Back to Journals » International Journal of General Medicine » Volume 18
Plasma Semaphorin 3A as a Potential Biomarker for Cognitive Impairment in Cerebral Small Vessel Disease
Authors Ma H, Li H, Fu X, Li X, Wei C, Ma A
Received 18 July 2025
Accepted for publication 25 October 2025
Published 5 November 2025 Volume 2025:18 Pages 6695—6708
DOI https://doi.org/10.2147/IJGM.S554075
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Redoy Ranjan
Haoran Ma, Hong Li, Xinyi Fu, Xuening Li, Chenchen Wei, Aijun Ma
Department of Neurology, The Affiliated Hospital of Qingdao University, Qingdao, People’s Republic of China
Correspondence: Aijun Ma; Chenchen Wei, Email [email protected]; [email protected]
Background: Sema3A plays an essential role in the nervous system and is thought to regulate the entire course of the inflammatory response. This study aimed to explore the potential association between plasma Sema3A levels and cerebral small vessel disease (CSVD).
Methods: This study enrolled patients with recent small subcortical infarct (RSSI) and 100 healthy controls (HCs). Sema3A levels were measured during the acute phase (≤ 3 days) and the subacute phase (14– 30 days) of RSSI. Cognitive function was assessed using the Montreal Cognitive Assessment (MoCA). Brain magnetic resonance imaging (MRI) was used to evaluate CSVD imaging features. The study analyzed the association between Sema3A, CSVD imaging features, and cognitive impairment.
Results: Plasma Sema3A levels during the acute phase were markedly higher in RSSI patients than in HCs (p = 0.001), with an area under the receiver operating characteristic curve (AUC) of 0.715 (p < 0.01) for differentiating RSSIs from HCs. Levels were higher in the acute than the subacute phase (p < 0.001) but showed no difference between the subacute phase and HCs (p = 0.367).Within the RSSI group, acute-phase Sema3A levels were significantly higher in the CI subgroup (p < 0.001). A strong negative correlation was found between Sema3A and MoCA scores (r = − 0.459, p < 0.001). The AUC for distinguishing CI from non-cognitive impairment (NCI) was 0.793 (p < 0.01). In addition, acute-phase plasma Sema3A levels were notably elevated in patients with a high total CSVD burden (p < 0.001), and were positively correlated with the total imaging burden score (r = 0.469, p < 0.001). Mediation analysis showed that white matter hyperintensities (WMH) and lacunar infarcts (LI) significantly mediated the relationship between acute-phase Sema3A and CI (p < 0.001).
Conclusion: Plasma Sema3A levels during the acute phase were elevated in RSSI patients and were linked to cognitive impairment through the mediating roles of WMH and LI.
Keywords: cerebral small vessel disease, recent small subcortical infarct, cognitive impairment, total burden score, Sema3A
Introduction
Cerebral small vessel disease (CSVD) is a syndrome involving clinical, imaging, and pathological changes in the brain’s small arteries, microarterioles, capillaries, microvessels, and small veins, arising from various causes. On MRI, CSVD typically appears as lacunar infarcts (LI), white matter hyperintensities (WMH), cerebral microbleeds, and enlarged perivascular spaces (EPVs), and cerebral atrophy.1 CSVD is a significant contributor to the global stroke burden, responsible for approximately 25% of all strokes and 20% of ischemic strokes. Moreover, a higher burden of CSVD on neuroimaging is independently associated with increased risks of functional disability, all-cause mortality, and stroke recurrence.2,3 Notably, disability develops in about 20% of CSVD patients,4 and the disease is a primary etiology of cognitive impairment following a stroke.5 Approximately 45% of vascular dementia cases are linked to CSVD,6 which often presents with impairments in attention, processing speed, executive function, and cognitive flexibility.7 A significant relationship has been reported between the total CSVD burden and cognitive decline.8 Therefore, early identification and intervention of CSVD are crucial for improving the long-term prognosis of stroke patients. Inflammation is a well-established factor in the pathogenesis of CSVD. Current evidence indicates that inflammation contributes to the pathogenesis of CSVD primarily by mediating endothelial dysfunction, disrupting tight junction proteins, promoting extracellular matrix remodeling, and activating glial cells9 represent. A cohort study showed a strong association between classical systemic inflammatory markers, such as C-reactive protein and IL-6, and both CMBs and RSSI. Elevated alpha 1-antichymotrypsin levels have also been found to correlate with severe WMH.10 Despite these findings, there are currently no specific biomarkers in routine clinical use for early CSVD diagnosis, and the exact link between inflammatory responses and CSVD-related cognitive decline remains unclear.
Sema3A (Semaphorin 3A) is a secreted glycoprotein initially identified for its chemorepellent effects in axon guidance.11 It plays an essential role in the nervous system, contributing to multiple aspects of neuronal development and function. Sema3A initiates intracellular signaling cascades and regulates axonal pathfinding, dendritic patterning, and cell migration by interacting with Neuropilin-1 and Plexin-A1 receptors.12–14 It is also recognized for its ability to regulate inflammatory processes and influence immune responses from initiation to resolution.13 Previous studies have indicated that Sema3A may intensify secondary neural injury after ischemic stroke through several pathological mechanisms, including inhibition of neurite regrowth, suppression of synaptic remodeling, and disruption of endogenous anti-inflammatory signaling pathways.15 Sema3A may cause a cascade reaction of inflammation through autocrine circulation, leading to an inflammatory storm.16 In addition, Sema3A can specifically recruit pro angiogenic mononuclear phagocytes to ischemic areas, promoting further development of inflammation.17 Sema3A aggravates ischemic brain injury by promoting neuronal apoptosis and amplifying immune-driven inflammation, leading to compromised blood-brain barrier integrity.18 More recently, experimental findings have shown that downregulation of Sema3A expression significantly reduced blood-brain barrier disruption, brain edema, and neurological deficits in mice with traumatic brain injury. It also alleviated endothelial barrier breakdown under hypoxic-ischemic conditions.19 Additionally, a recent review suggested that Sema3A may restrict structural plasticity in the human hippocampus by finely adjusting synaptic cleft dynamics. This mechanism could influence learning and cognitive performance.20 In summary, Sema3A exhibits a complex duality in the nervous system: on one hand, it may exert neuroprotective effects during development and under specific conditions; on the other hand, under pathological states such as ischemia and inflammation, its pro-inflammatory and neurotoxic roles may prevail, leading to secondary neural injury and vascular dysfunction. Taken together, As an indicator closely related to inflammation, Sema3A likely contributes to cerebrovascular damage and cognitive dysfunction. However, its specific role in CSVD and the clinical relevance of Sema3A in CSVD-associated CI remain largely unexplored.
