Back to Journals » Neuropsychiatric Disease and Treatment » Volume 22
Plasma Plasminogen Activator Inhibitor-1 as a Biomarker for Disease Activity and Pharmaco-Response Prediction in Pediatric Epilepsy: A Prospective Cohort Study
Authors Xiao X, Shi XY, Feng J, Zhou XY, Wang ML ![]() , Zhang BB
, Zhang BB ![]() , Xu C, Tang JH
, Xu C, Tang JH
Received 20 November 2025
Accepted for publication 2 March 2026
Published 12 March 2026 Volume 2026:22 583183
DOI https://doi.org/10.2147/NDT.S583183
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Taro Kishi
Xiao Xiao,* Xiao-Yan Shi,* Jun Feng, Xin-Yu Zhou, Man-Li Wang, Bing-Bing Zhang, Chen Xu, Ji-Hong Tang
Department of Neurology, Children’s Hospital of Soochow University, Suzhou, Jiangsu, 215025, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ji-Hong Tang, Department of Neurology, Children’s Hospital of Soochow University, Suzhou, Jiangsu, 215025, People’s Republic of China, Email [email protected]
Purpose: To investigate the clinical relevance of plasma plasminogen activator inhibitor-1 (PAI-1) in pediatric epilepsy, focusing on its associations with seizure severities and therapeutic outcomes.
Methods: We conducted a prospective cohort study to compare the plasma PAI-1 levels quantified by ELISA across children with active epilepsy, seizure-free patients, and healthy controls. Furthermore, subgroup analyses were conducted to assess the impact of AED treatment or long-term drug response to the plasma PAI-1 levels.
Results: PAI-1 levels were 2.1-fold higher in the seizure group than in the control group (p < 0.0001), and 1.3-fold higher than in remission patients (p < 0.0001). No significant difference was observed between the anti-epileptic drug-treated and untreated subgroups (p = 0.0689). Baseline PAI-1 levels predicted 12-month pharmaco-responses, with pharmaco-resistant patients showing 12% higher PAI-1 concentrations than responders (p = 0.0234).
Conclusion: Our findings establish plasma PAI-1 as a promising biomarker for identifying children at high risk for pharmaco-resistant epilepsy, thereby addressing a high-burden condition. The persistence of PAI-1 elevation hints at underlying inflammatory or synaptic pathologies that may be novel therapeutic targets beyond conventional AEDs.
Keywords: plasminogen activator inhibitor-1, PAI-1, pediatric epilepsy, drug-resistant epilepsy, biomarker, neuroinflammation
Introduction
Plasminogen activator inhibitor-1 (PAI-1), a critical regulator of fibrinolysis, has emerged as a multifunctional player in thrombosis, inflammation, and tissue remodeling.1,2 As a serine protease inhibitor, PAI-1 primarily inhibits tissue-type and urokinase-type plasminogen activators, thereby modulating plasmin generation and affecting extracellular matrix dynamics.3 Beyond its well-documented role in cardiovascular diseases, growing evidence implicates PAI-1 in neurological disorders such as post-traumatic stress disorders (PTSD), depression, stroke, Alzheimer’s disease, and multiple sclerosis.4–9 Its dual role in fibrinolysis and neuroinflammation suggests that PAI-1 is a key mediator at the intersection of vascular and inflammatory pathways in the central nervous system.10
Epilepsy represents a significant global health challenge, affecting an estimated 70 million individuals, and the majority have an onset in childhood.11 It is particularly noteworthy that the incidence of neonatal seizures (approx. 0.1–0.5%) is significantly higher than in other childhood periods.12 Recurrent neonatal seizures may cause impairment of cortical network structure and function, which can, in the long term, adversely affect cognition, behavior, and learning capacity, while also elevating the risk for drug-resistant epilepsy, which contributes an approximately of 30% of epilepsy.13 In this case, the pursuit of reliable circulating biomarkers to prognosticate disease trajectory and therapeutic efficacy represents a critical frontier in the personalized management of epilepsy. Beyond electroclinical assessments, molecular indicators could significantly refine patient stratification. Elevated plasma levels of both forms of the gut-brain peptides ghrelin and des-acyl ghrelin are associated with a favorable response to antiseizure medications.14 Simultaneously, inflammatory miRNAs are increasingly recognized as promising diagnostic biomarkers and therapeutic targets in epilepsy.15 These findings highlight the intricate link between systemic metabolism and neuronal excitability. In parallel, the fibrinolytic and inflammatory mediator PAI-1 has surfaced as a compelling candidate worthy of detailed clinical investigation.
Recent studies suggest that PAI-1 may contribute to epilepsy, a complex disorder characterized by recurrent seizures and aberrant neuronal activity.16 Abnormal PAI-1 expression in the brain has been linked to microthrombosis, blood–brain barrier (BBB) disruption, and neuroinflammation processes, which have now been recognized as critical to epileptogenesis.17–20 Specifically, based on the study by Thomas et al, elevated PAI-1 following status epilepticus impairs proBDNF cleavage to mBDNF by inhibiting tPA activity, thus promoting neuronal hyperexcitability and epileptogenesis in the developing brain.21 The results further connect neuroinflammation to seizure pathogenesis. Additionally, our preliminary cytokine profiling study extended these findings, demonstrating elevated PAI-1 levels in children with refractory epilepsy alongside IGFBP2 and CCL5,22 suggesting a direct link between PAI-1 and synaptic dysfunction.
