Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Plant-derived oleanolic acid ameliorates markers associated with non-alcoholic fatty liver disease in a diet-induced pre-diabetes rat model
Authors Gamede M ![]() , Mabuza L, Ngubane P
, Mabuza L, Ngubane P ![]() , Khathi A
, Khathi A
Received 7 June 2019
Accepted for publication 24 July 2019
Published 1 October 2019 Volume 2019:12 Pages 1953—1962
DOI https://doi.org/10.2147/DMSO.S218626
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Konstantinos Tziomalos
Mlindeli Gamede, Lindokuhle Mabuza, Phikelelani Ngubane, Andile Khathi
School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
Correspondence: Andile Khathi
Department of Human Physiology, School of Laboratory Medicine & Medical Sciences, University of KwaZulu-Natal, Room E3-408, Durban, South Africa
Tel +27 31 260 7585
Fax +27 31 260 7132
Email [email protected]
Background: The increased prevalence of non-alcoholic fatty liver disease (NAFLD) in type 2 diabetes mellitus (T2DM) patients is becoming a worldwide health burden. Studies have indicated, however, that the onset of NAFLD occurs during pre-diabetes, a condition that often precedes the onset of T2DM. Oleanolic acid has been reported to improve glucose homeostasis in diet-induced pre-diabetes; however, the effects of this triterpene on liver function have not been evaluated.
Purpose: This study was aimed at evaluating the therapeutic effects of oleanolic acid (OA) on selected markers of NAFLD in a pre-diabetes rat model.
Methods and materials: Pre-diabetes was induced by exposing Sprague Dawley rats to a high-fat high-carbohydrate diet for 20 weeks. The pre-diabetic rats were then treated with OA (80 mg/kg) or metformin (500 mg/kg) in the presence and absence of dietary interventions for a period of 12 weeks. The effects of OA were evaluated on parameters including plasma triglycerides (TGs), very low-density lipoprotein (VLDL) particles, bilirubin, AST, ALT, SREBP and antioxidant profile while the livers were collected for histological analysis.
Results: The findings of this study showed that the administration of OA to pre-diabetic rats ameliorated body/liver weights ratio and significantly decreased plasma triglycerides (TGs) and VLDL. Furthermore, OA also ameliorated hepatic oxidative stress, lowered the SREBP expression and intrahepatic TGs. In addition, OA administration decreased plasma concentrations of bilirubin and liver damage enzyme biomarkers.
Conclusion: The findings of the study suggest that OA ameliorates the risk of developing pre-diabetes-related NAFLD through the prevention of intrahepatic fat accumulation while also lowering hepatic inflammation.
Keywords: pre-diabetes, insulin resistance, NAFLD and oleanolic acid
Introduction
Non-alcoholic fatty liver disease (NAFLD) is conventionally defined as the spectrum of clinicopathologic conditions that are characterized by hepatocyte lipid infiltration without any excessive alcohol consumption.1 The prevalence of NAFLD worldwide is approximately 25% and in patients with type 2 diabetes mellitus (T2DM), it is about 67–80 %.2,3 However, there is barely any knowledge on the prevalence of this condition in pre-diabetes.3 This may be due to the invasive nature of diagnosis which requires a liver biopsy for accurate diagnosis.3–5 Some of the underlying risk factors that may lead to the development of NAFLD include dysregulation of hepatic glucose homeostasis accompanied by insulin resistance and hyperinsulinemia.5,6 Co-existence of these conditions are directly linked with the dysregulation of peripheral lipolysis and increased hepatic de-novo lipogenesis as a result of increased expression of sterol regulatory element-binding protein (SREBP1c) and subsequent accumulation of non-esterified fatty acids (NEFA).7 This may lead to increased hepatocellular free fatty acid flux and resultant accumulation of cytoplasmic diacylglycerol (DAG) in the hepatocytes.8 The hepatic accumulation of DAG and free fatty acids (FFA) is directly linked with an elevation of triglycerides (TG) through transesterification.9 Overall, this hepatic fat accumulation is linked with complications such as hepatocyte inflammation, hepatocellular ballooning degeneration, hepatocellular fibrosis and consequently cirrhosis without any history of excessive alcohol consumption.10,11 These complications are associated with hepatocyte injuries which lead to the release of transaminases enzymes such as alanine transaminase (ALT) and aspartate transaminase (AST) into the circulation and hence, the risk for developing NAFLD.4 Moreover, hepatic fat accumulation causes increased lipid peroxidation which is associated with compromised antioxidant capacity which leads to oxidative stress. Intrahepatic fat accumulation and oxidative stress are directly linked with the onset of steatohepatitis.12 In addition, insulin resistance and hepatic fat accumulation have been implicated in the reduction of circulating total bilirubin which is also associated with the progression of hepatic steatosis to fibrosis.12 Conventional therapeutic strategies for diabetes-associated NAFLD include the combination of lifestyle modifications with insulin-sensitizing drugs, and antioxidants.13 However, these are prescribed at advanced stages of NAFLD and are associated with side-effects that may counteract the amelioration of the disease.14 Previous studies in our laboratory have reported that oleanolic acid (OA) improves insulin sensitivity and has cardio-protective effects in the pre-diabetic state even in the absence of dietary modifications.15 However, no studies have been done to investigate the effects of hepato-protective effects of this compound during prediabetes. Hence, this study was designed to evaluate whether plant-derived OA can reverse the early stages of NAFLD and restore liver function in a diet-induced pre-diabetes rat model.
Methods and materials
Drugs and chemicals
All chemicals and reagents were sourced from standard pharmaceutical suppliers and were of analytical grade.
