Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Platelet activating factors are associated with depressive symptoms in coronary artery disease patients: a hypothesis-generating study
Authors Mazereeuw G, Herrmann N ![]() , Xu H, Blanchard A, Figeys D, Oh P, Bennett S, Lanctôt KL
, Xu H, Blanchard A, Figeys D, Oh P, Bennett S, Lanctôt KL ![]()
Received 22 April 2015
Accepted for publication 6 June 2015
Published 4 September 2015 Volume 2015:11 Pages 2309—2314
DOI https://doi.org/10.2147/NDT.S87111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Graham Mazereeuw,1,2,4 Nathan Herrmann,1,5 Hongbin Xu,3,4 Alexandre P Blanchard,3,4 Daniel Figeys,3,4 Paul I Oh,6 Steffany AL Bennett,3,4 Krista L Lanctôt1,2,4–6
1Hurvitz Brain Sciences Program, Sunnybrook Research Institute, Sunnybrook Health Sciences Centre, Toronto, ON, 2Department of Pharmacology and Toxicology, University of Toronto, Toronto, ON, Canada; 3Ottawa Institute of Systems Biology and Neural Regeneration Laboratory, Department of Biochemistry, Microbiology, and Immunology, University of Ottawa, Ottawa, ON, 4CIHR Training Program in Neurodegenerative Lipidomics, Department of Biochemistry, Microbiology, and Immunology, University of Ottawa, Ottawa, ON, 5Department of Psychiatry, University of Toronto, Toronto, ON, Canada; 6UHN Toronto Rehabilitation Institute, Toronto, ON, Canada
Introduction: Depression is a frequent complication of coronary artery disease (CAD) with an unknown etiology. Platelet activating factor (PAF) lipids, which are associated with CAD, have recently been linked with novel proposed etiopathological mechanisms for depression such as inflammation, oxidative/nitrosative stress, and vascular endothelial dysfunction.
Methods and results: This hypothesis-generating study investigated the relationships between various PAF species and depressive symptoms in 26 CAD patients (age: 60.6±9.2 years, 69% male, mean Hamilton Depression Rating Scale [HAM-D] score: 11.8±5.2, HAM-D range: 3–20). Plasma PAF analyses were performed using high performance liquid chromatography electrospray ionization mass spectrometry in precursor ion scan. Significant associations between depressive symptom severity (HAM-D score) and a greater plasma abundance of the PAFs phosphocholine (PC) PC(O-12:0/2:0) (r=0.49, P=0.01), PC(O-14:1/2:0) (r=0.43, P=0.03), PC(O-17:3/2:0) (r=0.44, P=0.04), and PC(O-18:3/2:0) (r=0.50, P=0.01) were observed. Associations between those PAFs and HAM-D score persisted after adjusting for age and sex.
Conclusion: These preliminary findings support the exploration of the PAF lipidome for depressive symptom biomarkers in CAD patients. Patients were recruited as part of the following clinical trial: NCT00981383.
Keywords: depression, lipidomics, cardiovascular, inflammation, oxidative stress, biomarker, vascular depression
Introduction
Up to 65% of coronary artery disease (CAD) patients will experience a major or minor depressive episode after an acute coronary syndrome,1 increasing their risk of mortality.2 As response rates to antidepressant interventions are generally modest in CAD patients,3,4 there is a clinical need to identify mechanistic and/or treatment response biomarkers that may clarify depressive symptom etiology and predict its course in those with CAD. Biomarker identification is, however, limited by a poor mechanistic understanding of the association between depression and CAD. Converging evidence implicates elevated inflammatory activity, activated oxidative and nitrosative stress pathways, and vascular endothelial pathophysiology as processes common to both depression and CAD.5,6 Persistent activation of those inflammatory, oxidative/nitrosative, and vascular pathways can lead to neurodegeneration and to the susceptibility to future depressive episodes.7 Circulating inflammatory mediators have been implicated as potential mechanistic biomarkers related to those pathways in depression;5 however, their nuances are not well understood. This warrants additional approaches to identify inflammatory, oxidative/nitrosative, and/or vascular biomarkers, which may provide mechanistic insight into depressive symptoms in CAD.