RSSI is a key imaging marker of CSVD, representing the acute phase of the disease.21 In this study, we selected RSSI patients as the research cohort. Our aim was to measure plasma Sema3A levels in CSVD patients, examine their relationship with cognitive performance, and assess the clinical significance of Sema3A in CSVD-related cognitive impairment.
Method
Study Participants
This study prospectively and consecutively enrolled patients hospitalized in the Department of Neurology at the Affiliated Hospital of Qingdao University who were diagnosed with RSSI between August 2023 and May 2024. Inclusion criteria were: (1) acute onset with focal neurological deficits; (2) cranial MRI performed during the acute phase (within 3 days of symptom onset); (3) high signal on diffusion-weighted imaging (DWI); and (4) infarct diameter less than 20 mm. Exclusion criteria were: (1) presence of other types of cerebral infarction as classified by TOAST criteria; (2) hemorrhagic stroke; (3) comorbid neurodegenerative diseases such as Parkinson’s disease or Alzheimer’s disease; (4) severe cardiac, hepatic, or renal dysfunction; and (5) inflammation-related conditions including rheumatic autoimmune diseases or malignant tumors. Age- and gender-matched individuals undergoing routine physical examinations, with no history of cognitive impairment, were recruited during the same period as healthy controls. All participants provided written informed consent. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University (QYFYWZLL29423).
Data Collection
Baseline clinical data, neuroimaging findings, and blood biomarkers relevant to cerebrovascular disease were collected for all participants. Baseline variables included age, gender, vital signs (systolic and diastolic blood pressure, heart rate), medical history (hypertension, diabetes mellitus, hyperlipidemia), lifestyle history (smoking, alcohol intake), and laboratory indices (fasting glucose, triglycerides, total cholesterol, low-density lipoprotein, high-density lipoprotein, white blood cell count, C-reactive protein, D-dimer, fibrinogen, uric acid, and homocysteine levels). All RSSI patients underwent cranial MRI, cervical vascular evaluation (via cervical ultrasound or carotid CTA), intracranial vascular evaluation (via MRA or cerebral CTA), and cardiac ultrasound. Healthy controls underwent head CT or MRI to exclude cerebrovascular abnormalities before inclusion. Cognitive assessments for RSSI patients were performed by a neurologist using a standardized procedure in a controlled setting. Cognitive function was evaluated using the Montreal Cognitive Assessment (MoCA), adapted according to Chinese MoCA norms.22 CI was defined based on education level as follows: for individuals with ≥7 years of education, a MoCA score ≤24; for those with 1–6 years of education, a score ≤19; and for illiterate participants, a score ≤13.
Blood Sample Collection and Processing
All RSSI patients and healthy controls had 5 mL of venous blood collected in EDTA anticoagulant tubes while fasting in the early morning after admission. For RSSI patients, a second blood sample was collected during the subacute phase (14–30 days post-onset). All blood draws were performed between 6:00 AM and 8:00 AM, following a fasting period of at least 8 hours. To reduce pre-analytical variation, samples were processed within 30 minutes of collection. Blood was centrifuged at 3000 r/min for 8 minutes at 4°C to isolate plasma. The resulting clear, yellowish supernatant was promptly transferred into pre-chilled EP tubes to prevent freeze-thaw cycles. Aliquots were stored at −80°C in a dedicated freezer with continuous temperature monitoring until further analysis.
Measurement of Plasma Sema3A
Plasma Sema3A concentrations were measured using a double antibody sandwich enzyme-linked immunosorbent assay (ELISA, Human SEMA3A ELISA Kit, Elabscience, Wuhan, China). The ELISA kit had a detection range of 0.16–10 ng/mL for plasma Sema3A, with a minimum detectable concentration of 0.01 ng/mL. The assay showed no significant cross-reactivity with related proteins. Inter-assay CV% was below 10%, and intra-assay CV% was below 15%.
Neuroimaging Analysis
All RSSI patients underwent brain imaging using a 3.0T MRI scanner, following the updated international STRIVE-2 guidelines for neuroimaging of CSVD published in 2023.23 Each radiological marker of CSVD, LI, WMH, CMBs, and EPVs was assessed independently. Imaging was interpreted separately by two trained neurologists. In cases of disagreement, a third neurologist reviewed the findings, and consensus was reached through joint discussion. The presence of LI and CMBs was recorded as either present or absent. WMH was evaluated using the Fazekas scale and graded as 0, 1, 2, or 3. EPVs were assessed using the Potter scale based on number: 0, 1–10, 11–20, 21–40, and ≥40, corresponding to Grades 0 through 4. For the total CSVD burden score, one point was assigned for each of the following: presence of LI, presence of CMBs, EPVs ≥11, and Fazekas score ≥2. A total score of 0–2 was classified as the low-burden group, and 3–4 as the high-burden group.
Statistical Analysis
The distribution of quantitative data was assessed using the Kolmogorov–Smirnov test. Variables with a normal distribution were expressed as mean ± standard deviation, and group comparisons were performed using the independent samples t-test. Non-normally distributed data were presented as median (interquartile range), with comparisons made using the Mann–Whitney U-test. Categorical variables were reported as percentages and compared using Pearson’s chi-square test. Comparisons between the acute and subacute phases were conducted using the paired-samples t-test. For all pairwise comparisons among the acute phase, subacute phase, and control group, Bonferroni correction was applied (α = 0.05/3 = 0.0167). Univariate analysis was initially performed. Variables with p < 0.05 were included in a multivariate logistic regression to construct Model 1. Vascular risk factors (hypertension, diabetes, hyperlipidemia) and demographic variables (age, gender) were added in Model 2 for further adjustment. This approach was used to identify independent risk factors in patients with RSSI. The same method was used to identify independent predictors of cognitive impairment and high CSVD burden within the RSSI cohort. Diagnostic accuracy of the tested indices was evaluated using receiver operating characteristic (ROC) curve analysis. The AUC was calculated, and the Youden index was used to determine the optimal cutoff point. Spearman correlation was used to assess the relationship between plasma Sema3A levels and both the MoCA score and total CSVD burden score. Mediation analysis was conducted in the RSSI group, adjusting for age, sex, and education level, to determine whether specific imaging markers mediated the relationship between plasma Sema3A levels and cognitive performance. All statistical analyses were performed using IBM SPSS 25.0 (IBM Corporation, Armonk, NY, USA).