Extraction method
The extraction of OA was done using a well-established protocol which is explained in detail in previous publications.15
Animal studies
Male Sprague-Dawley rats (130–160 g) used in this study were bred and housed in the Biomedical Research Unit of the University of KwaZulu-Natal. The animals were maintained under standard laboratory conditions of constant temperature (22±2°C), CO2 content (<5000 p.m.), relative humidity (55±5%) and illumination (12 hrs light/dark cycle, lights on at 07h00). The noise level was maintained at less than 65 decibels. The animals were allowed access to food and fluids ad libitum. All animal procedures and housing conditions were approved by the Animal Research Ethics Committee of the University of KwaZulu-Natal which subscribes to the principles and guidelines of Canadian Council on Animal Care (ethics no: AREC/035/016M). The animals acclimatized to their new environment for 1 week while consuming standard rat chow and tap water before exposure to a well-established experimental high-fat high-carbohydrate (HFHC) diet.
Experimental design
Experimental pre-diabetes was induced in male Sprague-Dawley rats (n=36) using a previously described protocol.16–18 The pre-diabetic animals were further sub-divided into six groups each group having six rats (n=6). The groups were as follows: pre-diabetic control group (PC) which were the pre-diabetic animals that continued with the experimental diet throughout the study period; metformin group (MET) which were the pre-diabetic animals that continued with the experimental diet but received metformin during treatment period; metformin and diet intervention group (MET + DI) which were the pre-diabetic animals that changed to a normal diet and received metformin during treatment period; oleanolic acid group (OA) which were the pre-diabetic animals that continued with experimental diet but received oleanolic acid during treatment period as well as OA and diet intervention group (OA + DI) which were the pre-diabetic animals that changed to a normal diet and received oleanolic acid during experimental period. Animals that served as normal controls (NPC) were those that were fed standard rat chow and diagnosed as without pre-diabetes.
Treatment of pre-diabetic animals
The treatment period lasted for 12 weeks. The animals were treated every third day where the MET and MET + DI groups received metformin (500 mg/kg p.o) while the OA and OA + DI groups were given oleanolic acid (80 mg/kg p.o). Parameters including total cholesterol and triglyceride concentrations were measured every fourth week in all groups for the duration of the treatment period.
Blood collection and tissue harvesting
At the end of the 12-week treatment period, the animals were sacrificed. For blood collection, all animals were anesthetized with Isofor (100 mg/kg) (Safeline Pharmaceuticals (Pty) Ltd, Roodeport, South Africa) via a gas anesthetic chamber (Biomedical Resource Unit, UKZN, Durban, South Africa) for 3 mins. Blood was collected by cardiac puncture and then injected into individual pre-cooled heparinized containers. The blood was then centrifuged (Eppendorf centrifuge 5403, Germany) at 4°C, 503 g for 15 mins. Plasma was collected and stored at −80°C in a Bio Ultra freezer (Snijers Scientific, Holland) until ready for biochemical analysis. Thereafter, the liver was removed, rinsed with cold normal saline solution and snap frozen in liquid nitrogen before storage in a Bio Ultra freezer (Snijers Scientific, Tilburg, Netherlands) at −80°C until further biochemical analysis.
Biochemical and histological analysis
SREBP were analyzed using their respective rat ELISA kits (Elabscience Biotechnology Co., Ltd) according to the manufacturer’s instructions. Liver damage biomarkers including alanine aminotransferase (ALT), aspartate aminotransferase (AST) and total bilirubin (TBIL) were measured in plasma using IDEXX instrument (IDEXX laboratories (PTY) Ltd. Pretoria, South Africa). Liver histology analysis was done using a previously described protocol.19
Hepatic oxidative stress
The concentrations of both GPx and SOD were measured using specific ELISA kits from Elabscience following the manufacturer’s instructions. Liver malondialdehyde (MDA) was measured using a well-established protocol explained by Gamede et al.
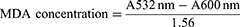
Plasma lipid analysis
Plasma samples were sent to Global Clinical and Viral laboratories (Amanzimtoti, South Africa) for the quantification of plasma triglycerides. Plasma VLDL concentrations were calculated using the Friedewald formula:
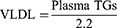
Statistical analysis
All data were expressed as means ± S.E.M. Statistical comparisons were performed with GraphPadInStat Software (version 5.00, Graph Pad Software, Inc., San Diego, California, USA) using two-way analysis of variance (ANOVA) followed by Bonferroni post hoc comparison test. A value of p<0.05 was considered statistically significant.
Results
Liver and body weights
At the end of the treatment period, the body weights of all experimental groups were measured before sacrifice. Following sacrifice, the livers were harvested and weighed. The PC group had significantly higher liver and body weights when compared to NPC. The OA-treated groups had significantly lower liver and body weights in comparison to PC (p<0.05) (Table 1).
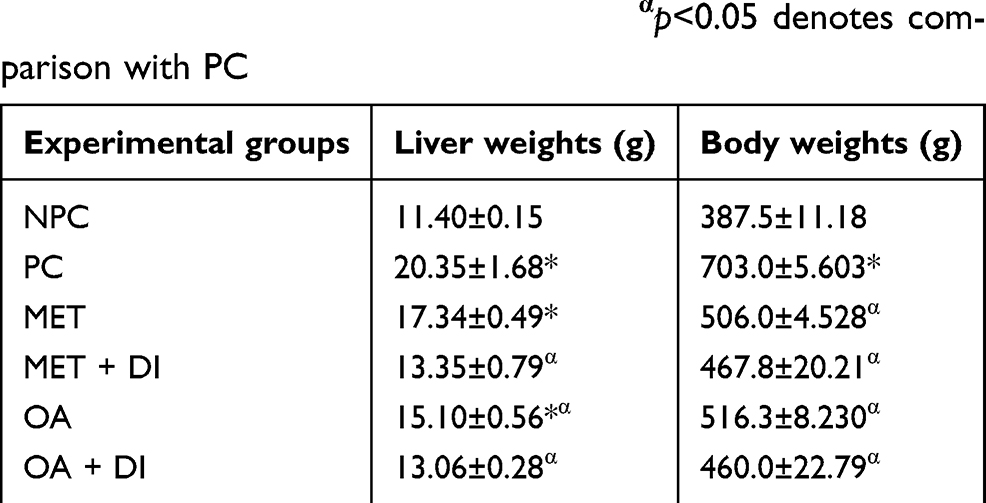 |
Table 1 Effects of OA (n=6, per group) on liver and body of pre-diabetic rats. Values are presented as standard deviation of mean ± SEM. Non-pre-diabetes control (NPC), pre-diabetes control (PC), metformin-treated (MET) and oleanolic acid-treated (OA). *p<0.05 denotes comparison with NPC; αp<0.05 denotes comparison with PC |
Very low-density lipoprotein (VLDL)
Plasma triglycerides (TGs) of all experimental groups were monitored every fourth week of the treatment period (weeks 0, 4, 8 and 12) and VLDL-cholesterol particles (VLDL-C) were calculated from those TGs. The results showed that from the beginning to the end of the treatment period, PC had a significantly higher TGs and VLDL-C concentration when compared to NPC (p<0.05). However, the animals that were treated with OA (OA and OA + DI) showed a progressive significant decrease in TGs and VLDL-C concentration when compared to PC (p<0.05) (see Figure 2A and B). The administration of metformin alone showed no significant improvement in TGs and VLDL-C particularly during the 12th week of the study. However, MET + DI showed a progressive significant decrease in TGs and VLDL-C by comparison to PC (p<0.05) (See Figure 1).