Platelet activating factors (PAFs) are a family of potent pro-inflammatory phospholipids with variable systemic actions including vessel dilation8 and the facilitation of long-term potentiation in the central nervous system (CNS)9 under normal conditions. However, PAF concentrations become elevated in response to inflammatory activity. The PAF metabolism pathway has therefore been associated with CAD and with CAD severity.10–14 PAFs may also play a key role in the mechanisms thought to underlie the onset of depressive symptoms in CAD patients, such as inflammation, oxidative/nitrosative stress, and vascular endothelial dysfunction,6,15–17 as well as long-term neurodegenerative damage from persistent activation of inflammatory and oxidative/nitrosative pathways in depression.17 As such, PAFs may be biomarkers reflecting the contributions of multiple pathophysiological mechanisms in depressed CAD patients. However, the relationships between circulating PAF levels and depressive symptoms in patients with CAD have yet to be investigated despite these mechanistic links. Furthermore, technological limitations had previously precluded investigation of PAFs as individual mediators. The purpose of this study was to use breakthrough lipidomics technology to generate hypotheses regarding the association between the PAF lipidome and depressive symptoms in CAD patients, given the proposed mechanistic relevance of PAFs.17
Materials and methods
Patients
Patients were approached at entry to a cardiac rehabilitation program at local centers in Toronto, Ontario. Each patient entered cardiac rehabilitation at a standardized time (8–10 weeks) after an acute coronary syndrome. Included patients had CAD (myocardial infarction, prior revascularization procedure, and/or angiographic evidence of at least 50% stenosis in a major coronary artery), were aged 45–80 years, spoke and understood English, and provided written and informed consent to participate. Once included, demographic information, a detailed medical history, and concomitant medications were recorded. The use of antidepressant medication was permitted on the condition of dose stability over at least 3 months. This study was approved by the research ethics boards of Sunnybrook Research Institute, University Health Network at Toronto Rehabilitation Institute, and Trillium Health Partners and was conducted according to the principles expressed in the Declaration of Helsinki.
Patients were excluded if they demonstrated significant cognitive impairment (Standardized Mini-Mental State Examination ≤24),18 had a history of a neurological condition, had a pre-morbid diagnosis of any Axis I psychiatric disorder other than depression, or had a significant acute medical illness (eg, autoimmune condition, uncontrolled diabetes mellitus, and uncontrolled hypothyroidism).
Design
Included patients were invited to a study interview within 2 weeks of initial contact. Depressive symptoms were assessed using the investigator-rated 17-item Hamilton Depression Rating Scale (HAM-D),19 a continuous measure that is highly reliable and sensitive in assessing depressive symptoms in CAD patients.20 The presence of a major depressive episode (MDE) was determined by trained study personnel using the Structured Clinical Interview for DSM (IV) Disorders.21 Depressive symptom severity and the presence of a depressive episode were assessed by a trained study associate under the guidance of an experienced psychiatrist. CAD patients both meeting MDE criteria and not meeting MDE criteria were included in this study to provide a range of depressive symptom severity.
Fasting blood was collected from all patients at the study interview. PAFs were analyzed using high performance liquid chromatography electrospray ionization mass spectrometry in precursor ion scan as we have previously described.22 Briefly, lipid species in the m/z range from 450 to 600 were analyzed using precursor ion scan for the diagnostic ion at m/z 184 for phosphocholine (PC).23,24 Only species identified as PAFs following bioinformatic prediction using VaLID v1.125 were analyzed further.
Statistics
Peak area of each lipid species, representing the area under the curve, in counts per second was established using Analyst v1.4.2 (AB Sciex, Concord, Canada). All peak areas were then normalized to the synthetic internal standard (PC[13:0/0:0]) spiked at time of lipid extraction from plasma and multiplied by 106 before log2 transformation, yielding the abundance. The association between PAF abundance and HAM-D score was investigated using linear regression (SPSS statistical software, version 13.0, SPSS Inc., Chicago, IL, USA), adjusting for the covariates age and sex in independent models as these have previously demonstrated associations with depressive symptoms and inflammatory activity in CAD patients.26–28 Results were not corrected for multiple comparisons in order to preserve potentially hypothesis generating findings.
