")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Plasmapheresis, Anti-ACE2 and Anti-FcγRII Monoclonal Antibodies: A Possible Treatment for Severe Cases of COVID-19
Authors Sedokani A , Feizollahzadeh S
Received 12 May 2020
Accepted for publication 26 June 2020
Published 6 July 2020 Volume 2020:14 Pages 2607—2611
DOI https://doi.org/10.2147/DDDT.S262491
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianbo Sun
Amin Sedokani,1 Sadegh Feizollahzadeh2
1Cardiology Department, Medical Faculty, Urmia University of Medical Sciences, Urmia, Iran; 2Medical Immunology, Laboratory Sciences, School of Allied Medical Sciences, Urmia University of Medical Sciences, Urmia, Iran
Correspondence: Amin Sedokani
Cardiology Department, Medical Faculty, Urmia University of Medical Sciences, 17 Sharivar St., Urmia 571478334, Iran
Tel +98 443237 5907
Fax +98 443 237 2917
Email [email protected]
Abstract: In March 2020, the WHO declared the COVID-19 disease as a pandemic disease. There have been studies on the COVID-19 to find a certain treatment, but yet, there is no certain cure. In this article, we present a possible way to treat severe cases of COVID-19. Based on the previous studies, there are similarities between the spike antigens of SARS-CoV and SARS-CoV-2 viruses. It is expected that these similarities (structural and affinity to the receptor of ACE2) can lead to the same pathophysiological activity of the virus by the use of ACE2 and FcγRII (the antibody-dependent enhancement mechanism). Therefore, we propose a way of washing out (by plasmapheresis) the possible antibodies against the spike protein of the virus out of patients’ plasma to stop the antibody-dependent enhancement (ADE)-mediated infection of the immune system cells at the first phase of the treatment and simultaneous use of the anti-ACE2 with anti-FcγRII monoclonal antibodies at the second phase. We propose these procedures for the patients that have no significant response for typical anti-viral, ARDS and conservative therapies, and the disease persists or progresses despite sufficient therapies.
Keywords: COVID-19, SARS-CoV-2, antibody-dependent enhancement, plasmapheresis, monoclonal antibodies
Introduction
December of 2019 has become a malign month for global health by emerging ofCOVID-19 (corona virus disease 2019), in mainland China, at present involving many other countries by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1–3 The WHO declared the situation as a pandemic due to the 118,000 cases reported globally in 114 countries on March 11, 2020.4
Recent studies on SARS-CoV-2 as well as past studies on the SARS-CoV virus demonstrated that the viruses use the angiotensin-converting enzyme receptor 2 (ACE2) as a receptor (by binding to the spike protein or S protein of the virus) to enter the cells. Also, studies have shown that SARS-CoV-2 and SARS-CoV bind with similar affinities to ACE2 with the same downstream (TMPRSS2 serine protease) activation to priming the S protein.5–9 Hoffman et al have shown that inhibiting the S protein of SARS-CoV-2 by the same antibodies of SARS-CoV can protect the cell against COVID-19. The mechanism was explained by Yip et al and indicated that anti-spike protein antibodies were responsible for the immune system cells infection.10–12
The effector mechanisms of the immune system to some viral or bacterial pathogens may lead to life-threatening consequences to the host. For example, the overexpression and release of cytokines (cytokine storm) following influenza virus infection can increase the severity of the disease.13 Also, it is known that several viral diseases are mediated by antibody-dependent enhancement (ADE), a mechanism in which antiviral antibodies facilitate host cell infections (mostly non-neutralizing proteins). With this process, the virus can infect the cells that have no typical receptor for it by intermediation of FcR (Fc Receptor).14–16 Jaume et al have shown the pathway of FcγRII (CD32) as a novel non-ACE2 mediated endosomal/lysosomal pathway for SARS-CoV infection in immune cells.12 Due to the similarity of the SARS-CoV and SARS-CoV-2,17 the study of Hoffman et al, proposing the use of TMPRSS2 Protease Inhibitor (the intracellular serine protease priming the spike-protein of SARS-CoV and SARS-CoV-2), cannot respond completely in clinical trials by considering the ADE pathway of FcγRII (CD32).