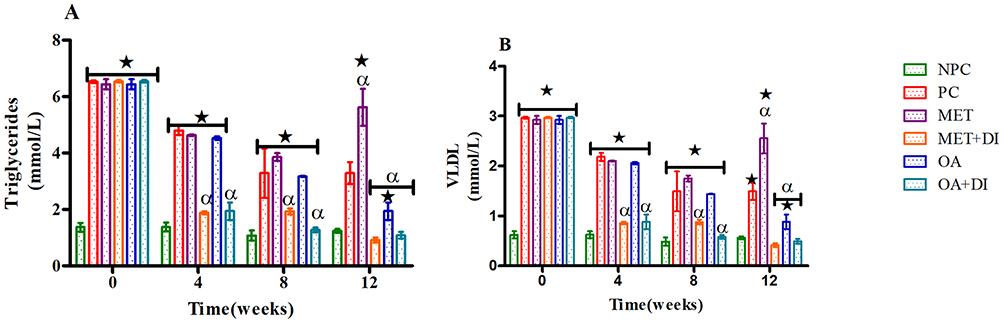 |
Figure 1 Effects of OA (n=6, per group) on plasma TGs (A) and VLDL-cholesterol particles (B) concentrations all experimental groups. Values are presented as standard deviation of mean ± SD. ⋆=p<0.05 denotes comparison with NC; αp<0.05 denotes comparison with PC. Abbreviations: NPC, non-pre-diabetes control; PC, pre-diabetes control; MET, metformin treated; MET + DI, metformin with diet intervention; OA, oleanolic acid treated; OA + DI, oleanolic acid. |
Intrahepatic triglycerides and sterol regulatory element-binding protein (SREBP)
All experimental groups were sacrificed at the end of the treatment period (week 12) and the plasma SREBP concentration was measured using the ELISA kit while hepatic triglycerides were measured using tissue triglycerides assay kit. The results showed that plasma SREBP concentration of PC was significantly higher than of NPC (p<0.05). Both OA-treated groups, ie, OA and OA + DI, had a significantly lower SREBP concentration when compared to PC. Similar results were obtained for the metformin-treated animals (MET and MET + DI) (Figure 2A). The intrahepatic triglycerides also followed the same trend as SREBP, ie, all the treated groups had a significant lower intrahepatic triglyceride in comparison to pre-diabetic control (PC) (Figure 2B)
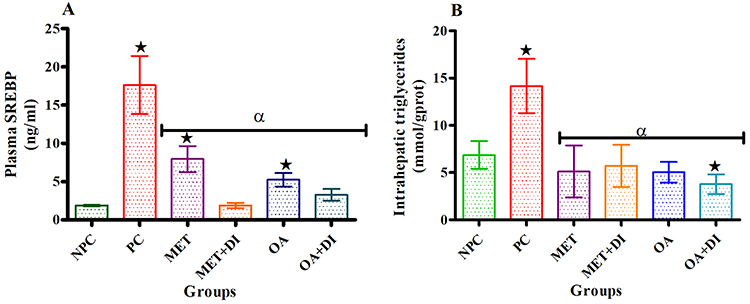 |
Figure 2 Effects of OA (n=6, per group) on plasma SREBP (A) and intrahepatic triglycerides (B) concentrations all experimental groups. Values are presented as standard deviation of mean ± SD. ⋆=p<0.05 denotes comparison with NPC; αp<0.05 denotes comparison with PC. Abbreviations: NPC, non-pre-diabetes control; PC, pre-diabetes control; MET, metformin treated; MET + DI, metformin with diet intervention; OA, oleanolic acid treated; OA + DI, oleanolic acid. |
Total bilirubin
All experimental groups were sacrificed at the end of the treatment period (week 12) and the plasma total-bilirubin concentration was measured. The results showed that the pre-diabetic control (PC) had a significantly lower plasma TBIL concentration in comparison to normal control (NPC) (p<0.05). OA treatment also resulted in a significant increase in TBIL levels when compared to PC (p<0.05). Similar results were obtained for the metformin-treated groups (Figure 3).
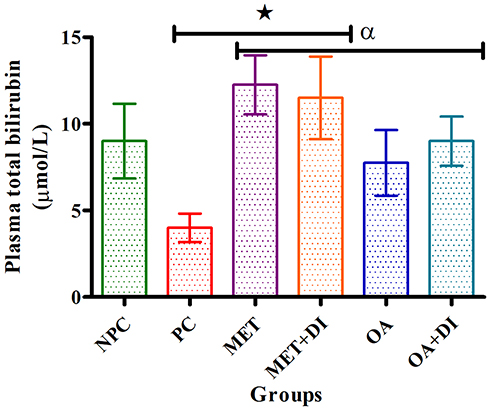 |
Figure 3 Effects of OA (n=6, per group) on total bilirubin of pre-diabetic rats. Values are presented as standard deviation of mean ± SD. ⋆=p<0.05 denotes comparison with NPC; αp<0.05 denotes comparison with PC. Abbreviations: NPC, non-pre-diabetes control; PC, pre-diabetes control; MET, metformin treated; OA, oleanolic acid treated. |
Liver transaminase enzymes
All experimental groups were sacrificed at the end of the treatment period (week 12) and the plasma transaminases were measured. The results showed that both plasma ALT and AST concentrations of the PC group were significantly higher when compared to the NPC group (p<0.05). However, the administration of OA (OA and OA + DI) showed a significant decrease in both ALT and AST when compared to PC (Figure 4A and B).