Result
Comparison of Clinical Characteristics Between RSSI Patients and HCs
This study included 122 patients diagnosed with RSSI, with a mean age of 60 years and 56.6% male, along with 100 HCs enrolled during the same period. Table 1 presents the comparison of baseline characteristics between the two groups. Compared to HCs, RSSI patients had significantly higher rates of hypertension, hyperlipidemia, and smoking history, as well as elevated levels of systolic blood pressure, diastolic blood pressure, white blood cell count, triglycerides, total cholesterol, low-density lipoprotein, homocysteine, and plasma Sema3A (p < 0.05). Plasma Sema3A was significantly higher in RSSI patients compared to HCs (7.70 ± 2.44 ng/mL vs 6.07 ± 1.82 ng/mL) (Figure 1A). After including all variables with p < 0.05 in a multivariate logistic regression and adjusting for demographic data and vascular risk factors, acute-phase plasma Sema3A was identified as an independent risk factor for RSSI, with an odds ratio (OR) of 1.433 (95% confidence interval [CI] = 1.148–1.789, p = 0.001) (Table 2).
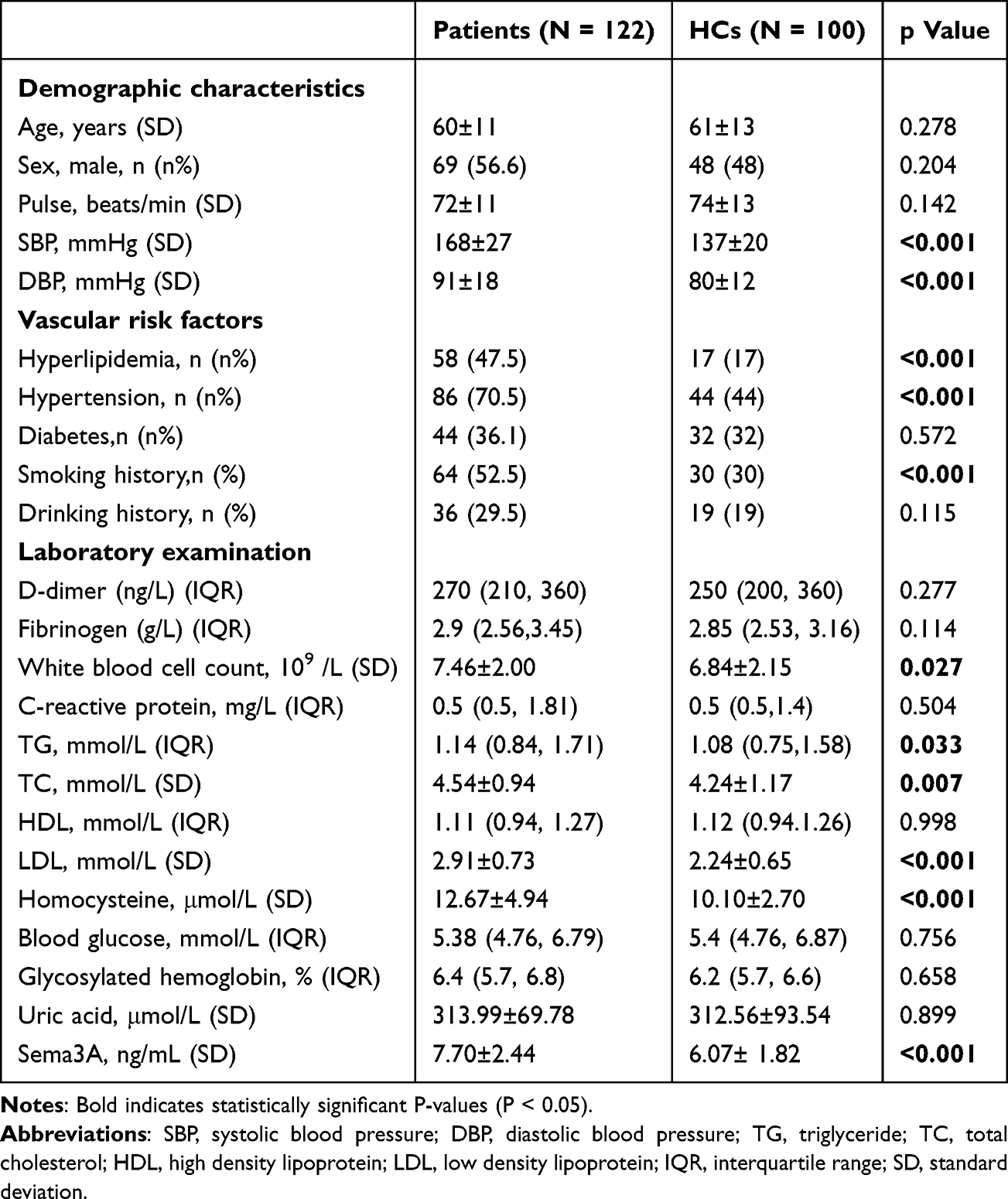 |
Table 1 Comparison of Clinical Baseline Data and Multivariate Logistic Regression Analysis. Comparison of Clinical Baseline Data Between RSSI Patients and HCs |
 |
Table 2 Comparison of Clinical Baseline Data and Multivariate Logistic Regression Analysis. Comparison. Multivariate Logistic Regression Analysis of RSSI Group and HCs Group |
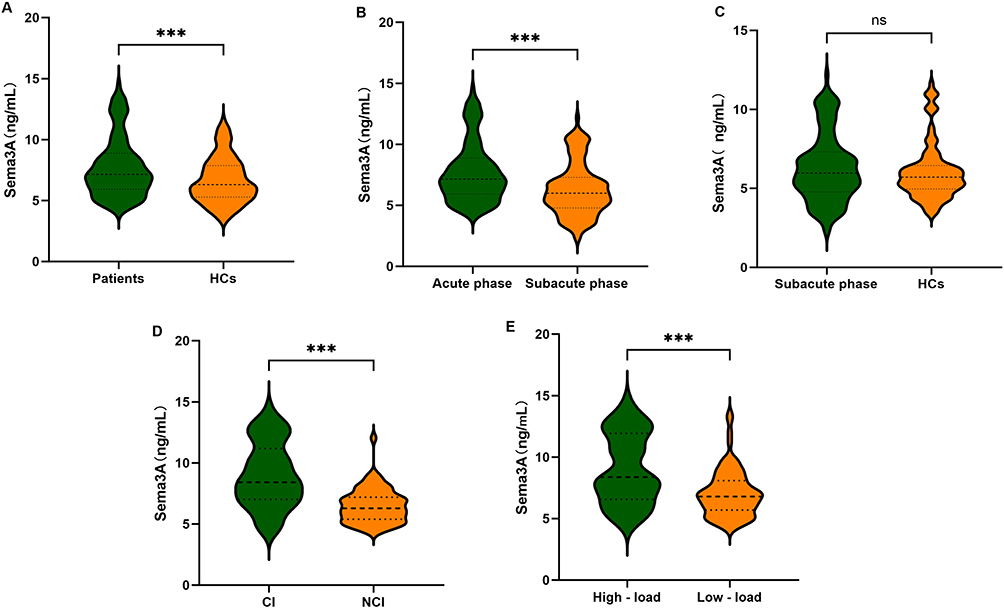 |
Figure 1 Violin plot comparing plasma Sema3A levels across diagnostic categories. (A) Patients vs HCs. (B) Acute phase vs subacute phase in RSSI patients. (C) Subacute phase vs HCs. (D) CI vs NCI subgroup in patients. (E) High-load vs low-load CSVD imaging score subgroups. Abbreviations: HC: healthy control; CI: cognitive impairment; NCI: non-cognitive impairment; CSVD: cerebral small vessel disease. *** P < 0.001.ns: not significant. |
A subgroup analysis within the HC group showed no significant difference in plasma Sema3A levels between individuals with and without vascular risk factors (Supplementary Figure 1). Given the known influence of sex and age on brain physiology, we also analyzed plasma Sema3A levels by sex and age subgroups within the RSSI group. No significant differences were observed between males and females (Supplementary Figure 2A) or across different age groups (Supplementary Figure 2B). We further divided HCs into four subgroups based on quartiles of plasma Sema3A levels. We compared the demographics and distribution of comorbidities among these quartile subgroups. The results revealed no statistically significant differences in any of the comparisons (all p > 0.05, Supplementary Table 1).