Despite these advances, the clinical use of PAI-1 as a biomarker and its therapeutic potential remain limited. One study indicated that the PAI-1 inhibitor could serve as a protector against doxorubicin-induced cellular senescence, as well as reduce oxidative stress.23 Another study found that Tiplaxtinin, another PAI-1 inhibitor, attenuated neointimal hyperplasia by promoting PAI-1 cleavage to an inactive form (CL-PAI-1) that enhanced VSMC apoptosis via the TWEAK/FN14 pathway.24 No findings of PAI-1 or its inhibitor were reported during epilepsy.
However, prospective clinical evidence linking plasma PAI-1 dynamics to epilepsy progression and drug resistance remains limited, particularly in pediatric populations. This study therefore aimed to evaluate the possible association between plasma PAI-1 levels and epilepsy severity in children, while characterizing the mechanism of PAI-1 in synaptic consolidation and neuronal excitability. Our findings may advance personalized management strategies for pediatric epilepsy.
Methods
Study Design and Participant Recruitment
A prospective cohort study was conducted at the Children’s Hospital Affiliated to Soochow University, from August 2022 to December 2023. In this study, pediatric participants were systematically categorized into three baseline groups following stringent eligibility protocols.
The active seizure group (n = 47, median age: 6 years; range: 0.217−16.2 years; male-to-female ratio: 25:22) included children with at least one seizure episode within 72 hours prior to enrollment. We excluded patients with identifiable epilepsy causes (determined through medical histories, physical examinations, blood tests assessing metabolic/inflammatory markers, and brain MRI/CT scans) and those with chronic conditions including autoimmune disorders, immunodeficiency syndromes, diabetes, malignancies, or severe neuropsychiatric comorbidities such as autism spectrum disorder with intellectual disabilities. Additional exclusions included individuals with acute systemic infections or recent antibiotic use within 14 days prior to serum sampling.
The remission group (n = 19; median age: 8 years; range: 1−15 years; male-to-female ratio: 14:5) was defined by seizure freedom for ≥6 consecutive months without normalized electrographic activity, while age- and sex-matched healthy controls (n = 21; median age: 1.417 years; range: 0.43−11 years; male-to-female ratio: 13:8) were recruited from the community, excluding individuals with personal or familial neurological histories.
In advance, to test whether serum PAI-1 levels were influenced by prior anti-epileptic drug (AED) use or could predict seizure severity, the active epilepsy cohort was twice divided into subgroups, first by medication history, then by clinical outcomes.
Participants were first divided into the AED-treated subgroup (n = 14) and untreated subgroup (n = 33). The former included patients receiving ≥3 months of guideline-adherent antiseizure pharmacotherapy per International League Against Epilepsy (ILAE) recommendations,25 while the latter subgroup included newly diagnosed patients without prior AED exposure.26
Subsequently, based on a telephone follow-up conducted 12 months after enrollment, the active seizure cohort was redefined as either pharmaco-responsive (n = 41), characterized by a ≥ 50% reduction in seizure frequency, or pharmaco-resistant (n = 6), meeting operational criteria for pharmaco-responsive, specifically persistent seizures, despite adequate trials of two appropriately selected antiseizure medications at therapeutic doses.
Sample Collection
Peripheral blood samples were collected within 4 hours of hospital admission using standardized phlebotomy protocols. Serum was immediately separated by centrifugation (3000 × g, 15 minutes at 4°C), aliquoted into cryovials, and stored at −80°C. All samples were batch-analyzed for post-study completion, to minimize interassay variability.
Ethical Considerations
This study received ethical approval from the Institutional Review Board of the Children’s Hospital Affiliated to Soochow University (Approval No. 2024CS085) and adhered to the principles of the Declaration of Helsinki. Written informed consent was obtained from all participants’ parents or legal guardians prior to enrollment. Patient identifiers were removed during data analysis to ensure confidentiality.
Sample Measurement
Serum concentrations of plasminogen activator inhibitor-1 (PAI-1) were quantified using a commercially available Human PAI-1 ELISA Kit (Enzyme Immunoassay; Jiangsu Meimian Industrial Co. Ltd., Jiangsu, China, Cat. No. MM-15240H1) following the manufacturer’s protocol, employing a sandwich ELISA methodology. Briefly, standards (2.580 ng/mL) and samples were dispensed in quadruplicate (50 μL/well) into antibody-coated microplates alongside blank controls. After a 60-minute incubation at 37°C with horseradish peroxidase-conjugated detection antibody (100 μL/well), plates underwent five automated wash cycles (Rayto RT-6100 washer) using 1× wash buffer (phosphate-buffered saline with 0.05% Tween®20) (350 μL/well), prepared by 1:19 dilution of a 20× concentrate, with deionized water. Following the addition of 50 μL each of substrate solutions of 3,3′,5,5′-tetramethylbenzidine (TMB), plates were incubated in darkness at 37°C for 15 minutes. Reactions were terminated with 50 μL/well stop solution, and the optical density was measured at 450 nm within 15 minutes, using a microplate reader. A six-point standard curve was generated for each assay, with sample concentrations calculated through four-parameter logistic regression modeling (GraphPad Prism 8.0). Quality control parameters demonstrated intraassay and interassay coefficients of variation below 10% and 15%, respectively. Hemolyzed or lipemic serum samples were systematically excluded to mitigate potential analytical interference, ensuring measurement reliability.