The mechanism of ADE-FcγRII can also be responsible for lymphopenia in COVID-19 patients. Also, our hypotheses for high mortality in the older patient are the high amounts of serum immunoglobins from other types of Coronaviridae family,18–20 with a wide spectrum of affinities, would trigger the ADE mechanism, cytokine release syndrome, and elevated IL-6, which leads to multi-organ failure (High clinical SOFA or Sequential Organ Failure Assessment scores in patients).21 Therefore, it is expected that the SARS-CoV-2 virus would be covered by antibodies that have produced against other viruses during the lifetime of the patient. Then, the virus can use either the ADE-FcγRII pathway (mostly in macrophages) or ACE2 pathway to enter cells by the spike protein. Moreover, chloroquine efficacy against COVID-19 may be attributed to the blocking of endocytosis dependent virus entry to macrophages.22 However, after several months of study on chloroquine/hydroxychloroquine efficacy on COVID-19, there are still controversies on it. Meanwhile, in a study published by the LANCET infectious disease, it has been indicated that after 2 weeks on symptom onset, serum antibodies were positive for 94% for anti-NP IgG, 88% anti-NP IgM, 100% for anti-RBD IgG, and 94% for anti-RBD IgM, but still the disease was active and severe in clinical assessment.23 So, the COVID-19 disease may not be an ordinary viral disease that could be cured by ordinary immune pathways (Figure 1).
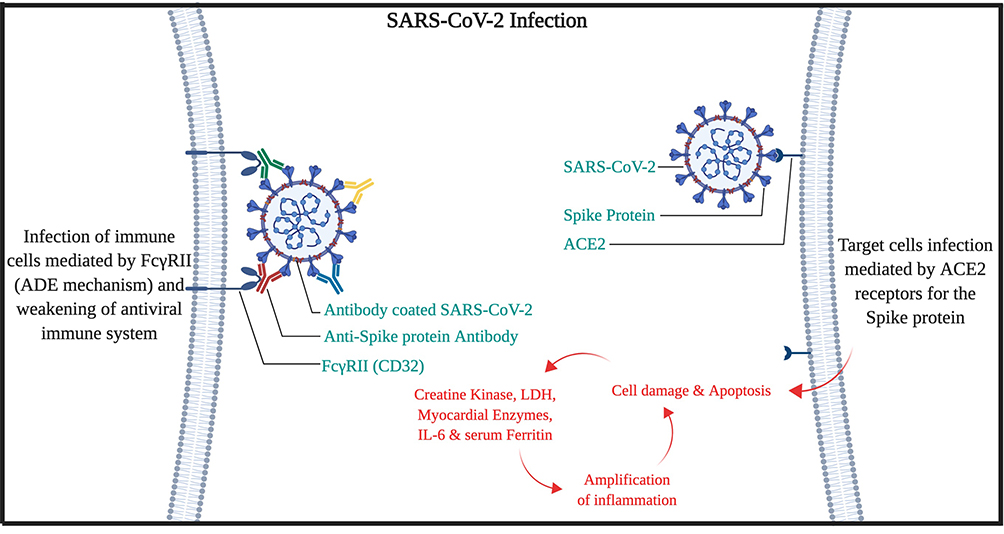 |
Figure 1 Cell entry and infection mechanism of SARS-CoV-2. |
Hypothesis
According to the mentioned subjects, hereby we propose clinical treatment for severe patients. At the first point, for stopping the virus proliferation cycle, we must eliminate all possible antibodies that could trigger the ADE-FcγRII pathway (endosomal/lysosomal pathway). The plasmapheresis would be a great choice for removing the plasma contents of cytokines, interleukins, antiviral proteins, γ-globin and antibodies, acute phase reactants, etc., with a privilege that there is no need to recognize specific low-affinity different antibodies that persist in the serum for long times. In addition to viremia removal, this process could stop or at least delay the ARDS Acute Respiratory Distress Syndrome injury and multi-organ failure process.24,25 The second phase must be the simultaneous use of the anti-ACE2 with Anti-FcγRII monoclonal antibodies (Figure 1). The reasons we emphasis on monoclonal antibodies rather than TMPRSS2 Protease Inhibitor alone are as follows:
- We should be certain that the downstream of the ACE2 pathway is completely blocked and the virus cannot use the three-dimensional molecular ACE2 as a target antigen for spike-protein.