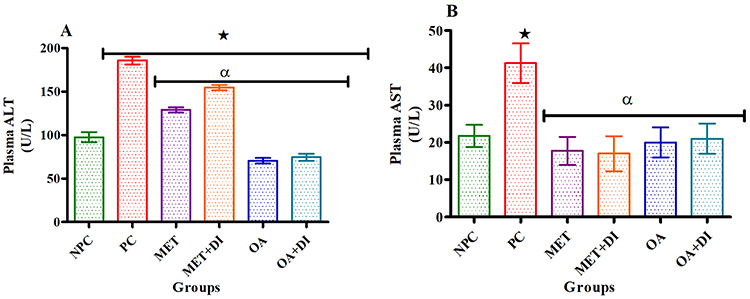 |
Figure 4 Effects of OA (n=6, per group) on ALT (A) and AST (B) of pre-diabetic rats. Values are presented as standard deviation of mean ± SD. ⋆=p<0.05 denotes comparison with NPC; αp<0.05 denotes comparison with PC. Abbreviations: NPC, non-pre-diabetes control; PC, pre-diabetes control; MET, metformin treated; OA, oleanolic acid treated. |
Liver oxidative status
All experimental groups were sacrificed at the end of the treatment period (week 12) and the liver oxidative stress markers including malondialdehyde (MDA) concentration and antioxidant enzyme activity, ie, GPx and SOD activity, were measured. The results showed that pre-diabetic animals (PC) had increased oxidative stress in comparison with NPC. This was evident by the high levels of MDA accompanied with decrease GPx and SOD activity in PC. However, the treatment with OA, with and without dietary intervention, resulted in improved antioxidant enzyme activity and the subsequent decrease in the hepatic MDA concentration when compared to PC (p<0.05) (Table 2). The trend observed in OA-treated animals was also seen in metformin-treated animals (MET and MET + DI) (p<0.05) (see Table 2).
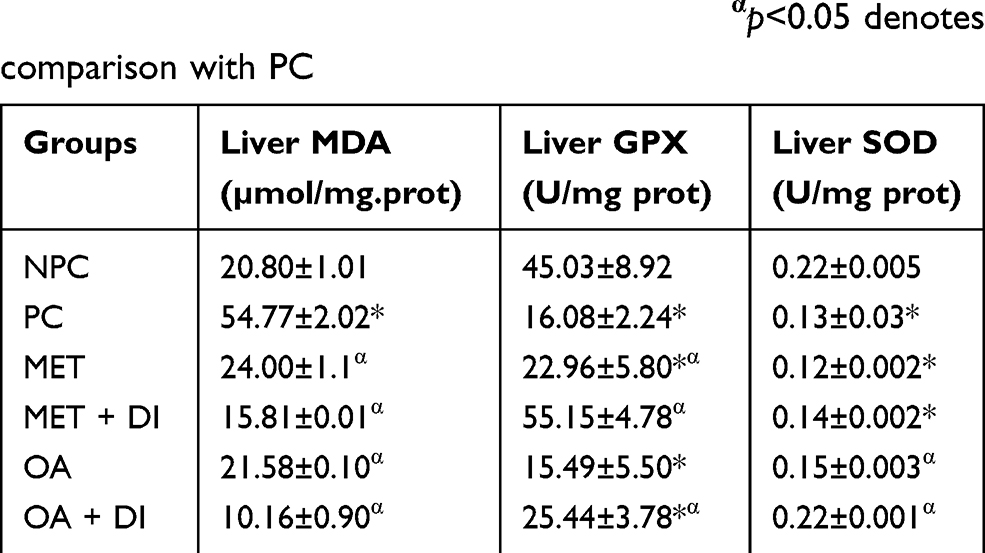 |
Table 2 Effects of OA (n=6, per group) on liver oxidative stress of pre-diabetic rats. Values are presented as standard deviation of mean ± SD. Non-pre-diabetes control (NPC), pre-diabetes control (PC), metformin-treated (MET) and oleanolic acid-treated (OA). *p<0.05 denotes comparison with NPC; αp<0.05 denotes comparison with PC |
Liver histology
Liver tissues obtained from all the experimental groups were processed for histo-analysis. The histology results for PC (B) were found to have derangements including hepatic lipid accumulation and hepatocytes injuries (microvesicular and nuclei pyknosis) in comparison to non-pre-diabetic controls (A). The treatment with OA alone (E) showed some symptoms of hepatocytes regeneration including multinuclei and hyperchromsia; however, some signs of degeneration such as nuclei pyknosis were still present. Interestingly, the treatment with OA + DI (F) was able to reverse the hepatic degeneration. The trend observed in OA-treated animals were also seen in metformin-treated animals (MET and MET + DI) (see Figure 5).