Results
Twenty-six CAD patients were included and assessed (Table 1). Fifteen (58%) met DSM-IV criteria for a MDE, and HAM-D total scores ranged from 3 to 20 points. Only two patients (8%) were using an antidepressant, all were using statins, all were using a platelet inhibitor (acetylsalicylic acid or clopidogrel), and one (4%) was using a non-steroidal anti-inflammatory medication. No significant relationships between patient characteristics and depressive symptom severity were observed (Table 1).
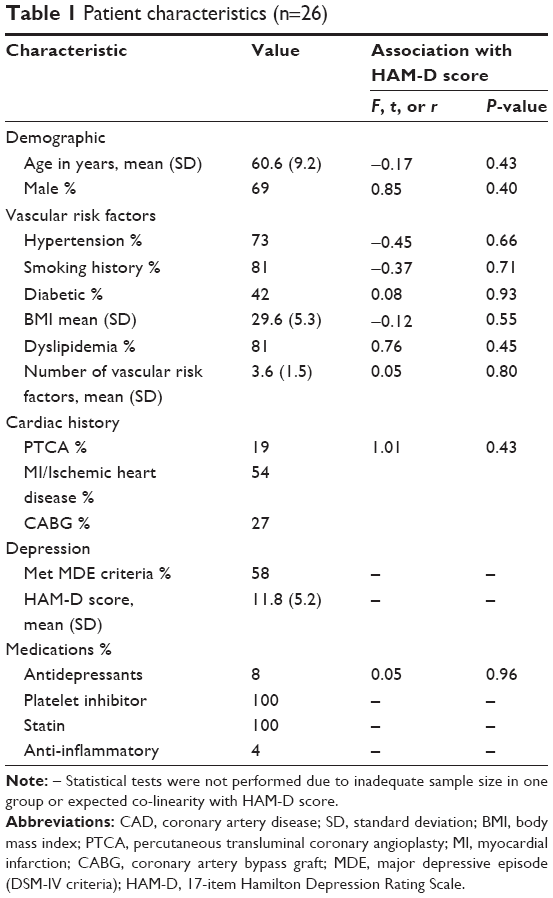 | Table 1 Patient characteristics (n=26) |
Seventy-two PC messenger species with m/z between 450 and 600 were detected in patient’s plasma. Of these, 20 species were identified as PAFs. Four PAF species demonstrated a significant association with HAM-D score in our sample. In linear regression, a greater abundance of the PAFs PC(O-12:0/2:0), PC(O-14:1/2:0), PC(O-17:3/2:0), and PC(O-18:3/2:0) was significantly associated with greater HAM-D scores in CAD patients, while the PAF PC(O-13:0/2:0) trended with HAM-D score (Table 2).