- ACE2 is not the only target of the virus to infect cells. In macrophages FcγRII endosomal/lysosomal pathway had been spotted as a great infection gap for the immune system itself that tries to neutralize and ingest it by antibody-mediated opsonization. Besides, neutrophils also may use this pathway to neutralize the virus, which plays a great role in ARDS, alveoli injury and respiratory failure.26 The important point is that the anti-ACE2 monoclonal antibody must be against the 723 lengths amino acid topological domain that is extracellular (position of 18–740).27
In addition, we should say, the plasmapheresis may cause the patient to undergo an immuno-compromised state that might make them susceptible to concomitant viral or other types of infections. But there are two points that we may consider; 1. The plasmapheresis must be used just for specific severe cases that do not show significant clinical response to typical anti-viral, ARDS and conservative therapies. 2. The plasmapheresis effects minimally on the cellular immunity, but again after the lowering the disease severity, the humoral immunity against specific disease (due to the patient, region, disease history, etc.) must be supported to prevent concomitant viral or other types of infections till the effect of anti-ACE2 with Anti-FcγRII monoclonal antibodies would be gone and humoral immunity, reconstruct itself.
The risk for activation/inhibition of angiotensin-converting enzyme 2 must be acceptable. Because we are aiming the 3-dimensional extracellular structure of ACE2 to prevent the virus usage of it, and typically, it should not target the enzymatic purpose of ACE2. But still, in the case of lowering the blood pressure by inhibition of ACE2 enzyme, lower afterload for the heart with well-supported cardiopulmonary ventilation may have positive effects on the patients, especially patients with hypertension, heart failure, etc. that are great mortality risk factors for COVID-19.28 However, the in-vivo, primary and preclinical studies must identify the potential clinical risks and confirm the effect of used antibodies on human ACE2 (hACE2). We propose these procedures for the patients that have no significant response for typical anti-viral, ARDS and conservative therapies and the disease persists or progresses despite sufficient therapies. In addition to ACE2 and possible pathway of FcγRII, there are novel proposed pathways to SARS-CoV-2 infection such as CD147 and CD209.29,30 However, more specific studies are expected before judge about plasmapheresis and FcγRII, CD147, and CD209 targeting (Figure 2).
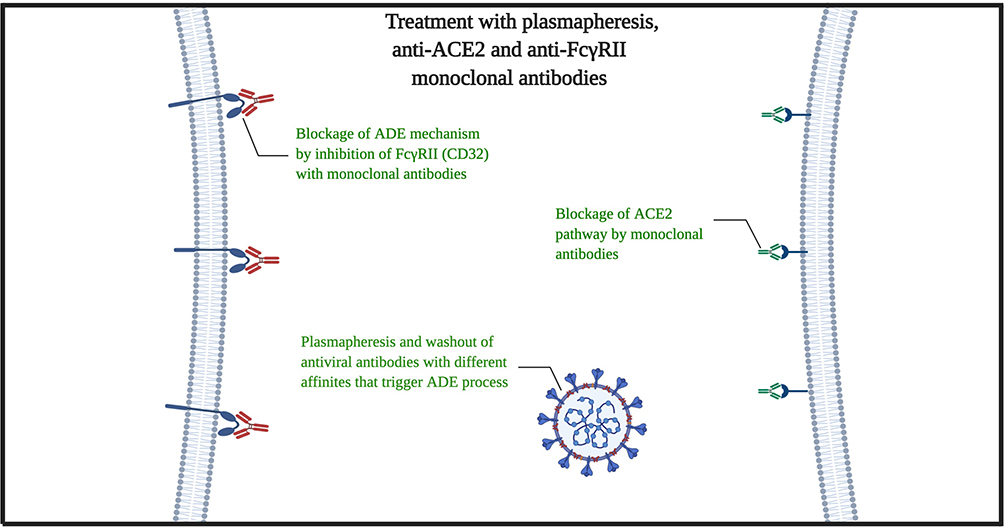 |
Figure 2 Mechanism of plasmapheresis and monoclonal antibodies (anti-ACE2 with anti-FcγRII). |
Acknowledgment
We like to appreciate the BioRender team for providing the opportunity of creating a visual abstract for this article.
Disclosure
Authors report no conflicts of interest in this work.
References
1. Gorbalenya AE. Severe acute respiratory syndrome-related coronavirus–The species and its viruses, a statement of the Coronavirus Study Group. BioRxiv. 2020.