Comparison of Plasma Sema3A Levels Between the Acute and Subacute Phases in RSSI Patients
Plasma Sema3A levels were significantly higher during the acute phase than in the subacute phase in RSSI patients (7.70 ± 2.44 ng/mL vs 6.31 ± 2.15 ng/mL, p < 0.001) (Figure 1B). No significant difference was observed between the subacute phase and the healthy controls (6.31 ± 2.15 ng/mL vs 6.07 ± 1.82 ng/mL, p = 0.367) (Figure 1C).
Comparison of Clinical Characteristics Between the CI Subgroup and the NCI Subgroup of Patients with RSSI
Among the 122 patients with RSSI, 59 were categorized into the CI group and 63 into the NCI group based on MoCA scores. Patients in the CI group were older and showed significantly higher levels of white blood cell count, fasting glucose, homocysteine, and acute-phase plasma Sema3A compared with those in the NCI group. The total CSVD burden score was also significantly higher in the CI group (all p < 0.05, Supplementary Table 2). Acute-phase plasma Sema3A levels were significantly higher in the CI group than in the NCI group (9.00 ± 2.66 ng/mL vs 6.49 ± 1.40 ng/mL, p < 0.001) (Figure 1D). After adjusting for demographic and vascular risk factors in a multivariate logistic regression model, acute-phase plasma Sema3A remained an independent risk factor for CI, with an OR of 1.838 (95% CI = 1.313–2.573, p < 0.001) (Table 3). In addition, in the comparison of CI, NCI, and HCs,the results showed a highly significant statistical difference in Sema3A levels among the three groups (p < 0.001, Supplementary Table 3).
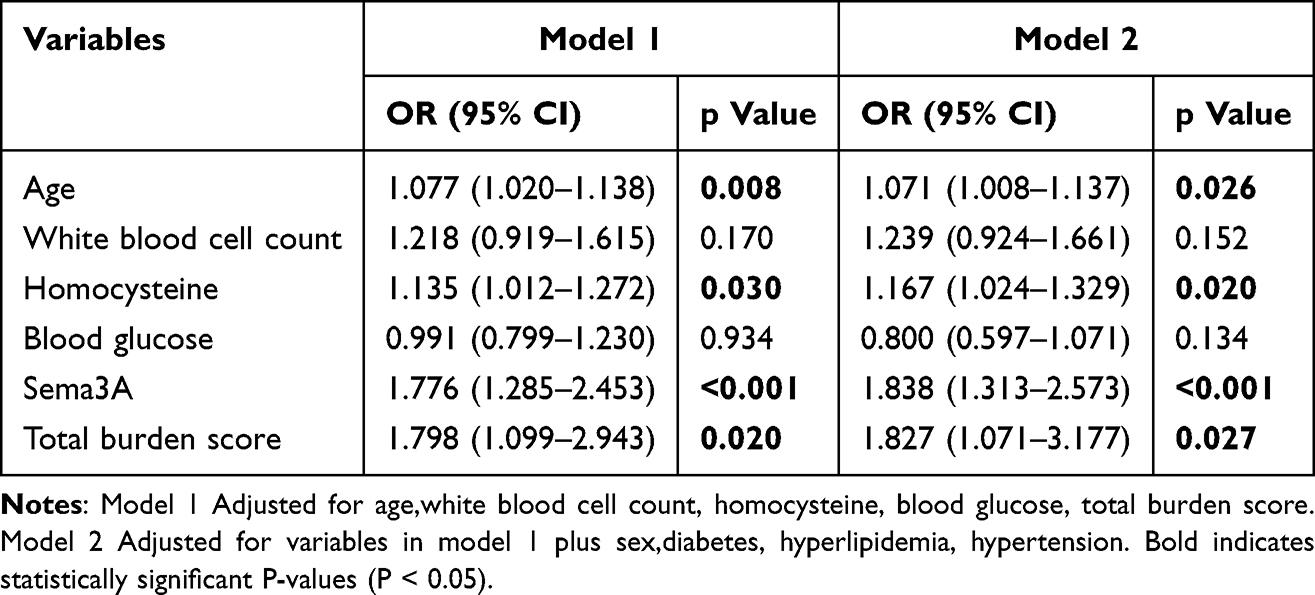 |
Table 3 Multivariate Logistic Regression Analysis of CI Subgroup and Non-CI Subgroup |
Correlation Between Plasma Sema3A Levels in the Acute Phase and Cognitive Function in Patients with RSSI
Spearman correlation analysis showed a significant negative correlation between acute-phase plasma Sema3A levels and MoCA scores (r = –0.459, p < 0.001) (Figure 2A). Further correlation analysis of individual cognitive domains within the MoCA revealed a significant negative association with acute-phase plasma Sema3A levels across all domains (Table 4).