Statistical Analysis
The Shapiro–Wilk test indicated that the data in all groups were normally distributed, and parametric tests were therefore applied (Table 1). Group comparisons were analyzed via a one-way analysis of variance followed by Tukey’s post hoc test for multiple comparisons among the three main groups (control, remission, seizure), with significance defined as p < 0.05. For comparisons between two independent subgroups within the seizure cohort (ie, AED-treated vs. untreated, and pharmaco-responsive vs. pharmaco-resistant), statistical significance was assessed using unpaired two-tailed Student’s t-tests, with p-values adjusted for multiple comparisons using the Bonferroni method (corrected α = 0.05/2 = 0.025). Data are presented as the mean ± standard error of the mean. Statistical analyses and graphical representations were conducted using GraphPad Prism 8 (GraphPad). Unless otherwise specified, error bars represent the standard deviation, with an asterisk (*) indicating statistical significance at p < 0.05, while four asterisks (****) indicated p < 0.0001.
 |
Table 1 Shapiro–Wilk Test Results of the Seizure, Remission, and Control Groups |
Results
Participants: As previously described, the seizure group included 47 pediatric patients who had experienced at least one epileptic seizure within 72 hours (regardless of antiepileptic treatment status), and nineteen patients who had received standardized antiepileptic therapy and maintained seizure-free status for a minimum of 6 months constituted the remission group, along with 21 healthy children serving as the control group. No statistically significant intergroup difference was detected in age distribution or sex ratios among the three cohorts (see Table 2 and Figure 1). The clinical characteristics of the seizure and remission groups are shown in Table 3, along with the AEDs they used shown in Table 4.