2. Lu H, Stratton CW, Tang YW. Outbreak of pneumonia of unknown etiology in Wuhan China: the mystery and the miracle. J Med Virol. 2020.
3. Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. The Lancet. 2020;395(10223):470–473. doi:10.1016/S0140-6736(20)30185-9
4. WHO. WHO Director-General’s opening remarks at the media briefing on COVID-19: World Health Organization; 2020. Available from: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19—11-march-2020.
5. Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi:10.1038/nature02145
6. Li W, Zhang C, Sui J, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi:10.1038/sj.emboj.7600640
7. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi:10.1126/science.1116480
8. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2). doi:10.1016/j.cell.2020.02.052.
9. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi:10.1038/s41586-020-2012-7
10. Yip MS, Leung NHL, Cheung CY, et al. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J. 2014;11(1):82. doi:10.1186/1743-422X-11-82
11. Wang S-F, Tseng S-P, Yen C-H, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem Biophys Res Commun. 2014;451(2):208–214. doi:10.1016/j.bbrc.2014.07.090
12. Jaume M, Yip MS, Cheung CY, et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH-and cysteine protease-independent FcγR pathway. J Virol. 2011;85(20):10582–10597. doi:10.1128/JVI.00671-11
13. Peiris JS, Cheung CY, Leung CY, Nicholls JM. Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol. 2009;30(12):574–584. doi:10.1016/j.it.2009.09.004
14. Morens DM. Antibody-dependent enhancement of infection and the pathogenesis of viral disease. Clin Infect Dis. 1994;19(3):500–512. doi:10.1093/clinids/19.3.500
15. Sullivan NJ. Antibody-mediated enhancement of viral disease. Curr Top Microbiol Immunol. 2001;260:145–169. doi:10.1007/978-3-662-05783-4_8
16. Takada A, Kawaoka Y. Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev Med Virol. 2003;13(6):387–398. doi:10.1002/rmv.405
17. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Structure VD. Function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. doi:10.1016/j.cell.2020.02.058
18. Geller C, Varbanov M, Duval RE. Human coronaviruses: insights into environmental resistance and its influence on the development of new antiseptic strategies. Viruses. 2012;4(11):3044–3068. doi:10.3390/v4113044
19. Glowacka I, Bertram S, Muller MA, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85(9):4122–4134. doi:10.1128/JVI.02232-10
20. Malik YA. Properties of Coronavirus and SARS-CoV-2. Malays J Pathol. 2020;42(1):3–11.
21. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet. 2020;395(10229):1054–1062. doi:10.1016/S0140-6736(20)30566-3
22. Hu TY, Frieman M, Wolfram J. Insights from nanomedicine into chloroquine efficacy against COVID-19. Nat Nanotechnol. 2020;15(4):247–249. doi:10.1038/s41565-020-0674-9
23. To KK, Tsang OT, Leung WS, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect Dis. 2020;20(5):565–574. doi:10.1016/S1473-3099(20)30196-1
24. Douzinas EE, Markakis K, Karabinis A, Mandalaki T, Bilalis D, Fessas P. Early plasmapheresis in patients with thrombotic thrombocytopenic purpura. Crit Care Med. 1992;20(1):57–61. doi:10.1097/00003246-199201000-00017
25. Kohli RS, Bleibel W, Shetty A, Dhanjal U. Plasmapheresis in the treatment of hypertriglyceridemic pancreatitis with ARDS. Dig Dis Sci. 2006;51(12):2287–2291. doi:10.1007/s10620-006-9315-x
26. Zemans RL, Matthay MA. What drives neutrophils to the alveoli in ARDS? Thorax. 2017;72(1):1–3. doi:10.1136/thoraxjnl-2016-209170
27. UniProtKB - Q9BYF1 (ACE2_HUMAN): UniProt. Available from: https://www.uniprot.org/uniprot/Q9BYF1.
28. Jordan RE, Adab P, Cheng KK. Covid-19: risk factors for severe disease and death. BMJ. 2020;368:m1198. doi:10.1136/bmj.m1198
29. Wang K, Chen W, Zhou Y-S, et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv. 2020;2020:988345.
30. Chan KY, Xu MS, Ching JC, et al. Association of a single nucleotide polymorphism in the CD209 (DC-SIGN) promoter with SARS severity. Hong Kong Med J. 2010;16(5 Suppl 4):37–42.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.