4161/auto.27750
38. Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi:10.1038/nature04724
39. Pua HH, Dzhagalov I, Chuck M, et al. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204(1):25–31. doi:10.1084/jem.20061303
40. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi:10.1056/NEJMra1205406
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.