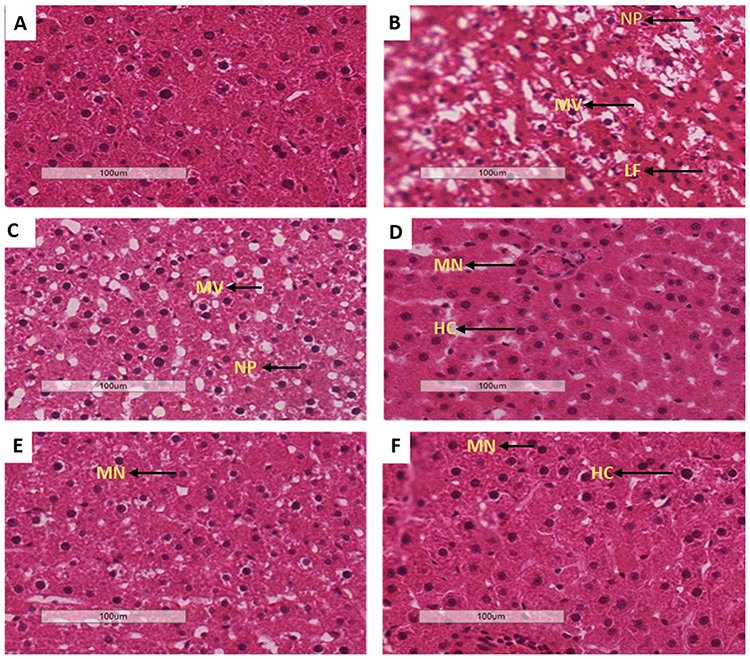 |
Figure 5 Effects of OA (n=6, per group) on liver oxidative stress of pre-diabetic rats on hepatocyte degeneration. Magnification of 20x100 µm, H&E staining, A-NPC, B-PC, C-MET, D-MET + DI, E-OA and F-OA + DI. Abbreviations: NP, nuclei pyknosis; HC, hyperchromatic nucleus; MN, multinuclear; LF, liver fibrosis; MV, microvesicular steatosis. |
Discussion
Over the last few decades, the prevalence of metabolic disorders such as non-alcoholic fatty liver disease (NAFLD) has been increasing.20,21 This may be attributed to chronic consumption of high caloric diets and sedentary lifestyles.22 Various diet-induced animal models have been developed to study the pathogenesis of diabetes and the associated metabolic disorders.23 Recent studies in our laboratory have reported that prolonged consumption of a high-fat high-carbohydrate (HFHC) diet leads to the development of pre-diabetes.17 This condition is characterized by complications including hypertriglyceridemia, insulin resistance and hyperinsulinemia which are risk factors for the development of NAFLD.16,24 The conventional management of NAFLD involves the combination of insulin sensitizers and lifestyle modification therapy which include exercise and dietary modification.25 However, pharmacological therapy is normally prescribed during T2DM while low patient compliance has been reported with regard to lifestyle modifications.26 Therefore, new drugs that will be able to promote glycemic control as well as have hepatoprotective effects in both the absence and presence of lifestyle modification are needed. The current study investigated the effects of plant-derived oleanolic acid with and without dietary intervention on NAFLD arising from diet-induced pre-diabetes in a rat model.
In this study, the untreated pre-diabetic animals were found to have increased liver weights. The increase in liver weights may be attributed to the accumulation of lipids in hepatocytes as was demonstrated by the presence of histologic microvesicular steatosis that was observed in the untreated pre-diabetic animals.27 Microvesicular steatosis results from increased intrahepatic fat deposition which may be attributed to the increased hepatic FFA influx from various sources including a HFHC diet, lipolysis or de novo lipogenesis.28,29 This may be symbolic of the early stages of hepatomegaly which is known to precede the onset of NAFLD.30 Moreover, the untreated pre-diabetic animals also had increased body weight which may suggest the pre-diabetic animals had ectopic fats which are highly susceptible to lipolysis and the subsequent increased hepatic FFA influx.31 However, the administration of OA with and without diet intervention resulted in normalization of both liver and body weights as well as elevation of histologic microvesicular steatosis. This may suggest that OA was able to lower total body fat content and eventually lower the hepatic FFA influx. These findings may be attributed to the previously reported effects of OA which suggest that OA lowers caloric intake by restoring the coordination between ghrelin and leptin to regulate food intake and the resultant total body fat content.16
Moreover, OA has been reported to improve insulin sensitivity in peripheral organs such as skeletal muscle and thus attenuate hyperglycemia in a pre-diabetic rat model.17 This lowers the hepatic glucose influx which subsequently results in a decrease in hepatic lipogenesis.16,32 However, OA + DI had more pronounced effects which may suggest that diet intervention may have also contributed in the reduction of liver weight and total body fat content. This may happen through minimizing the daily caloric intake which lowers the amount of energy that is stored in the form of fats, hence, a decrease in hepatic FFA influx.33 The derangements in lipid metabolism as a result of increased caloric intake, adipose tissue insulin resistance or lipolysis are highly associated with increased circulating FFA and low-density lipoprotein cholesterol particles (LDL-C) in the liver.34 The current study also observed that pre-diabetic animals had sustained a high level of circulating TGs and VLDL-C particles in pre-diabetic animals which also correlated with increased expression of SREBP and intrahepatic triglycerides concentration. The observed high circulating TGs and VLDL particles may also be attributed to increased FFA production from lipolysis of adipose tissue due to insulin resistance.35,36 The insulin resistance is associated with activation of hormone-sensitive lipase (HSL) and as a result it increases circulating FFA. The fate of plasma FFA include being esterified and stored in the liver as triglycerides as well as being carried by VLDL-C to the tissues for production of energy. The reduced insulin sensitivity in peripheral organs such as skeletal muscle and adipose tissue may lead to elevation of circulating glucose.37 During pre-diabetes or insulin resistance, the fate of circulating glucose results in increased influx in organs such as liver that can take up glucose independently of insulin.38,39 The increase in hepatic glucose influx may result in glucotoxicity and increased hepatic de novo lipogenesis from glucose metabolites such as of dihydroxy acetone phosphate (DHAP). DHAP is one of the precursors of TGs synthesis that comes from the hepatic glycolysis pathway; thus, an increase in DHAP might result in intrahepatic accumulation.40 Hepatic de novo lipogenesis is regulated by proteins including sterol regulatory element-binding proteins (SREBP) and its isoforms (SREBP-1c and SREBP-2).41 An increase of SREBP expression enhances the process of hepatic de novo lipogenesis.42 Hyperinsulinemia also exacerbates the expression of SREPB and its isoform (SREBP-1c) and consequently leads to intrahepatic lipid accumulation resulting in hepatocyte degeneration.43 Therefore, the high levels of SREBP and the concurrent intrahepatic triglycerides observed in this study may be attributed to the co-existence of hyperglycemia and hyperinsulinemia.38,39 Interestingly, the treatment with OA with and without dietary intervention resulted in decreased circulating TGs and VLDL-C. Furthermore, OA treatment also caused decreased SREBP and intrahepatic triglycerides concentrations. Previous studies in our laboratory have shown that the administration of OA is associated with improvement of insulin sensitivity and glucose uptake by the peripheral cells.15,44 This may suggest that OA is able to sensitize the cells for insulin and suppress the hormone-sensitive lipase (HSL) which inhibit adipose tissue lipolysis and consequently decreasing circulating TGs and