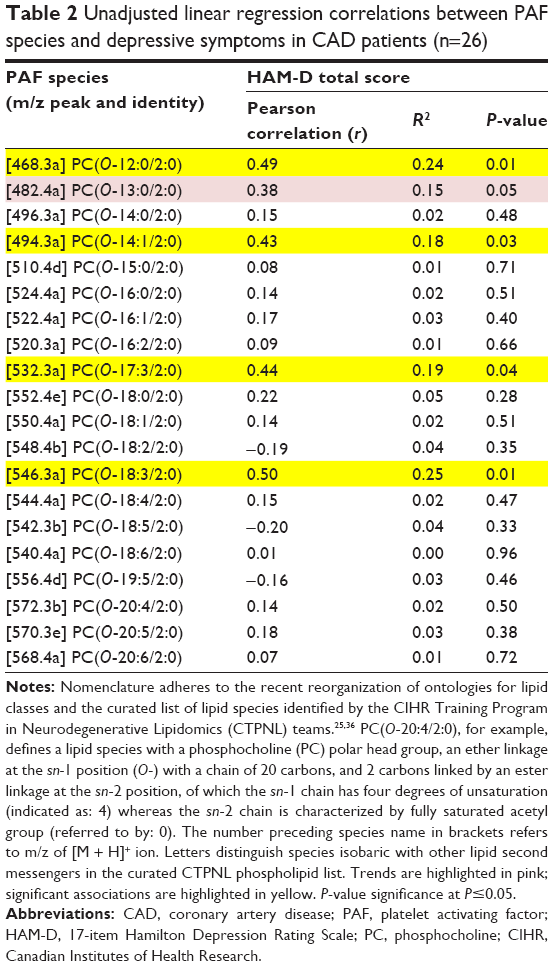 | Table 2 Unadjusted linear regression correlations between PAF species and depressive symptoms in CAD patients (n=26) |
Those associations were then investigated in independent age- and sex-adjusted models. In the age-adjusted models, PC(O-12:0/2:0) (β=0.47, R2=0.24, P=0.02), PC(O-14:1/2:0) (β=0.43, R2=0.22, P=0.03), PC(O-17:3/2:0) (β=0.44, R2=0.20, P=0.04), and PC(O-18:3/2:0) (β=0.51, R2=0.25, P=0.01) remained significantly associated with HAM-D score. In the sex-adjusted models, PC(O-12:0/2:0) (β=0.49, R2=0.27, P=0.01), PC(O-14:1/2:0) (β=0.45, R2=0.23, P=0.02), PC(O-17:3/2:0) (β=0.48, R2=0.29, P=0.02), and PC(O-18:3/2:0) (β=0.52, R2=0.29, P=0.01) remained significantly associated with HAM-D score. There were no interactions between patient characteristics and the identified PAFs (not shown), thus no additional covariate-adjusted models were analyzed. Due to consistent, significant associations with HAM-D score in both covariate-adjusted models, PAFs PC(O-12:0/2:0), PC(O-14:1/2:0), PC(O-17:3/2:0), and PC(O-18:3/2:0) were identified as key species of potential relevance to depressive symptoms in CAD patients.
Discussion
The findings of this study suggest that certain PAFs may be associated with depressive symptoms in CAD patients, thus identifying PAF species of potential interest for future studies, and generally implicating the PAF lipidome as potentially relevant to the search for biomarkers of depressive symptoms in CAD.
To our knowledge, this is the first study to explore the association between depressive symptom severity and the plasma abundance of PAFs in any patient population, and therefore comparisons with other studies are not possible. However, these findings support a role for PAFs as inflammatory and neurodegenerative lipid messengers associated with depressive symptoms in CAD patients as previously hypothesized.17 These findings are also in line with previously reported associations between PC-derived lipid mediators and depressive symptoms29 or depressive symptom-like behavior.30 For example, lower circulating concentrations of the PC(O-36:4) were associated with greater depressive symptom severity in a community-based cohort study. That negative relationship may be relevant to PAFs as membrane phospholipids such as PC(O-36:4) may be cleaved to produce PAFs and arachidonic acid (20:4) during periods of increased inflammation, leading to reduced circulating concentrations of the precursor lipid. As such, the lipidomic approach used for the present study may complement previous lipidomic approaches by identifying PAFs that may be derived from membrane phospholipids such as PC(O-36:4). More generally, the findings of the present study support the exploration of lipidomic biomarkers for clarifying etiological mechanisms of depression in those with CAD.
The PAFs suggested as depressive symptom-related in the present study may reflect the particular composition of PC precursor lipids in peripheral leukocytes or vascular endothelial cell membranes. As such, the identified PAFs may be related not only to the activation of PAF metabolism, but may indicate the presence of different membrane lipid compositions in depression. The identified PAFs may also reflect specific signaling properties necessary to participate in the peripheral inflammatory, oxidative/nitrosative, and vascular mechanisms relevant to depression in CAD. For example, pre-clinical studies indicate that PAFs have differential signaling effects not only at the widely-expressed PAF receptor, but also at the PAF receptor independently;31 therefore, each species identified in our plasma analysis may have a unique signaling effect in the periphery, depending on its relative potency at the PAF receptor, the PAF receptor landscape in various tissues, and the PAF receptor-independent pathways that it can activate. Interestingly, the PAFs PC(O-16:0/2:0) and PC(O-18:0/2:0) did not demonstrate a significant association with depressive symptom severity in our sample despite their consistent characterization as highly potent PAFs with various pro-apoptotic effects on neurons when elevated in the CNS.31 While it should be stressed that these findings are preliminary and that associations between those species and depressive symptoms may be revealed in a larger sample, it is important to consider that PAFs may have different actions in the CNS than in the periphery and that peripheral depression-related PAFs may not align with PAFs relevant to depression-related CNS processes. Collectively, the PAF species examined in this study may shed light on the mechanisms relevant to depression in CAD patients.