 |
Table 2 Demographic Characteristics of the Seizure, Remission, and Control Groups |
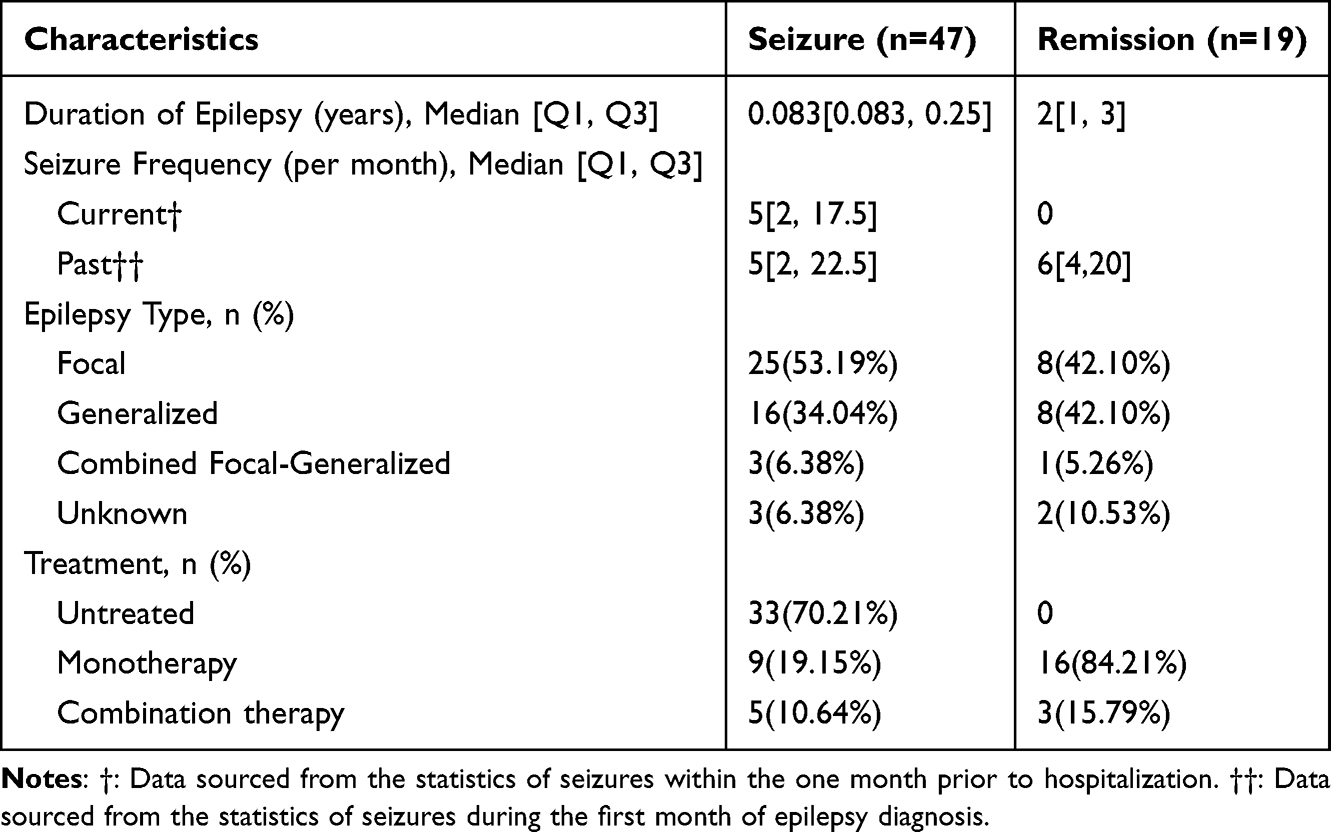 |
Table 3 Clinical Characteristics of the Seizure and Remission Groups |
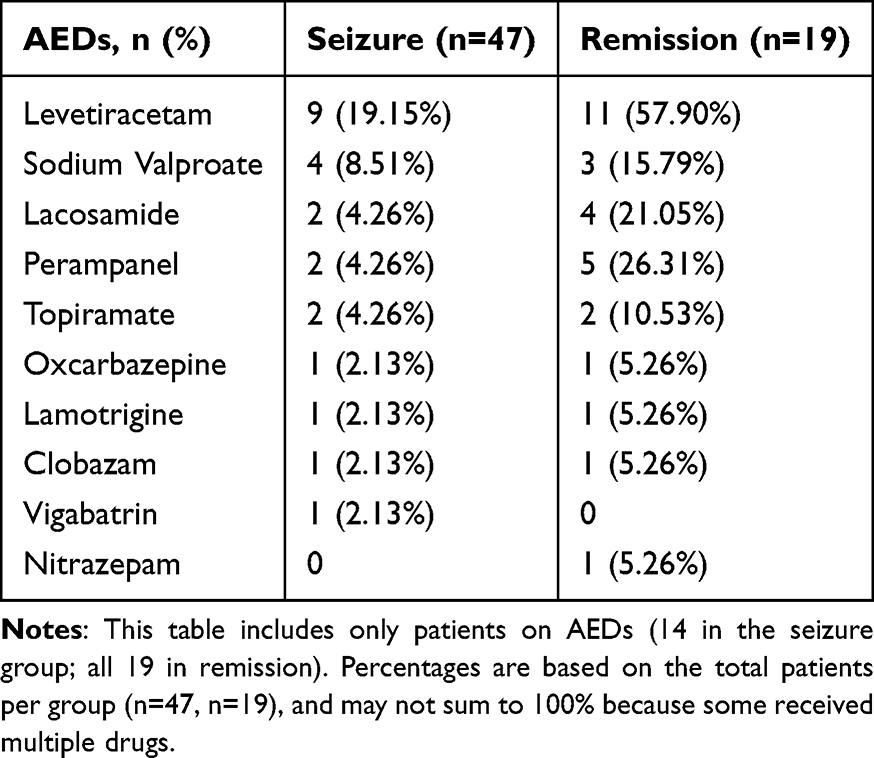 |
Table 4 Anti-Epileptic Drugs (AEDs) Used in the Seizure and Remission Groups |
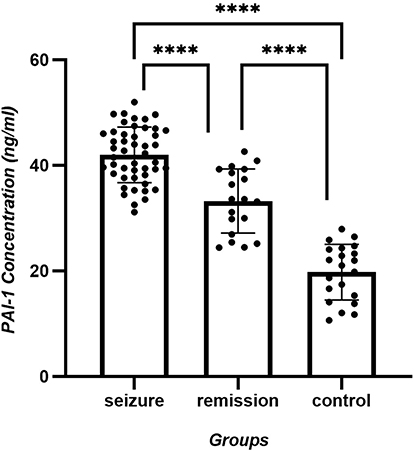 |
Figure 1 PAI-1 concentration (ng/mL) of seizure, remission and control group. Highly significant differences is shown between the control and remission groups (p < 0.0001), the control and active phase groups (p < 0.0001), as well as between the remission and active phase groups (p < 0.0001). |
Statistical analysis of human PAI-1 concentration data across the control (n = 21), remission (n = 19), and seizure groups (n = 47) was conducted. Tukey’s multiple comparison test showed highly significant differences between the control and remission groups (p < 0.0001), the control and active phase groups (p < 0.0001), as well as between the remission and active phase groups (p < 0.0001). Detailed data are presented in Table 5 and Figure 1.
 |
Table 5 Plasma Plasminogen Activator Inhibitor-1 Concentrations of the Seizure, Remission, and Control Groups |
To determine whether antiepileptic drugs had any effect on the serum PAI-1 concentration, the seizure group was further stratified into two subgroups based on prior exposure to antiepileptic therapy, involving the seizure treated (n = 14) and seizure untreated (n = 33) subgroups. Subsequent analysis of serum PAI-1 levels during the ictal phase showed no statistically significant (p = 0.0698; corrected significance threshold α = 0.025) difference between these two subgroups, as shown in Figure 2 and Table 6.
