")
Back to Journals » OncoTargets and Therapy » Volume 7
PKN1 modulates TGFß and EGF signaling in HEC-1-A endometrial cancer cell line
Authors Attarha S, Saini R, Andersson S, Mints M, Souchelnytskyi S
Received 28 March 2014
Accepted for publication 14 May 2014
Published 4 August 2014 Volume 2014:7 Pages 1397—1408
DOI https://doi.org/10.2147/OTT.S65051
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Sanaz Attarha,1,2 Ravi Kanth Rao Saini,3 Sonia Andersson,2 Miriam Mints,2 Serhiy Souchelnytskyi1,4,5
1Department of Oncology–Pathology, 2Department of Women's and Children's Health, Karolinska Institutet, Stockholm, 3Department of Biomedicine, Sahlgrenska Cancer Center, University of Gothenburg, Gothenburg, Sweden; 4OCD-AB, Uppsala, Sweden; 5Neurocentrum, Karolinska University Hospital, Solna, Sweden
Background: The response of cells to TGFβ and EGF is mediated by a network of various intracellular regulators. The signaling crosstalk between different regulators is of key importance for tumorigenesis. The crosstalk may explain the modulation of cellular responses to the same regulator by another signaling molecule. As PKN1 – a serine/threonine kinase implicated in tumorigenesis – was identified as potential crosstalk node for TGFβ and EGF signaling, the cellular functions that may be affected by PKN1 in a crosstalk of TGFβ and EGF were explored.
Methods: To investigate the contribution of PKN1 to TGFβ and EGF signaling, transiently PKN1-transfected HEC-1-A endometrial cancer cells were generated and subjected to treatment with TGFβ1, EGF, and their combination. Proliferation, apoptosis, invasion, wound healing, and migration assays were performed. The impact of PKN1 on the expression and phosphorylation of intracellular proteins was monitored by immunoblotting.
Results: It was demonstrated that PKN1 modulated the responses of HEC-A-1 endometrial cancer cells to TGFβ1 and EGF. PKN1 had an inhibitory effect on the stimulation of cell migration, and PKN1 kinase activity was required for the inhibitory effect of TGFβ and EGF on cell proliferation and invasiveness. It was observed that phosphorylation of Smad2, FAK, and Erk1/2 correlated with responses of the cells to TGFβ1 and EGF.
Conclusion: PKN1 modulates TGFβ- and EGF-dependent regulation of cell proliferation, migration, and invasiveness, and therefore is a component of the network signaling downstream of TGFβ and EGF.
Keywords: PKN1 kinase, TGFβ, EGF, cell migration, proliferation, invasiveness
Introduction
PKN1, also known as PAK1, is a serine/threonine kinase implicated in formation of mammary gland tumors and premalignant lesions in animal models, although with a long latency.1 It has been reported that overexpression of PKN1 correlates with aggressive ovarian,2 colorectal,3 and prostate cancers.4 Other findings suggested that PKN1 has a role in the development of invasive phenotypes of breast5 and gastric6 cancer cells.
PKN1 contains three highly conserved regions: 1) a unique regulatory domain in the amino terminus; 2) a catalytic domain homologous to PKC in carboxyl terminal region; and 3) a so-called D-region located between the regulatory and catalytic domains.4,7 The amino terminal region plays a critical role in activation of PKN1.7 It provides the activation loop of PKN1 that is important for serine/threonine kinase activity.2 Once PKN1 has been activated it mediates downstream signaling events involved in cytoskeletal reorganization, cell motility, apoptosis, and transformation. PKN1 activates a number of signaling pathways, including p38 MAPK, extracellular signal-regulated protein kinase, Jun N-terminal kinase, and NF-kB.8
TGFβ was found to have a double role in tumorigenesis. In the early stage of cancer, TGFβ has a tumor suppressor role that results in growth inhibition, cell cycle arrest, and apoptosis. In the advanced stage of cancer, TGFβ promotes tumorigenesis. Cancer cells may lose responsiveness to TGFβ and may acquire aberrant TGFβ signaling followed by promotion of survival, proliferation, epithelial–mesenchymal transition, and increased motility and invasiveness of the cells.9 Numerous signaling pathways converge with the TGFβ pathway to modulate its effects, including signaling induced by EGF.10 EGF is a potent regulator of cell functions, with a predominantly pro-mitogenic role in tumorigenesis.11 EGF also promotes cell survival, angiogenesis, and differentiation. Deregulation of EGF pathways by its overexpression or constitutive activation of EGF signaling can promote tumorigenesis and is associated with a poor prognosis in many human malignancies.12
The signaling crosstalk between different regulators is of key importance for tumorigenesis. The crosstalk may explain modulation of cellular responses to the same regulator by another signaling molecule. As PKN1 was identified as potential crosstalk node for TGFβ and EGF signaling, the cellular functions that may be affected by PKN1 in a crosstalk of TGFβ and EGF were explored. Here, it is reported that PKN1 modulates TGFβ- and EGF-dependent regulation of cell proliferation, migration, and invasiveness, and therefore is a component of the network signaling downstream of TGFβ and EGF.