 |
Table 4 Correlation Analysis of Plasma Sema3A Levels in the Acute Phase with MoCA Scores and Various Cognitive Domains in RSSI Patients |
 |
Figure 2 Correlation analysis of plasma Sema3A levels in the acute phase. (A) Correlation between plasma Sema3A levels and MoCA score in RSSI patients. (B) Correlation between plasma Sema3A levels and total burden score in RSSI patients. |
ROC Analysis of the Plasma Sema3A Levels in the Diagnosis of RSSI and CI in the Acute Phase
A ROC curve was constructed to evaluate the diagnostic performance of acute-phase plasma Sema3A levels for identifying RSSI and related CI (Figure 3). The AUC for distinguishing RSSI patients from HCs was 0.715 (p < 0.01, 95% CI: 0.647–0.784) (Figure 3A). The optimal threshold was 6.52 ng/mL, yielding a sensitivity of 63.1%, specificity of 79.0%, and a maximum Youden index of 0.483. Similarly, the AUC for differentiating between CI and NCI within RSSI patients was 0.793 (p < 0.01, 95% CI: 0.711–0.875), with an optimal threshold of 8.01 ng/mL. At this threshold, sensitivity was 61.0%, specificity was 87.3%, and the maximum Youden index was also 0.483 (Figure 3B).
 |
Figure 3 ROC analysis of plasma Sema3A levels. (A) ROC curve comparing RSSI patients and HCs. (B) ROC curve comparing CI and NCI subgroups in RSSI patients. |
Correlation Analysis of Plasma Sema3A Levels in the Acute Phase with CSVD Total Burden Score
Based on the total CSVD score, the 122 RSSI patients were divided into a low-load group (0–2 points, n = 81) and a high-load group (3–4 points, n = 41). Patients in the high-load group were older and had significantly higher levels of white blood cell count, homocysteine, and plasma Sema3A (Supplementary Table 4). After including all variables with p < 0.05 in a multivariate logistic regression and adjusting for demographic and vascular risk factors, acute-phase plasma Sema3A remained significantly elevated in the high-load group compared to the low-load group (9.02 ± 2.85 ng/mL vs 7.04 ± 1.91 ng/mL) (Figure 1E). Sema3A was also identified as an independent risk factor for high CSVD burden, with an odds ratio (OR) of 1.382 (95% CI = 1.249–1.663, p < 0.001) (Table 5). Spearman correlation analysis showed that acute-phase plasma Sema3A levels were positively correlated with the total CSVD burden score (r = 0.469, p < 0.001) (Figure 2B).
 |
Table 5 Multivariate Logistic Regression Analysis of High Load Group Versus Low Load Group Based on Total CSVD Burden Score |
The Mediating Analysis Between Sema3A, CSVD Total Burden Score, and Cognitive Function
Spearman correlation analysis showed that acute-phase plasma Sema3A levels were negatively associated with overall MoCA scores and positively associated with total CSVD burden scores. Based on these findings, we conducted a mediation analysis to examine whether the CSVD burden mediated the relationship between Sema3A and cognitive function. A conceptual model of the mediation structure is shown in Figure 4A. Age, sex, and education were included as covariates in the model. The analysis revealed that the CSVD total burden score significantly mediated the negative effect of elevated acute-phase Sema3A levels on MoCA scores (indirect effect = –0.2858, 95% CI = –1.979 to –0.0374) (Figure 4B). To further clarify the contribution of individual imaging markers, we separately analyzed the components of the total CSVD burden score LI, WMH, CMBs, and EPVs. The presence of LI mediated a significant indirect negative effect of acute-phase Sema3A on MoCA scores (indirect effect = –0.2207, 95% CI = –2.4430 to –0.1747) (Figure 4C). Increased WMH also contributed to this effect (indirect effect = –0.1456, 95% CI = –0.2917 to –0.0255) (Figure 4F). Although CMBs (p < 0.001) and EPVs (p < 0.01) were each directly associated with lower MoCA scores, they did not mediate the impact of elevated Sema3A levels on cognitive function (Figure 4D and E).
 |
Figure 4 Mediation analysis of plasma Sema3A, CSVD total burden score, and cognitive function. (A) Conceptual diagram of mediation analysis with single mediator (M). Total effect of X on Y (c) = indirect effect through M (a × b) + direct effect (c’). (B) Sema3A (X), total burden score (M), MoCA (Y). (C) Sema3A (X), LI (M), MoCA (Y). (D) Sema3A (X), CMBs (M), MoCA (Y). (E) Sema3A (X), EPVs (Potter score) (M), MoCA (Y). (F) Sema3A (X), WMH (Fazekas score) (M), MoCA (Y). Covariates: age, sex, education.***P < 0.001. |
Discussion
This study examined the association between plasma Sema3A levels and clinical, cognitive, and imaging features in patients with RSSI. Plasma Sema3A levels were significantly elevated in RSSI patients compared to HCs, with the highest levels observed during the acute phase. No significant difference was found between subacute-phase Sema3A levels and those of HCs. In cognitive subgroup analysis, acute-phase plasma Sema3A levels were significantly higher in patients with CI than in those without (NCI). These levels were negatively correlated with total MoCA scores and with each cognitive domain, indicating a broad impact on cognitive performance. In imaging subgroup comparisons, acute-phase plasma Sema3A levels were significantly higher in the high CSVD burden group than in the low-burden group. Sema3A levels were also positively correlated with total CSVD burden scores. Mediation analysis showed that the association between acute-phase plasma Sema3A levels and cognitive decline was mediated by the total CSVD burden score. Among the imaging markers, LI and WMH played a significant mediating role in this relationship.