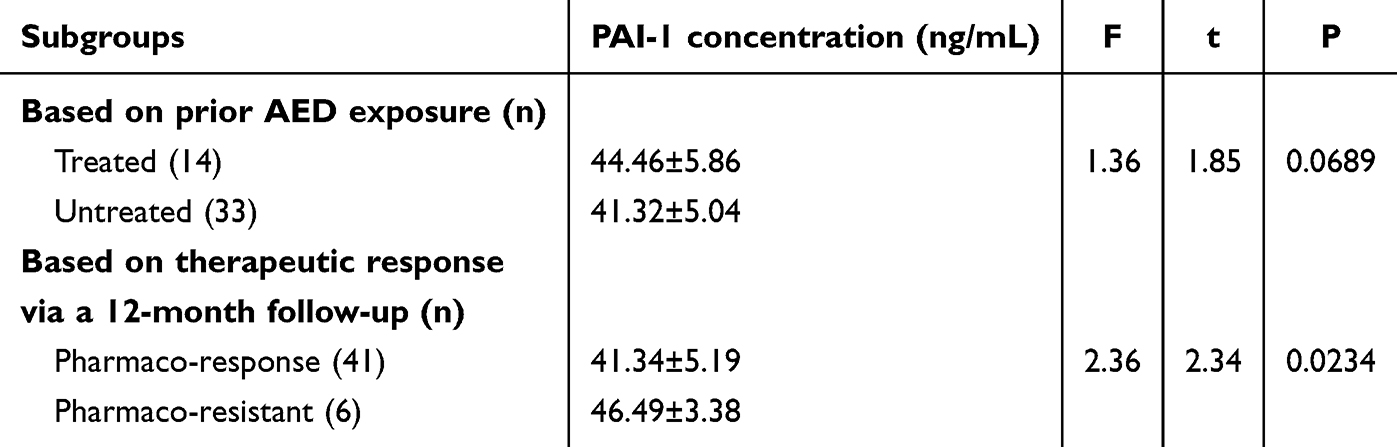 |
Table 6 Plasma Plasminogen Activator Inhibitor-1 Concentrations in the Epileptic Seizure Subgroups Stratified by Anti-Epileptic Drug (AED) Exposure History and 12-Month Therapeutic Response |
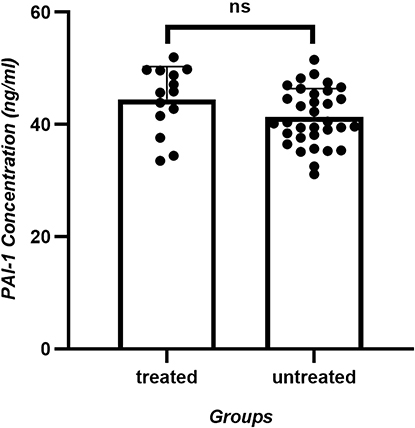 |
Figure 2 PAI-1 concentration (ng/mL) of seizure treated and seizure untreated subgroups. Serum PAI-1 levels during the ictal phase showed no statistically significant (p= 0.0698) difference between these two subgroups. |
To determine if baseline serum PAI-1 levels were early indicators of epileptic trajectory, we conducted a telephone follow-up assessment of the seizure-active cohort, 1 year after blooding sampling. Based on therapeutic responses to AED and seizure control status, patients were reclassified into two subgroups: the pharmaco-responsive subgroup (being seizure free for at least 6 months prior to follow-up with monotherapy, n = 41) and the pharmaco-resistant subgroup (requiring polytherapy with ≥2 AEDs or exhibiting ≥1 seizure recurrences within 6 months prior to follow-up, n = 6). Subsequent repeat analysis of seizure group serum PAI-1 levels showed statistically significant differences between these refined therapeutic response subgroups even after Bonferroni correction (p = 0.0234 < 0.025), as shown in Figure 3 and Table 6.
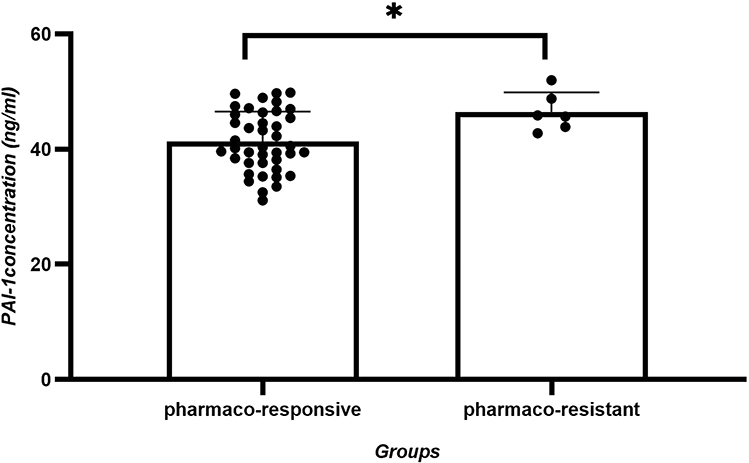 |
Figure 3 PAI-1 concentration (ng/mL) of pharmaco-responsive and pharmaco-resistant subgroups. Seizure group serum PAI-1 levels showed statistically significant (p= 0.0234) differences between these refined therapeutic response subgroups. |
Discussion
This case–control study provided novel clinical evidence linking elevated plasma PAI-1 levels to pediatric epilepsy progression and therapeutic outcomes. Three key findings emerged. First, PAI-1 concentrations showed significant stratification across disease states, with active epilepsy patients exhibiting 2.1-fold higher levels than controls (p < 0.0001), while the remission group levels remained 1.6-fold elevated. Second, no statistically significant difference (p = 0.0689) was observed in PAI-1 levels between AED-treated and untreated subgroups within the active seizure cohort (44.46 vs. 41.32 ng/mL, respectively). Third, baseline PAI-1 levels predicted therapeutic responses, with pharmaco-resistant patients having 12% higher concentrations than responders after a 12-month follow-up (p = 0.0234).