Limitations
This study used a lipidomic approach to explore the range of PAFs detectible in plasma and, in turn, generate hypotheses for their clinical relevance to depressive symptoms in CAD patients. As such, the study results were not corrected for multiple comparisons in order to highlight potential species of interest for future studies. This study was limited by the small sample size, which permitted the use of only one covariate per analysis. Thus, we were unable to include age and sex together in the same statistical model. However, we were able to approximate the combined influence of those covariates by investigating the association between depressive symptom severity and PAF abundance in two independent covariate-adjusted models, with the interpretation of our findings based on these collective analyses. These findings may have also been limited by relatively mild depressive symptom severity among CAD patients meeting DSM-IV depressive episode criteria, although the wide range of depressive symptom severity on the HAM-D (3–20 points) likely provided sufficient range for exploratory correlations with PAF abundance. To that point, depressive symptoms tend to be milder in those with CAD than in those with major depressive disorder,32 and as such, the studied population was representative of depressive symptoms in CAD. Finally, these findings are limited to PAFs derived from alkylacylglycerophosphocholines; however, as mentioned, these are highly abundant PAFs and are therefore appropriate for this first-step exploration.
Future directions
The relationships between the identified PAFs and depressive symptoms ought to be confirmed in a study of larger sample size. It may also be advantageous to explore the relationships between PAFs and depressive symptoms in CAD over time using a repeated-measure analysis. Such an approach would clarify the temporal relationship between PAFs and the worsening or resolution of depressive symptoms.
The association between depression and CAD is complex and likely heterogeneous; various psychosocial factors33,34 and/or pathophysiological contributions from CAD34 are among the numerous contributors to the onset of depressive symptoms in those with CAD. However, current evidence continues to support a role for etiopathology related to aberrant inflammatory activity in the presence and persistence of depression in CAD,7,35 with particular relevance to the progressive neurodegeneration often associated with persistent depression.7 Thus, the preliminary results of this study support the continued investigation of PAFs as a new group of inflammatory mediators potentially contributing to depressive symptoms in CAD patients. Additional mechanistic models incorporating conventional inflammatory, vascular, and genetic risk factors for depression in CAD may be needed to confirm the relevance of PAFs. Should future studies implicate PAFs as depressive symptom biomarkers in CAD, the generalizability of PAFs to other patient populations, such as major depressive disorder, may be worthwhile to investigate.
Conclusion
The findings of this study suggest that further investigation of PAF species, possibly the PAFs PC(O-12:0/2:0), PC(O-14:1/2:0), PC(O-17:3/2:0), and PC(O-18:3/2:0), as markers of depressive symptoms in CAD patients is warranted. As PAFs are elevated in CAD, are important mediators of vascular pathology and neurodegeneration, and are implicated in several of the leading mechanisms proposed to underlie the association between depression and CAD, these findings are potentially clinically relevant. Larger, longitudinal studies of PAFs in depressed CAD patients may confirm their relevance and elucidate their clinical utility.
Acknowledgments
The authors acknowledge operating funds from the Ontario Mental Health Foundation and the Canadian Institutes of Health Research (CIHR) MOP 89999 to DF and SALB, MOP 114913 to KLL and NH, and a Strategic Training Initiative in Health Research (STIHR) CIHR Training Program in Neurodegenerative Lipidomics (CTPNL) and Institute of Aging TGF 96121 to KLL, DF, and SALB. GM and APB received CTPNL graduate scholarships as well as OGS and FRSQ graduate scholarships, respectively. HX was supported by a CTPNL and MITACS post-doctoral fellowship.