VLDL-C particles.45 This may also result in the reduction of hepatic microvesicular steatosis thus preventing the onset of NAFLD.46,47 Moreover, OA has also been reported to reduce the risk of developing cardiovascular disease by lowering LDL-C particles while increasing high-density lipoprotein cholesterol (HDL-C) particles.16,17 These findings correlated with the previous study that reported that derivatives of OA such as 3-acetyl-oleanolic acid have also been shown to lower intrahepatic total cholesterol (TC), TGs and macrovesicular steatosis in high-fat diet-fed rats.16,18,48 While the observed decrease in plasma TGs and VLDLs may partly be attributed to dietary intervention, it may also be ascribed to the insulin-sensitizing effects of OA. Many studies have reported that OA improves insulin sensitivity in animal models of metabolic syndrome and diabetes.49,50 A recent study in our laboratory reported that this model of prediabetes has high levels of systemic ROS as a result of lipotoxicity.17 In addition, augmented FFA influx into the liver is also associated with hepatocyte degeneration including ballooning and inflammation which subsequently lead to non‐alcoholic steatohepatitis (NASH).51 The current findings also demonstrated that during pre-diabetes there is augmented circulating and intrahepatic triglycerides which is directly associated with lipotoxicity and generation of reactive oxygen species (ROS).28 Under normal physiological conditions, ROS play an important role as a defense mechanism against microorganisms.49,52 In pre-diabetes however, there is an overproduction of ROS leading to mitochondrial dysfunction.53
Hepatic oxidative stress is implicated in the progression of hepatic steatosis to cirrhosis.54 The current study observed that pre-diabetic animals had increased levels of malondialdehyde (MDA) and compromised antioxidant levels. MDA is a reactive metabolite produced by the reaction between the ROS and saturated fatty acids found on the cell membrane lipid bilayers, a process called lipid peroxidation.55 The increase in MDA levels correlated with a decrease in hepatic SOD and GPx concentrations in the untreated pre-diabetic animals. However, treatment with OA and dietary intervention resulted in normalization of hepatic MDA and improved SOD and GPx concentrations. Another recent study in our laboratory reported that OA is able to ameliorate pre-diabetes-induced high lipid peroxidation and oxidative stress.16 These findings correlated with the results reported by Yi et al, that OA and betulinic acid have hepatoprotective effects.56 This study stated that the possible mechanism involved in the antioxidant activity of these triterpenes might include the suppression of the liver-damaging enzyme activity while increasing the production of the antioxidant enzymes such as GPx and SOD.56 The current study also observed that pre-diabetic animals also had low levels of circulating total bilirubin. Bilirubin is a by-product of the catabolism of red blood cells and is also synthesized by hepatocytes.57 The correlation between the increased antioxidant capacity and low levels of plasma transaminases enzymes with high levels of circulating total bilirubin have been reported.58 Furthermore, bilirubin has been reported to be inversely associated with NAFLD and steatohepatitis.57,59 The administration of OA with and without dietary intervention resulted in the elevation of circulating total bilirubin. This is attributed to the antioxidant activity and cytoprotective effects of bilirubin.60 This could explain the attenuation of hepatocytes inflammation observed through elevation of steatotic inflammatory histological features such as nuclei pyknosis.61 Moreover, bilirubin has also been reported to have protective effects against cardiovascular diseases such as atherogenesis, coronary artery disease, as well as peripheral arterial diseases.62 Other clinical markers used in the detection of liver damage include aminotransferase enzymes such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST). The increase of these liver enzymes in the circulation of untreated pre-diabetic rats may be attributed to hepatocyte damage as a result of intrahepatic lipid accumulation and peroxidation.63 The elevation of these enzymes has been widely associated with intrahepatic FFA accumulation and hepatocytes injury; hence, they are used for the diagnosis of NAFLD.64 Therefore, the decrease of plasma ALT and AST following OA administration may suggest that OA can protect against hepatocyte injury.42,65 Some of these effects of OA were comparable with the previously reported therapeutic properties of metformin such as reduction of hepatic glucose production and increasing peripheral utilization of glucose which are strongly dependent on life-style modification.25,66 Furthermore, the reduction in SREBP, intrahepatic TGs and oxidative stress that was observed in this study are mostly associated with thiazolidinediones that need to be used in conjunction with dietary intervention.67 This may suggest that use of OA in both the presence and absence of dietary intervention reduces the circulating carbohydrates and lipids that would have led to NAFLD.
Conclusion
The findings of this study provide evidence that the administration of OA with and without dietary intervention has hepato-protective properties in pre-diabetes through the reduction of circulating triglycerides and protecting against oxidative stress. This treatment also was shown to protect against hepatic structural changes such as ballooning degeneration and inflammation. These pharmacological properties of OA may suggest that OA may potentially provide another avenue for the treatment of pre-diabetes-associated NAFLD; however, further studies are required to elucidate the associated mechanisms of OA at the molecular and genetical level including genes that are involved in the fatty acid synthesis, oxidation and regulation.
Acknowledgment
The authors are grateful to the Biomedical Resource Unit, University of KwaZulu-Natal for the supply of animals, Mrs Shoohana Singh for her technical expertise and the National Research Foundation (South Africa), National Research Foundation (South Africa) (Grant number 106041).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published and agree to be accountable for all aspects of the work. Mlindeli Gamede and Lindokuhle Mabuza carried out experiments, study design, analysis of data, writing of manuscript. Andile Khathi and Phikelelani Ngubane were involved in conceptualization, carried out experiments, study design, analysis of data, were involved in the writing of the manuscript and provided funding.