These findings were consistent with former experimental models implicating PAI-1 in epileptogenesis,21 while advancing the possibility of clinical translations. Our results also demonstrated that the PAI-1 vascular and inflammatory effects may persist, even during remission. The incomplete PAI-1 normalization in seizure-free patients (33.25 vs.19.75 ng/mL, p < 0.0001) may be related with residual synaptic consolidation or subclinical inflammation.
Integrating our findings with the mechanistic framework outlined in the introduction, elevated plasma PAI-1 likely reflects and may perpetuate the key epileptogenic processes of neuroinflammation and blood–brain barrier (BBB) disruption.17–20 The persistent intermediate elevation of PAI-1 in remission-phase patients suggests a sustained pro-inflammatory state or subclinical BBB dysfunction, which could explain the ongoing risk of relapse. Furthermore, the association between higher baseline PAI-1 and subsequent pharmaco-resistance implies that individuals with a more robust inflammatory-vascular pathology at disease onset are less likely to respond to conventional antiseizure medications, which primarily target neuronal excitability rather than these upstream mechanisms.
Importantly, we found no significant reduction in PAI-1 levels in AED-treated patients during active seizures. We propose two non-exclusive explanations for this finding. First, as noted, a numerical trend toward higher values was even shown in the treated subgroup. It likely reflected inherent selection bias in our cohort design. The treated subgroup (n = 14) only included patients with ongoing seizures, despite ≥3 months of guideline-adherent therapy, which was a population potentially enriched for pharmaco-resistance. Thus, the observed trend toward higher PAI-1 levels in treated patients may indicate intrinsic drug resistance, rather than therapeutic inefficacy. This interpretation was consistent with our follow-up data showing that PAI-1 was significantly higher in pharmaco-resistant patients (46.49 vs. 41.34 ng/mL in responders). Second, and more fundamentally, the primary mechanisms of action of most AEDs (modulating ion channels or synaptic transmission) may not directly intersect with the PAI-1-driven neuroinflammatory and vascular pathways.17–20 Consequently, even effective seizure control might not normalize PAI-1 levels if the underlying inflammatory-vascular pathology persists. This disconnect highlights a potential therapeutic gap for a subgroup of patients.
The incomplete normalization of PAI-1 levels in remission-phase patients (33.25 vs. 19.75 ng/mL in controls, p < 0.0001) supported preclinical models where PAI-1 is related to epileptogenesis. Residual elevations may reflect subclinical impairment of synaptic consolidation through proBDNF/mBDNF imbalance,21 which are processes not fully reversed by control of seizures. This persistence suggested that PAI-1 could serve as a biomarker for monitoring ongoing epileptogenic vulnerability.
The clinical use of PAI-1 as a biomarker and its therapeutic potential remain limited and deserve close attention. One study indicated that the PAI-1 inhibitor could serve as a protector against doxorubicin-induced cellular senescence, as well as reduce oxidative stress.23 Another study found that Tiplaxtinin, another PAI-1 inhibitor, attenuated neointimal hyperplasia by promoting PAI-1 cleavage to an inactive form (CL-PAI-1) that enhanced VSMC apoptosis via the TWEAK/FN14 pathway.24 These findings elucidated PAI-1’s contribution to neural injury and epileptogenesis, supporting further investigation of PAI-1 inhibitors as adjunctive therapies.
Study limitations of the present study included the small size of the treated subgroup (n = 14), which limited statistical power, and the need for cautious interpretation due to multiple comparisons, despite our application of correction methods. In addition, the focus on single blood samples, preventing analysis of how PAI-1 changed over time. By excluding children who responded well to AEDs (who were included in the remission group), the treated subgroup was biased toward resistant cases, potentially excluding AED benefits during milder epilepsy. The potential mechanisms underlying the different AED target and its affection in PAI-1-related pathological pathways should also be analyzed distinguished in the future. Furthermore, attrition or incomplete data during follow-up, due to refusal or inability to provide more detailed assessments, may also affect the comprehensiveness of the findings. Future research should therefore track PAI-1 levels repeatedly during treatments and introduce more objective indicators such as electroencephalogram, seizure logs to better characterize the PAI-1 relationships with drug responses and epilepsy prognosis. Given the close association between age and epilepsy onset in children, future larger-scale studies with age-stratified analyses are warranted to further elucidate this relationship.
Another key translational gap identified in this study is the lack of a defined clinical cutoff for plasma PAI-1. Future studies with larger sample sizes are required to perform robust ROC curve analyses to determine the optimal threshold that maximizes predictive accuracy for pharmaco-resistance. This threshold must be validated in independent cohorts and its positive/negative predictive values assessed in real-world clinical settings to guide therapeutic decision-making.