Our study found that higher homocysteine, LDL and SBP are independent risk factors for RSSI. Additionally, we observed that patients with smoking are at greater risk for RSSI. Higher homocysteine level is related with the neuroimaging burdens and cognitive impairment in CSVD patients, which is consistent with previous studies on inhibition24 Elevated Hcy levels can disrupt the integrity of the blood-brain barrier, enhance vascular inflammatory response, and downregulate vascular endothelial growth factor, leading to endothelial cell apoptosis. Elevated Hcy can exacerbate arteriosclerosis through cerebral vascular atrophy, further leading to chronic diffuse low brain tissue damage, and causing changes such as white matter degeneration in brain tissue ultimately leading to cognitive impairment.25 LDL is an important factor for arteriosclerosis at the small perforating artery opening, leading to occlusion of the perforating artery and RSSI. And LDL induced chronic ischemia can cause ischemic changes in white matter, leading to the occurrence of CSVD and affecting cognition to a certain extent.26,27 Similarly, long-term smoking can also lead to changes in the microstructure of cerebral blood vessels and white matter, as well as chronic cerebral hypoperfusion, which in turn affects the occurrence of CSVD.28 We found that increasing age is an independent risk factor for cognitive impairment and high burden of CSVD imaging, which is consistent with previous studies.21,29 A higher CSVD total load score is associated with an increased risk of mild cognitive impairment in middle-aged and elderly individuals, and the higher the total load score, the more severe the cognitive impairment may be; Especially severe CSVD burden can cause damage to multiple cognitive domains in the brain, which may be related to the increased probability of developing cerebrovascular disease risk factors with age,30 such as hypertension. Hypertension is an independent risk factor for CVSD,31 which can cause damage to the blood-brain barrier, endothelial dysfunction, and damage to oligodendrocytes, leading to white matter lesions such as demyelination, axonal loss, and decreased glial density.32
Plasma Sema3A levels were significantly elevated in patients with RSSI during the acute phase compared to HCs in this study. Previous research suggests that Sema3A may contribute to ischemic brain injury by disrupting endothelial cell integrity and increasing vascular permeability.33 Overexpression of Sema3A may weaken neuronal tissue and its connections, making neurons more vulnerable to additional injury.34 Besides, we found that the physiological variation of Sema3A levels in a healthy population is not associated with these common demographics and vascular comorbidities. Consequently, the elevated Sema3A levels are more likely to be specifically related to the pathological process of CSVD rather than being driven by underlying physiological conditions or comorbidities. It is speculated that Sema3A may interact with the progression of atherosclerosis and inflammatory cascades under pathological conditions, contributing to disease development. In assessing its diagnostic value, ROC analysis showed that plasma Sema3A had an AUC of 0.715, with a threshold of 6.52 ng/mL, for distinguishing acute-phase RSSI from HCs. Multivariate analysis confirmed that acute-phase plasma Sema3A was an independent indicator of RSSI after adjusting for vascular risk factors and demographic characteristics, suggesting that its role may be more directly tied to CSVD pathophysiology, possibly through mechanisms such as endothelial dysfunction, rather than simply reflecting vascular comorbidities. Our results further demonstrated that Sema3A expression was highest during the acute phase of RSSI and decreased as the condition entered the subacute phase. This acute-phase increase may reflect a dual response involving both neuroprotection and inflammatory regulation. We propose that Sema3A might support neuronal survival and help suppress inflammation early in the disease by binding to Neuropilin-1/Plexin-A4 receptors, thereby stabilizing microtubules in ischemic neurons and potentially reducing Wallerian degeneration during acute ischemia.35–37 However, Sema3A expression seems to be downregulated in later stages through feedback inhibition mechanisms,38 which may explain the observed reduction in expression during the subacute phase. Taken together, these findings suggest that acute-phase plasma Sema3A is a disease-associated molecule relevant to the pathophysiology of CSVD and warrants further study. Our findings suggest that acute-phase plasma Sema3A elevation in RSSI could represent a potential time window for early intervention, thus providing a rationale for future development of anti-Sema3A targeted therapies or the selection of existing anti-inflammatory strategies. Nonetheless, its precise neurophysiological role remains unclear and should be clarified in future research.
This study identified a strong association between plasma Sema3A levels and cognitive performance in patients with RSSI. Elevated plasma Sema3A levels during the acute phase were observed in patients with CI compared to those without, consistent with prior research on cognitive decline.39,40 Moreover, plasma Sema3A levels showed a substantial negative correlation with all cognitive domains assessed by the MoCA scale, including memory, attention, language, and executive function, suggesting a broad impact rather than domain-specific impairment. We suggest that overexpression of Sema3A may compromise neuronal structure and connectivity.34 Its role in vascular remodeling and inflammatory responses could further disrupt brain network function, contributing to widespread cognitive decline. Thus, elevated Sema3A may reflect diffuse neuronal damage and degenerative changes across multiple cognitive systems. ROC analysis showed that Sema3A had an AUC of 0.793 for distinguishing CI from non-CI patients, with a threshold of 8.01 ng/mL. These findings suggest that acute-phase plasma Sema3A elevation in RSSI could represent a potential time window for early intervention. Plasma Sema3A measurement may also serve as a useful complement to standard cognitive evaluation tools such as neuroimaging or performance-based scales. However, additional multicenter studies are necessary to validate its clinical applicability and establish standardized cutoff values.
Correlation investigation between Sema3A and CSVD total burden score revealed a positive correlation between acute-phase plasma Sema3A levels and the total CSVD burden score in RSSI patients. Sema3A levels were significantly higher in the high-burden group compared to the low-burden group. This finding aligns with earlier studies reporting that circulating inflammatory markers are closely linked to the severity and progression of CSVD imaging features.10 Mediation analysis further indicated that the increase in total CSVD burden significantly mediated the negative effect of elevated Sema3A levels on MoCA scores. This result is consistent with previous research showing that the CSVD total burden score reliably reflects disease severity as visualized on MRI and is inversely associated with cognitive performance.41–43 When we analyzed the specific contributions of each of the four imaging components of the total burden score, we found that the severity of WMH and the presence of LI contributed most strongly to the adverse impact of Sema3A on MoCA performance. Prior studies have shown that WMH and LI are associated with elevated levels of endothelial inflammatory markers.44,45 These markers may damage the vascular endothelium, raising the risk of WMH and LI. WMH may disrupt white matter–gray matter pathways, interfering with neural transmission and information processing, which can lead to cognitive impairment.46 LI can affect cerebrovascular microcirculation and cause ischemic injury, particularly in the frontal and parietal lobes, where disrupted connectivity contributes to cognitive dysfunction.47 We hypothesize that elevated plasma Sema3A may increase vascular permeability by damaging endothelial cells and altering their structure.33,48,49 This process could contribute to the development of LI and more severe WMH, ultimately impairing cognitive function. Our findings also indicated that the presence of CMBs and EPVs significantly reduced MoCA scores, consistent with previous studies.50,51 However, these two markers did not show a significant mediating effect between Sema3A and cognitive outcomes in our data. We propose that the relationship between plasma Sema3A and EPVs or CMBs may be parallel rather than sequential, and that brain tissue injury in cognitively intact individuals may still be in a mild or early stage. Future research with larger sample sizes is needed to test this hypothesis.
This study has several limitations. It was a single-center study conducted in a northern Chinese population, with a relatively small sample size, which may limit the generalizability of the findings. Additionally, the analysis focused only on plasma Sema3A levels; cerebrospinal fluid was not examined, which may have provided further insight into central nervous system involvement. Only plasma Sema3A levels were assessed during the subacute phase, and no concurrent laboratory indicators were evaluated. Therefore, the possibility of additional confounding variables cannot be excluded. Besides, patients in this study were all sampled after taking antiplatelet drugs, which prevented us from conducting efficacy analysis of antiplatelet drugs. Future research should address two key directions: (1) investigating the therapeutic relevance of Sema3A in CSVD and associated cognitive decline, and (2) conducting dynamic monitoring of Sema3A together with inflammatory cytokines throughout the acute and subacute stages of RSSI. Developing a combined predictive model may improve diagnostic precision. To confirm these results, larger, multicenter, prospective longitudinal studies and mechanistic experiments are needed.