Disclosure
The authors report no conflict of interest in this work.
References
1. Giorda C, Forlani G, Manti R, et al. Occurrence over time and regression of nonalcoholic fatty liver disease in type 2 diabetes. Diabetes Metab Res Rev. 2017;33(4):e2878. doi:10.1002/dmrr.2878
2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease – meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi:10.1002/hep.28431
3. Saponaro C, Gaggini M, Gastaldelli A. Nonalcoholic fatty liver disease and type 2 diabetes: common pathophysiologic mechanisms. Curr Diab Rep. 2015;15(6):34. doi:10.1007/s11892-015-0607-4
4. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi:10.1002/hep.20701
5. Liver E.A.f.S.o. Easl-aleh clinical practice guidelines: non-invasive tests for evaluation of liver disease severity and prognosis. J Hepatol. 2015;63(1):237.
6. Jung UJ, Choi M-S. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15(4):6184–6223. doi:10.3390/ijms15046184
7. Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003;29(5):478–485.
8. Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci. 2016;61(5):1282–1293. doi:10.1007/s10620-016-4054-0
9. Labrie M, Lalonde S, Najyb O, et al. Apolipoprotein D transgenic mice develop hepatic steatosis through activation of PPARγ and fatty acid uptake. PLoS One. 2015;10(6):e0130230.
10. Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149(2):389–97. e10. doi:10.1053/j.gastro.2015.04.043
11. Imajo K, Kessoku T, Honda Y, et al. Magnetic resonance imaging more accurately classifies steatosis and fibrosis in patients with nonalcoholic fatty liver disease than transient elastography. Gastroenterology. 2016;150(3):626–37.e7. doi:10.1053/j.gastro.2015.11.048
12. Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):424–431. doi:10.1053/j.gastro.2007.11.038
13. Mazzotti A, Caletti MT, Marchignoli F, Forlani G, Marchesini G. Which treatment for type 2 diabetes associated with non-alcoholic fatty liver disease? Digestive Liver Dis. 2017;49(3):235–240. doi:10.1016/j.dld.2016.12.028
14. Kunkel SD, Elmore CJ, Bongers KS, et al. Ursolic acid increases skeletal muscle and brown fat and decreases diet-induced obesity, glucose intolerance and fatty liver disease. PLoS One. 2012;7(6):e39332. doi:10.1371/journal.pone.0039332
15. Gamede M, Mabuza L, Ngubane P, Khathi A. The effects of plant-derived oleanolic acid on selected parameters of glucose homeostasis in a diet-induced pre-diabetic rat model. Molecules. 2018;23(4):794. doi:10.3390/molecules23040794
16. Gamede M, Mabuza L, Ngubane P, Khathi A. Plant-derived oleanolic acid (oa) ameliorates risk factors of cardiovascular diseases in a diet-induced pre-diabetic rat model: effects on selected cardiovascular risk factors. Molecules. 2019;24(2):340. doi:10.3390/molecules24020340
17. Mabuza L, Gamede M, Maikoo S, Booysen I, Ngubane P, Khathi A. Effects of a ruthenium schiff base complex on glucose homeostasis in diet-induced pre-diabetic rats. Molecules. 2018;23(7):1721. doi:10.3390/molecules23071721
18. Luvuno M, Khathi A, Mabandla M. Voluntary ingestion of a high-fat high-carbohydrate diet: a model for prediabetes. Ponte Int Sci Res J. 2018;74(5):119–143.
19. Mzimela NC, Ngubane PS, Khathi A. The changes in immune cell concentration during the progression of pre-diabetes to type 2 diabetes in a high-fat high-carbohydrate diet-induced pre-diabetic rat model. Autoimmunity. 2019;52(1):1–10.
20. Lu Z-Y, Shao Z, Li Y-L, Wulasihan M, Chen X-H. Prevalence of and risk factors for non-alcoholic fatty liver disease in a chinese population: an 8-year follow-up study. World J Gastroenterol. 2016;22(13):3663. doi:10.3748/wjg.v22.i37.8314
21. Kohli R, Sunduram S, Mouzaki M, et al. Pediatric nonalcoholic fatty liver disease: a report from the expert committee on nonalcoholic fatty liver disease (econ). J Pediatr. 2016;172:9–13. doi:10.1016/j.jpeds.2015.12.016
22. Romero-Gómez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67(4):829–846. doi:10.1016/j.jhep.2017.05.016
23. Asgharpour A, Cazanave SC, Pacana T, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol. 2016;65(3):579–588. doi:10.1016/j.jhep.2016.05.005
24. Fernández-Real JM, McClain D, Manco M. Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care. 2015;38(11):2169–2176. doi:10.2337/dc14-3082
25. Mazza A, Fruci B, Garinis GA, Giuliano S, Malaguarnera R, Belfiore A. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2011;(2012)1–14.
26. Sullivan S. Implications of diet on nonalcoholic fatty liver disease. Curr Opin Gastroenterol. 2010;26(2):160. doi:10.1097/MOG.0b013e3283358a58
27. Angulo P. Obesity and nonalcoholic fatty liver disease. Nutr Rev. 2007;65:S57–S63. doi:10.1111/j.1753-4887.2007.tb00329.x
28. Brunt EM, Wong VW-S, Nobili V, et al. Nonalcoholic fatty liver disease. Nature Rev Dis Primers. 2015;1:15080. doi:10.1038/nrdp.2015.80
29. Marino L, Jornayvaz FR. Endocrine causes of nonalcoholic fatty liver disease. World J Gastroenterol. 2015;21(39):11053.
30. Granér M, Nyman K, Siren R, et al. Ectopic fat depots and left ventricular function in nondiabetic men with nonalcoholic fatty liver diseaseclinical perspective. Circulation. 2015;8(1):e001979.
31. Heilbronn L, Smith S, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type ii diabetes mellitus. Int J Obes. 2004;28(S4):S12.