On a clinical level, measuring PAI-1 could earlier identify high-risk patients. However, completely blocking PAI-1 is risky, because it plays important roles in blood clotting.1,2 In summary, while standard AEDs may not fully normalize PAI-1 in patients with resistant epilepsy, the strong link of PAI-1 to disease severity and treatment failure makes it a valuable biomarker for prognostication. Given the multifactorial pathogenesis of epilepsy, a panel combining multiple biomarkers may offer superior predictive power and mechanistic insight compared to any single marker. For instance, ghrelin and des-acyl ghrelin have shown association with drug response in epilepsy, highlighting the potential of metabolic markers.14,27 Integrating PAI-1 with such candidates, as well as with other inflammatory mediators like IGFBP2 and CCL5 identified in our preliminary screen,22 could yield a composite score that more accurately stratifies patients by risk and underlying pathophysiology. Larger studies tracking PAI-1 during treatment could therefore refine its use in personalized care. Future longitudinal studies are warranted to develop and validate such multi-analyte models, and to determine whether biomarker-guided interventions can ultimately improve clinical outcomes.
Conclusion
This prospective cohort study showed that plasma PAI-1 levels served as a dynamic biomarker reflecting epilepsy activities and therapeutic outcomes in pediatric patients. Key findings included the following: (1) PAI-1 concentrations showed disease-state stratification, with active epilepsy patients exhibiting 2.1-fold higher levels than controls (41.99 vs.19.75 ng/mL, p < 0.0001), while remission-phase patients maintained intermediate elevations (33.25 ng/mL). (2) Baseline PAI-1 levels predicted 12-month pharmaco-resistance, with pharmaco-resistant patients having 12% higher concentrations than responders (46.49 vs. 41.34 ng/mL, p = 0.0234). (3) Prior AED exposure did not significantly alter PAI-1 levels during active seizures (44.46 vs. 41.32 ng/mL, p = 0.0689), suggesting intrinsic disease mechanisms, rather than treatment effects, which could drive increases in PAI-1 levels.
These clinical observations were consistent with experimental evidence of PAI-1’s role in impeding proBDNF cleavage to mBDNF via tPA inhibition,21 which disrupts TrkB-mediated synaptic consolidation and promotes p75NTR-dependent apoptosis. The incomplete PAI-1 normalization in remission-phase patients (1.7-fold higher than controls) suggested that persistent subclinical neuroinflammation or vascular pathology, as reflected by elevated PAI-1, could serve as a biomarker for ongoing epileptogenic vulnerability and increased risk of relapse. While limited by its single-center design and medication history confounders, the relatively small sample size of the pharmaco-resistant subgroup (n=6) necessitates caution in interpreting the predictive stability of the PAI-1 threshold, and findings require validation in larger, independent cohorts. Nevertheless, this study established PAI-1 levels as a promising biomarker for monitoring epileptogenic processes and personalizing anti-inflammatory therapies. For future clinical translation, patients identified with high baseline PAI-1 levels, indicative of a robust inflammatory-vascular pathology, might represent a subgroup that could benefit from personalized treatment strategies, such as adjunctive anti-inflammatory therapies, alongside conventional AEDs. Future longitudinal studies should therefore determine whether PAI-1-guided interventions improve seizure control in high-risk patients.
Highlight Statements
A prospective cohort linked plasma PAI-1 elevation to pediatric epilepsy severity. Seizure patients showed a 2.1-fold higher PAI-1 vs. controls. Baseline PAI-1 predicted 12-month drug resistance in epilepsy. AED treatment failed to normalize elevated plasma PAI-1 levels. PAI-1 served as a biomarker for epilepsy activity and risk.
Data Sharing Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Acknowledgments
This project was supported by the Science and Technology Program of Suzhou No. SKY2022007 and No. MSXM2025020.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gils A, Declerck PJ. Plasminogen activator inhibitor-1. Curr Med Chem. 2004;11(17):2323–10. PMID: 15379715. doi:10.2174/0929867043364595
2. Morrow GB, Mutch NJ. Past, present, and future perspectives of plasminogen activator inhibitor 1 (PAI-1). Semin Thromb Hemost. 2023;49(3):305–313. PMID: 36522166. doi:10.1055/s-0042-1758791
3. Andreasen PA, Kjoller L, Christensen IJ, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int, J, Cancer. 1997;72(1):1–22. doi:10.1002/(SICI)1097-0215(19970328)72:1<1::AID-IJC1>3.0.CO;2-B
4. Bouarab C, Roullot-Lacarrière V, Vallée M, et al. PAI-1 protein is a key molecular effector in the transition from normal to PTSD-like fear memory. Mol Psychiatry. 2021;26(9):4968–4981. PMID: 33510345; PMCID: PMC8589667. doi:10.1038/s41380-021-01024-1