Conclusion
In summary, this study demonstrated that plasma Sema3A levels during the acute phase were significantly associated with acute CSVD and influenced cognitive function through the mediating roles of white matter hyperintensities and lacunar infarcts. These findings provide new insight into the pathogenesis of CSVD and its cognitive consequences, suggesting that acute-phase plasma Sema3A may serve as an early clinical indicator of CSVD. Further large-scale prospective interventional trials are necessary to validate these findings and explore the role of Sema3A in CSVD treatment.
Data Sharing Statement
All data generated or analyzed during this study are included in this article and its Supplementary Material Files. Further enquiries can be directed to either corresponding author.
Statement of Ethics
This study was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University, approval number (QYFYWZLL29423).
Acknowledgments
The work presented was carried out in collaboration between all colleagues in our department, here is the thanks given for their help.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (81971111, 82371331 and 82401545); and Shandong Provincial Natural Science Foundation (ZR2022QH363).
Disclosure
The authors have no conflicts of interest to declare for this work.
References
1. Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. 2013;12(5):483–497. doi:10.1016/S1474-4422(13)70060-7
2. Nam KW, Kwon HM, Lim JS, et al. The presence and severity of cerebral small vessel disease increases the frequency of stroke in a cohort of patients with large artery occlusive disease. PLoS One. 2017;12(10):e0184944. doi:10.1371/journal.pone.0184944
3. Zhu S, Xu T, Xu T, et al. White Matter Hyperintensity, Immediate Antihypertensive Treatment, and Functional Outcome After Acute Ischemic Stroke. Stroke. 2020;51(5):1608–1612. doi:10.1161/STROKEAHA.119.028841
4. Zhu H, Li Z, Lv J, et al. Effects of cerebral small vessel disease on the outcome of patients with ischemic stroke caused by large artery atherosclerosis. Neurological Res. 2018;40(5):381–390. doi:10.1080/01616412.2018.1446283
5. Teng Z, Dong Y, Zhang D, et al. Cerebral small vessel disease and post-stroke cognitive impairment. Int J Neurosci. 2017;127(9):824–830. doi:10.1080/00207454.2016.1261291
6. Feigin VL, Roth GA, Naghavi M, et al. Global burden of stroke and risk factors in 188 countries, during 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016;15(9):913–924. doi:10.1016/S1474-4422(16)30073-4
7. Zhào H, Wei W, Do EY, et al. Assessing Performance on Digital Clock Drawing Test in Aged Patients With Cerebral Small Vessel Disease. Front Neurol. 2019;10:1259. doi:10.3389/fneur.2019.01259
8. Mu R, Qin X, Guo Z, et al. Prevalence and Consequences of Cerebral Small Vessel Diseases: a Cross-Sectional Study Based on Community People Plotted Against 5-Year Age Strata. Neuropsychiatr Dis Treat. 2022;18:499–512. doi:10.2147/NDT.S352651
9. Grochowski C, Litak J, Kamieniak P, et al. Oxidative stress in cerebral small vessel disease. Role of reactive species. Free Radical Research. 2018;52(1):1–13. doi:10.1080/10715762.2017.1402304
10. Gu Y, Gutierrez J, Meier IB, et al. Circulating inflammatory biomarkers are related to cerebrovascular disease in older adults. Neurology(R) Neuroimmunology & Neuroinflammation. 2019;6(1):e521. doi:10.1212/NXI.0000000000000521
11. Luo Y, Raible D, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1993;75(2):217–227. doi:10.1016/0092-8674(93)80064-L
12. Roth L, Koncina E, Satkauskas S, et al. The many faces of semaphorins: from development to pathology. Cell Mol Life Sci. 2009;66(4):649–666. doi:10.1007/s00018-008-8518-z
13. Suzuki K, Kumanogoh A, Kikutani H. Semaphorins and their receptors in immune cell interactions. Nat Immunol. 2008;9(1):17–23. doi:10.1038/ni1553
14. Rehman M, Tamagnone L. Semaphorins in cancer: biological mechanisms and therapeutic approaches. Semin Cell Dev Biol. 2013;24(3):179–189. doi:10.1016/j.semcdb.2012.10.005
15. Hira K, Ueno Y, Tanaka R, et al. Astrocyte-Derived Exosomes Treated With a Semaphorin 3A Inhibitor Enhance Stroke Recovery via Prostaglandin D(2) Synthase. Stroke. 2018;49(10):2483–2494. doi:10.1161/STROKEAHA.118.021272
16. Wen H, Lei Y, Eun SY, et al. Plexin-A4-semaphorin 3A signaling is required for Toll-like receptor- and sepsis-induced cytokine storm. J Exp Med. 2010;207(13):2943–2957. doi:10.1084/jem.20101138
17. Dejda A, Mawambo G, Cerani A, et al. Neuropilin-1 mediates myeloid cell chemoattraction and influences retinal neuroimmune crosstalk. J Clin Invest. 2014;124(11):4807–4822. doi:10.1172/JCI76492
18. Yang L, Wang L, Wang J, et al. Long non-coding RNA Gm11974 aggravates oxygen-glucose deprivation-induced injury via miR-122-5p/SEMA3A axis in ischaemic stroke. Metab Brain Dis. 2021;36(7):2059–2069. doi:10.1007/s11011-021-00792-7
19. Yang M, Wang X, Fan Y, et al. Semaphorin 3A Contributes to Secondary Blood-Brain Barrier Damage After Traumatic Brain Injury. Front Cell Neurosci. 2019;13:117. doi:10.3389/fncel.2019.00117
20. Carulli D, De Winter F, Verhaagen J. Semaphorins in Adult Nervous System Plasticity and Disease. Front Synaptic Neurosci. 2021;13:672891. doi:10.3389/fnsyn.2021.672891
21. Cannistraro RJ, Badi M, Eidelman BH, et al. CNS small vessel disease: a clinical review. Neurology. 2019;92(24):1146–1156. doi:10.1212/WNL.0000000000007654
22. Lu J, Li D, Li F, et al. Montreal cognitive assessment in detecting cognitive impairment in Chinese elderly individuals: a population-based study. J Geriatric Psychiatry Neurol. 2011;24(4):184–190. doi:10.1177/0891988711422528
23. Duering M, Biessels GJ, Brodtmann A, et al. Neuroimaging standards for research into small vessel disease-advances since 2013. Lancet Neurol. 2023;22(7):602–618. doi:10.1016/S1474-4422(23)00131-X
24. Ji Y, Li X, Teng Z, et al. Homocysteine is Associated with the Development of Cerebral Small Vessel Disease: retrospective Analyses from Neuroimaging and Cognitive Outcomes. J Stroke Cerebrovascular Dis. 2020;29(12):105393. doi:10.1016/j.jstrokecerebrovasdis.2020.105393
25. Pařízková M, Andel R, Lerch O, et al. Homocysteine and Real-Space Navigation Performance among Non-Demented Older Adults. JAD. 2017;55(3):951–964. doi:10.3233/JAD-160667
26. Duong MT, Nasrallah IM, Wolk DA, et al. Cholesterol, Atherosclerosis, and APOE in Vascular Contributions to Cognitive Impairment and Dementia (VCID): potential Mechanisms and Therapy. Front Aging Neurosci. 2021;13:647990. doi:10.3389/fnagi.2021.647990
27. Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313–2330. doi:10.1093/eurheartj/ehz962
28. Gons RA, Van Norden AG, De Laat KF, et al. Cigarette smoking is associated with reduced microstructural integrity of cerebral white matter. Brain. 2011;134(Pt 7):2116–2124. doi:10.1093/brain/awr145
29. Calabrese P, Sitek EJ, Korczyn AD, et al. The assessment of cognitive and behavioural disturbances in vascular cognitive impairment (VCI) - recommendations of an expert working group. Neurologia i neurochirurgia polska. 2021;55(4):333–345. doi:10.5603/PJNNS.a2021.0035
30. Zhi N, Zhang L, Wang Y, et al. Modified cerebral small vessel disease score is associated with vascular cognitive impairment after lacunar stroke. Aging. 2021;13(7):9510–9521. doi:10.18632/aging.202438
31. Hainsworth AH, Markus HS, Schneider JA. Cerebral Small Vessel Disease, Hypertension, and Vascular Contributions to Cognitive Impairment and Dementia. Hypertension. 2024;81(1):75–86. doi:10.1161/HYPERTENSIONAHA.123.19943
32. Goto M, Hagiwara A, Fujita S, et al. Influence of Mild White Matter Lesions on Voxel-based Morphometry. MRMS. 2021;20(1):40–46.