Disclosure
The authors report no conflicts of interest in this work.
References
Celano CM, Huffman JC. Depression and cardiac disease: a review. Cardiol Rev. 2011;19:130–142. | ||
Lesperance F, Frasure-Smith N. Depression in patients with cardiac disease: a practical review. J Psychosom Res. 2000;48:379–391. | ||
Dowlati Y, Herrmann N, Swardfager WL, Reim EK, Lanctot KL. Efficacy and tolerability of antidepressants for treatment of depression in coronary artery disease: a meta-analysis. Can J Psychiatry. 2010;55:91–99. | ||
Blumenthal JA, Sherwood A, Babyak MA, et al. Exercise and pharmacological treatment of depressive symptoms in patients with coronary heart disease: results from the UPBEAT (Understanding the Prognostic Benefits of Exercise and Antidepressant Therapy) study. J Am Coll Cardiol. 2012;16:1053–1063. | ||
Stapelberg NJC, Neumann DL, Shum DHK, McConnell H, Hamilton-Craig I. A topographical map of the causal network of mechanisms underlying the relationship between major depressive disorder and coronary heart disease. Aust NZ J Psychiatry. 2011;45:351–369. | ||
Baune BT, Stuart M, Gilmour A, et al. The relationship between subtypes of depression and cardiovascular disease: a systematic review of biological models. Transl Psychiatry. 2012;13:e92. | ||
Moylan S, Maes M, Wray NR, Berk M. The neuroprogressive nature of major depressive disorder: pathways to disease evolution and resistance, and therapeutic implications. Mol Psychiatry. 2013;18(5):595–606. | ||
Kuebler WM, Yang Y, Samapati R, Uhlig S. Vascular barrier regulation by PAF, ceramide, caveolae, and NO – an intricate signaling network with discrepant effects in the pulmonary and systemic vasculature. Cell Physiol Biochem. 2010;26:29–40. | ||
Arai A, Lynch G. Antagonists of the platelet-activating factor receptor block long-term potentiation in hippocampal slices. Eur J Neurosci. 1992;4:411–419. | ||
Chen H, Zheng P, Zhu H, et al. Platelet-activating factor levels of serum and gingival crevicular fluid in nonsmoking patients with periodontitis and/or coronary heart disease. Clin Oral Investig. 2010;14:629–636. | ||
Winkler K, Winkelmann BR, Scharnagl H, et al. Platelet-activating factor acetylhydrolase activity indicates angiographic coronary artery disease independently of systemic inflammation and other risk factors: the Ludwigshafen Risk and Cardiovascular Health Study. Circulation. 2005;111:980–987. [Erratum in Circulation. 2005;111(24):e444]. | ||
Samsamshariat S, Basati G, Movahedian A, Pourfarzam M, Sarrafzadegan N. Elevated plasma platelet-activating factor acetylhydrolase activity and its relationship to the presence of coronary artery disease. J Res Med Sci. 2011;16:674–679. | ||
Wilensky RL, Macphee CH. Lipoprotein-associated phospholipase A(2) and atherosclerosis. Curr Opin Lipidol. 2009;20:415–420. | ||
Winkler K, Hoffmann MM, Winkelmann BR, et al. Lipoprotein-associated phospholipase A2 predicts 5-year cardiac mortality independently of established risk factors and adds prognostic information in patients with low and medium high-sensitivity C-reactive protein (the Ludwigshafen risk and cardiovascular health study). Clin Chem. 2007;53:1440–1447. | ||
Klabunde RE, Anderson DE. Role of nitric oxide and reactive oxygen species in platelet-activating factor-induced microvascular leakage. J Vasc Res. 2002;39:238–245. | ||
Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. J Neurochem. 2007;101:577–599. | ||
Mazereeuw G, Herrmann N, Bennett SAL, et al. Platelet activating factors in depression and coronary artery disease: A potential biomarker related to inflammatory mechanisms and neurodegeneration. Neurosci Biobehav Rev. 2013;37:1611–1621. | ||
Molloy DW. Standardized Mini Mental State Examination. Troy, ON, Canada: New Grange Press; 1999. | ||