5. Jiang H, Li X, Chen S, et al. Plasminogen activator inhibitor-1 in depression: results from animal and clinical studies. Sci Rep. 2016;6:30464. PMID: 27456456; PMCID: PMC4960524. doi:10.1038/srep30464
6. Griemert EV, Schwarzmaier SM, Hummel R, et al. Plasminogen activator inhibitor-1 augments damage by impairing fibrinolysis after traumatic brain injury. Ann Neurol. 2019;85(5):667–680. PMID: 30843275; PMCID: PMC6593843. doi:10.1002/ana.25458
7. Chan SL, Bishop N, Li Z, Cipolla MJ. Inhibition of PAI (Plasminogen Activator Inhibitor)-1 improves brain collateral perfusion and injury after acute ischemic stroke in aged hypertensive rats. Stroke. 2018;49(8):1969–1976. PMID: 29991657; PMCID: PMC6202199. doi:10.1161/STROKEAHA.118.022056
8. Aging LRM. Cellular senescence, and alzheimer’s disease. Int J Mol Sci. 2022;23(4):1989. PMID: 35216123; PMCID: PMC8874507. doi:10.3390/ijms23041989
9. Jeon H, Kim JH, Kim JH, et al. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J Neuroinflammation. 2012;9(1):149. doi:10.1186/1742-2094-9-149
10. Bajou K, Peng H, Laug WE, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14(4):324–334. doi:10.1016/j.ccr.2008.08.012
11. Löscher W, Potschka H, Sisodiya SM, Vezzani A. Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev. 2020;72(3):606–638. PMID: 32540959; PMCID: PMC7300324. doi:10.1124/pr.120.019539
12. Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11–21. PMID: 21985866. doi:10.1016/j.expneurol.2011.09.033
13. Kalilani L, Sun X, Pelgrims B, Noack-Rink M, Villanueva V. The epidemiology of drug-resistant epilepsy: a systematic review and meta-analysis. Epilepsia. 2018;59(12):2179–2193. PMID: 30426482. doi:10.1111/epi.14596
14. Marchiò M, Roli L, Giordano C, et al. High plasma levels of ghrelin and des-acyl ghrelin in responders to antiepileptic drugs. Neurology. 2018;91(1):e62–e66. PMID: 29802169. doi:10.1212/WNL.0000000000005741
15. Yousefi MJ, Rezvanimehr A, Saleki K, et al. Inflammation-related microRNA alterations in epilepsy: a systematic review of human and animal studies. Rev Neurosci. 2025;36(8):901–923. PMID: 40755381. doi:10.1515/revneuro-2025-0041
16. Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology. 2013;69:16–24. doi:10.1016/j.neuropharm.2012.04.004
17. Lee TW, Tsang VW, Loef EJ, Birch NP. Physiological and pathological roles of tissue plasminogen activator and its inhibitor neuroserpin in the nervous system. Front Cell Neurosci. 2015;9:396. doi:10.3389/fncel.2015.00396
18. Goldberg E, Coulter D. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci. 2013;14:337–349. doi:10.1038/nrn3482
19. Shmakova AA, Rubina KA, Anokhin KV, et al. The role of plasminogen activator system in the pathogenesis of epilepsy. Biochemistry. 2019;84:979–991. doi:10.1134/S0006297919090013
20. Pitkänen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011;10(2):173–186. doi:10.1016/S1474-4422(10)70310-0
21. Thomas AX, Cruz Del Angel Y, Gonzalez MI, et al. Rapid increases in proBDNF after pilocarpine-induced status epilepticus in mice are associated with reduced proBDNF cleavage machinery. eNeuro. 2016;3(1):ENEURO.0020–15.2016. PMID: 27057559; PMCID: PMC4814566.. doi:10.1523/ENEURO.0020-15.2016
22. Du Y, Xiao X, You HZ, et al. Association of high plasma levels of SerpinE1, IGFBP2, and CCL5 with refractory epilepsy in children by cytokine profiling. Clin Pediatr. 2024;63(7):953–962. doi:10.1177/00099228231201245
23. Ghosh AK, Rai R, Park KE, et al. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget. 2016;7(45):72443–72457. PMID: 27736799; PMCID: PMC5341920. doi:10.18632/oncotarget.12494
24. Simone TM, Higgins SP, Archambeault J, et al. A small molecule PAI-1 functional inhibitor attenuates neointimal hyperplasia and vascular smooth muscle cell survival by promoting PAI-1 cleavage. Cell Signal. 2015;27(5):923–933. PMID: 25617690; PMCID: PMC4361315. doi:10.1016/j.cellsig.2015.01.009
25. Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the international league against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):522–530. 28276060. doi:10.1111/epi.13670
26. Mesraoua B, Brigo F, Lattanzi S, et al. Drug-resistant epilepsy: definition, pathophysiology, and management. J Neurol Sci. 2023;452:120766. PMID: 37597343.. doi:10.1016/j.jns.2023.120766
27. Guerra A, Biagini G, Giordano C, Trenti T, Guerra A, Biagini G. Decreased ghrelin and des-acyl ghrelin plasma levels in patients affected by pharmacoresistant epilepsy and maintained on the ketogenic diet. Clin Nutr. 2019;38(2):954–957. doi:10.1016/j.clnu.2018.03.009
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Elevated Serum IL-6/IL-10 Ratio as a Promising Diagnostic and Disease Grading Biomarker for Cerebral Small Vessel Disease
Li L, Chen Y, He G, Gao Y, Cheng Y, Chen J, Sun D, Shi M
Journal of Inflammation Research 2026, 19:532686
Published Date: 15 January 2026