33. Hou ST, Nilchi L, Li X, et al. Semaphorin3A elevates vascular permeability and contributes to cerebral ischemia-induced brain damage. Sci Rep. 2015;5(1):7890. doi:10.1038/srep07890
34. Ferretti G, Romano A, Sirabella R, et al. An increase in Semaphorin 3A biases the axonal direction and induces an aberrant dendritic arborization in an in vitro model of human neural progenitor differentiation. Cell Biosci. 2022;12(1):182. doi:10.1186/s13578-022-00916-1
35. Majed HH, Chandran S, Niclou SP, et al. A novel role for Sema3A in neuroprotection from injury mediated by activated microglia. The Journal of Neuroscience. 2006;26(6):1730–1738. doi:10.1523/JNEUROSCI.0702-05.2006
36. Rienks M, Carai P, Bitsch N, et al. Sema3A promotes the resolution of cardiac inflammation after myocardial infarction. Basic Res Cardiol. 2017;112(4):42. doi:10.1007/s00395-017-0630-5
37. Sumi C, Hirose N, Yanoshita M, et al. Semaphorin 3A Inhibits Inflammation in Chondrocytes under Excessive Mechanical Stress. Mediators Inflammation. 2018;2018:5703651. doi:10.1155/2018/5703651
38. Liang Y, Wang W, Huang J, et al. Potential Role of Semaphorin 3A and Its Receptors in Regulating Aberrant Sympathetic Innervation in Peritoneal and Deep Infiltrating Endometriosis. PLoS One. 2015;10(12):e0146027. doi:10.1371/journal.pone.0146027
39. Good PF, Alapat D, Hsu A, et al. A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. Journal of Neurochemistry. 2004;91(3):716–736. doi:10.1111/j.1471-4159.2004.02766.x
40. Yan YN, Williams JP, Niu K, et al. Intraperitoneal ozone injection prevents REM sleep deprivation - induced spatial learning and memory deficits by suppressing the expression of Sema3A in the hippocampus in rats. Iran J Basic Med Sci. 2022;25(8):980–988. doi:10.22038/IJBMS.2022.63994.14112
41. Tao W, Liu J, Ye C, et al. Relationships between cerebral small vessel diseases markers and cognitive performance in stroke-free patients with atrial fibrillation. Front Aging Neurosci. 2022;14:1045910. doi:10.3389/fnagi.2022.1045910
42. Zhang X, Liang C, Feng M, et al. Aberrant brain structural-functional connectivity coupling associated with cognitive dysfunction in different cerebral small vessel disease burdens. CNS Neurosci Ther. 2024;30(9):e70005. doi:10.1111/cns.70005
43. Jansen MG, Griffanti L, Mackay CE, et al. Association of cerebral small vessel disease burden with brain structure and cognitive and vascular risk trajectories in mid-to-late life. J Cerebral Blood Flow Metabo. 2022;42(4):600–612. doi:10.1177/0271678X211048411
44. Van Dijk EJ, Prins ND, Vermeer SE, et al. C-reactive protein and cerebral small-vessel disease: the Rotterdam Scan Study. Circulation. 2005;112(6):900–905. doi:10.1161/CIRCULATIONAHA.104.506337
45. Satizabal CL, Zhu YC, Mazoyer B, et al. Circulating IL-6 and CRP are associated with MRI findings in the elderly: the 3C-Dijon Study. Neurology. 2012;78(10):720–727. doi:10.1212/WNL.0b013e318248e50f
46. O’sullivan M, Morris RG, Huckstep B, et al. Diffusion tensor MRI correlates with executive dysfunction in patients with ischaemic leukoaraiosis. J Neurol Neurosurg. 2004;75(3):441–447. doi:10.1136/jnnp.2003.014910
47. Liu C, Shi L, Zhu W, et al. Fiber Connectivity Density in Cerebral Small-Vessel Disease Patients With Mild Cognitive Impairment and Cerebral Small-Vessel Disease Patients With Normal Cognition. Front Neurosci. 2020;14:83. doi:10.3389/fnins.2020.00083
48. Li C, Zhao Y, Li F, et al. Semaphorin3A Exacerbates Cardiac Microvascular Rarefaction in Pressure Overload-Induced Heart Disease. Adv Sci. 2023;10(21):e2206801. doi:10.1002/advs.202206801
49. Maione F, Capano S, Regano D, et al. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest. 2012;122(5):1832–1848. doi:10.1172/JCI58976
50. Chung CP, Chou KH, Chen WT, et al. Strictly Lobar Cerebral Microbleeds Are Associated With Cognitive Impairment. Stroke. 2016;47(10):2497–2502. doi:10.1161/STROKEAHA.116.014166
51. Javierre-Petit C, Kontzialis M, Leurgans SE, et al. Quantitative assessment of enlarged perivascular spaces via deep-learning in community-based older adults reveals independent associations with vascular neuropathologies, vascular risk factors and cognition. Brain Comm. 2024;6(4):fcae252. doi:10.1093/braincomms/fcae252
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.