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. | ||
Strik JJ, Honig A, Lousberg R, Denollet J. Sensitivity and specificity of observer and self-report questionnaires in major and minor depression following myocardial infarction. Psychosomatics. 2001;42:423–428. | ||
First MB, Spitzer RL, Gibbon M, et al. Structured clinical interview for DSM-IV diagnoses (SCID): Clinician and research versions. New York, NY: Biometrics Research Department, Columbia University; 1996. | ||
Mazereeuw G, Herrmann N, Xu H, et al. Platelet-activating factors are associated with cognitive deficits in depressed coronary artery disease patients: a hypothesis-generating study. J Neuroinflammation. 2014;11:119. | ||
Whitehead SN, Hou W, Ethier M, et al. Identification and quantitation of changes in the platelet activating factor family of glycerophospholipids over the course of neuronal differentiation by high-performance liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chem. 2007;79:8539–8548. | ||
Ryan SD, Whitehead SN, Swayne LA, et al. Amyloid-β42 signals tau hyperphosphorylation and compromises neuronal viability by disrupting alkylacylglycerophosphocholine metabolism. Proc Natl Acad Sci U S A. 2009;106:20936–20941. | ||
Blanchard AP, McDowell GSV, Valenzuela N, et al. Visualization and Phospholipid Identification (VaLID): online integrated search engine capable of identifying and visualizing glycerophospholipids with given mass. Bioinformatics. 2013;29:284–285. | ||
Dowlati Y, Herrmann N, Swardfager W, et al. Relationship between hair cortisol concentrations and depressive symptoms in patients with coronary artery disease. Neuropsychiatr Dis Treat. 2010;7:393–400. | ||
Todaro JF, Shen B-J, Niaura R, Tilkemeier PL. Prevalence of depressive disorders in men and women enrolled in cardiac rehabilitation. J Cardiopulm Rehabil. 2005;25:71–75; quiz 76–77. | ||
Madjid M, Awan I, Willerson JT, Casscells SW. Leukocyte count and coronary heart disease: implications for risk assessment. J Am Coll Cardiol. 2004;44:1945–1956. | ||
Demirkan A, Isaacs A, Ugocsai P, et al. Plasma phosphatidylcholine and sphingomyelin concentrations are associated with depression and anxiety symptoms in a Dutch family-based lipidomics study. J Psychiatr Res. 2013;47:357–362. | ||
Faria R, Santana MM, Aveleira CA, et al. Alterations in phospholipidomic profile in the brain of mouse model of depression induced by chronic unpredictable stress. Neuroscience. 2014;273:1–11. | ||
Ryan SD, Harris CS, Carswell CL, Baenziger JE, Bennett SAL. Heterogeneity in the sn-1 carbon chain of platelet-activating factor glycerophospholipids determines pro- or anti-apoptotic signaling in primary neurons. J Lipid Res. 2008;49:2250–2258. | ||
Groenewold NA, Doornbos B, Zuidersma M, et al. Comparing cognitive and somatic symptoms of depression in myocardial infarction patients and depressed patients in primary and mental health care. PLoS One. 2013;8:e53859. | ||
Ketterer MW, Mahr G, Goldberg AD. Psychological factors affecting a medical condition: ischemic coronary heart disease. J Psychosom Res. 2000;48:357–367. | ||
Sato S, Yeh TL. Challenges in treating patients with major depressive disorder: the impact of biological and social factors. CNS Drugs. 2013;27(Suppl 1):S5–S10. | ||
Maes M, Ruckoanich P, Chang YS, Mahanonda N, Berk M. Multiple aberrations in shared inflammatory and oxidative & nitrosative stress (IO&NS) pathways explain the co-association of depression and cardiovascular disorder (CVD), and the increased risk for CVD and due mortality in depressed patients. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011;35:769–783. | ||
Fahy E, Cotter D, Sud M, Subramaniam S. Lipid classification, structures and tools. Biochim Biophys Acta. 2011;1811:637–647. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.