32. Hannah WN, Harrison SA. Lifestyle and dietary interventions in the management of nonalcoholic fatty liver disease. Dig Dis Sci. 2016;61(5):1365–1374.
33. Kumar S, Kelly AS. Review of childhood obesity: from epidemiology, etiology, and comorbidities to clinical assessment and treatment.
34. Cali AM, Zern TL, Taksali SE, et al. Intrahepatic fat accumulation and alterations in lipoprotein composition in obese adolescents: a perfect pro-atherogenic state. Diabetes Care. 2007.30(12):3093–3098
35. Huang C-Z, Tung Y-T, Hsia S-M, Wu C-H, Yen G-C. The hepatoprotective effect of phyllanthus emblica l. Fruit on high fat diet-induced non-alcoholic fatty liver disease (NAFLD) in SD rats. Food Funct. 2017;8(2):842–850.
36. Shimabukuro M. Leptin resistance and lipolysis of white adipose tissue: an implication to ectopic fat disposition and its consequences. J Atheroscler Thromb. 2017;24(11):1088–1089.
37. Ngubane PS, Masola B, Musabayane CT. The effects of syzygium aromaticum-derived oleanolic acid on glycogenic enzymes in streptozotocin-induced diabetic rats. Ren Fail. 2011;33(4):434–439.
38. Woo S-L, Xu H, Li H, et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS One. 2014;9(3):e91111.
39. Yang L, Miura K, Zhang B, et al. Trif differentially regulates hepatic steatosis and inflammation/fibrosis in mice. Cell Mol Gastroenterol Hepatol. 2017;3(3):469–483.
40. Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441.
41. Ashraf N, Sheikh T. Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic Res. 2015;49(12):1405–1418.
42. Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP‐1c. Diabetes Obesity Metab. 2010;12:83–92.
43. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1049–1061.
44. Wang X, Liu R, Zhang W, et al. Oleanolic acid improves hepatic insulin resistance via antioxidant, hypolipidemic and anti-inflammatory effects. Mol Cell Endocrinol. 2013;376(1–2):70–80.
45. Morigny P, Houssier M, Mouisel E, Langin D. Adipocyte lipolysis and insulin resistance. Biochimie. 2016;125:259–266.
46. de Melo CL, Queiroz MGR, Fonseca SG, et al. Oleanolic acid, a natural triterpenoid improves blood glucose tolerance in normal mice and ameliorates visceral obesity in mice fed a high-fat diet. Chem Biol Interact. 2010;185(1):59–65.
47. Wang X, Li YL, Wu H, et al. Antidiabetic effect of oleanolic acid: a promising use of a traditional pharmacological agent. Phytother Res. 2011;25(7):1031–1040.
48. Ou-Yang Q, Xuan C-X, Wang X, et al. 3-acetyl-oleanolic acid ameliorates non-alcoholic fatty liver disease in high fat diet-treated rats by activating ampk-related pathways. Acta Pharmacol Sin. 2018.39(8):1284–1293.
49. Hauck AK, Bernlohr DA. Oxidative stress and lipotoxicity. J Lipid Res. 2016;57(11):1976–1986.
50. Law BA, Liao X, Moore KS, et al. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. Faseb J. 2018;32(3):1403–1416.
51. Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology. 2016;150(8):1769–1777.
52. Houldsworth A, Hodgkinson A, Demaine A, Millward A. Catalase polymorphism may influence the pathogenesis of diabetes mellitus. J Endocrinol Diab. 2016;3(1):1–7.
53. Marseglia L, Manti S, D’Angelo G, et al. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. 2015;16(1):378–400.
54. Pijls KE, Jonkers A, Daisy M, et al. Is intestinal oxidative stress involved in patients with compensated liver cirrhosis? Ann Hepatol. 2016;15(3):402–409.
55. Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: mechanisms and therapeutic potential. Pharmacol Ther. 2017;172:50–62.
56. Yi J, Xia W, Wu J, et al. Betulinic acid prevents alcohol-induced liver damage by improving the antioxidant system in mice. J Vet Sci. 2014;15(1):141–148.
57. Kwak M-S, Kim D, Chung GE, et al. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol. 2012;18(4):383.
58. Dani C, Poggi C, Pratesi S. Bilirubin and oxidative stress in term and preterm infants. Free Radic Res. 2019;53(1):2–7.
59. Hjelkrem M, Morales A, Williams C, Harrison S. Unconjugated hyperbilirubinemia is inversely associated with non‐alcoholic steatohepatitis (nash). Aliment Pharmacol Ther. 2012;35(12):1416–1423.
60. Tian J, Zhong R, Liu C, et al. Association between bilirubin and risk of non-alcoholic fatty liver disease based on a prospective cohort study. Sci Rep. 2016;6:31006.
61. Kim S-M, Grenert JP, Patterson C, Correia MA. Chip−/−-mouse liver: adiponectin-ampk-foxo-activation overrides cyp2e1-elicited jnk1-activation, delaying onset of nash: therapeutic implications. Sci Rep. 2016;6:29423.
62. Liu W, Baker SS, Baker R. D, Zhu L. Antioxidant mechanisms in nonalcoholic fatty liver disease. Curr Drug Targets. 2015;16(12):1301–1314.
63. Koneru M, Sahu BD, Kumar JM, et al. Fisetin protects liver from binge alcohol-induced toxicity by mechanisms including inhibition of matrix metalloproteinases (mmps) and oxidative stress. J Funct Foods. 2016;22:588–601.
64. Dowman JK, Tomlinson J, Newsome P. Systematic review: the diagnosis and staging of non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis. Aliment Pharmacol Ther. 2011;33(5):525–540.
65. Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism. 2014;63(7):895–902.
66. Caldwell SH, Argo CK, Al-Osaimi AM. Therapy of nafld: insulin sensitizing agents. J Clin Gastroenterol. 2006;40:S61–S66.
67. Li Y, Liu L, Wang B, Wang J, Chen D. Metformin in non-alcoholic fatty liver disease: a systematic review and meta‑analysis. Biomed Rep. 2013;1(1):57–64.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.