Material and methods
Cell culture
HEC-1-A cells were obtained from American Type Culture Collection (Manassas, VA, USA) and were cultured in McCoy’s 5A modified medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Sigma-Aldrich, St Louis, MO, USA). HEC-1-A cells are recognized as an accepted model to study endometrial cancer.13,14 Human recombinant TGFβ1 and EGF were obtained from PeproTech (Rocky Hill, NJ, USA).
Cell transfection
A day before transfection, HEC-1-A cells were cultured to reach 50% confluence for transfection. HEC-1-A cells were transfected with pcDNA3 control vector, full length of human PKN1-FLAG in pRc/CMV (wild type [WT]), full length of human PKN1 (T774A)-FLAG in pRc/CMV (kinase negative [KN]), and catalytic domain of human PKN1-FLAG in pRc/CMV (constitutively active [CA]) in twelve-well plates using GeneJuice® transfection reagent, as recommended by the supplier (EMD Millipore, Billerica, MA, USA). The expression constructs were kindly provided by Dr Hideyuki Mukai. The medium was changed 6 hours after transfection and cells were incubated in complete medium for 48 hours prior to treatment. Transfection efficiency was monitored by detection of levels of PKN1 constructs and endogenous PKN1 (Figure S1).
Cell proliferation assay
Cell proliferation in response to TGFβ1 and EGF treatment was measured by using CellTiter 96® AQueous One Solution Cell assay (Promega Corporation, Mudison, WI, USA) according to the manufacturer’s recommendations. Cells were grown in McCoy’s 5A modified medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin with or without TGFβ1 (5 ng/mL) and EGF (10 ng/mL) for 24 hours.
Cell apoptosis assay
Cell apoptosis was determined by using Cell Death Detection ELISAPLUS (Hoffman-La Roche Ltd, Basel, Switzerland). Briefly, cell lysates were placed in a streptavidin-coated microplate. A mixture of anti-histone–biotin and anti-DNA–peroxidase was added and incubated for 2 hours at 25°C. After removal of the unbound antibodies by washing steps, the peroxidase was determined photometrically at 405 nm with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) as the substrate. As a control, the presence of apoptotic cells and apoptotic bodies upon transfection and treatment of cells was monitored (Figure S2).
Wound healing assay
Cells were grown in culture media containing 10% fetal bovine serum for 48 hours, until the cells reached confluence. Monolayers of confluent cultures were scratched with a 20 μL pipette tip, and images of the scratched areas were taken under a microscope. TGFβ1 (5 ng/mL) and EGF (10 ng/mL) were added, and the cells were incubated for 24 hours. After 24 hours, images of the scratched areas were taken. Quantification was done by measuring the open wound area, which is the fraction of open image area at a later time point compared to the initial time point, given as a percentage using TScratch software (CSElab, Zurich, Switzerland).15
Migration assay
Cells were seeded on the membranes of the 96-well plate ChemoTx® chemotaxis system (116-8; Neuro Probe, Inc., Gaithersburg, MD, USA) in a culture medium containing the growth factors as indicated for treatment. After 24 hours, the membrane was washed twice in phosphate-buffered saline and fixed with 70% ethanol. The nonmigrated cells were removed by cotton swab from the upper side of the membrane. The membrane was stained with 0.5% crystal violet, and subsequently visualized and quantified by using ImageJ software (National Institutes of Health, Bethesda, MD, USA).16
Invasion assay
Membranes of the 96-well plate ChemoTx chemotaxis system were covered with 3% gelatin, and cells were seeded on the membranes in a culture medium containing 5 ng/mL TGFβ1 and 10 ng/mL EGF. After 24 hours, the membrane was washed twice in phosphate-buffered saline and fixed with 70% ethanol. The noninvaded cells were removed by cotton swab from the upper side of the membrane. The membrane was stained with 0.5% crystal violet, and subsequently visualized and quantified by using ImageJ software.16
Immunoblotting
Cell lysates were resolved in 10% sodium dodecyl sulfate polyacrylamide mini gels (Mini-PROTEAN® Tetra Cell; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and transferred onto Whatman® Protran® nitrocellulose membranes (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Membranes were blocked with 5% (weight/volume [w/v]) bovine serum albumin in Tris-buffered saline with Tween 20 for 1 hour, incubated with primary antibodies against target proteins, and then incubated with horseradish peroxidase-conjugated secondary antibody (GE Healthcare). The proteins were visualized using Western blotting Luminol reagent (Santa Cruz Biotechnology Inc., Dallas, TX, USA). The following antibodies were used: anti-PKN1 (sc-7161), anti-Smad2/3 (sc-6032), anti-pSmad2 (sc-135644), anti-ERK1/2 (sc-135900), anti-pERK1/2 (sc-7383), vimentin (sc-7557), E-cadherin (sc-7870), anti-FAK (sc-558), actin (sc-8432) (Santa Cruz Biotechnology), and anti-FAK (phospho Y397) (ab4803; Abcam, Cambridge, UK).
Systematic analysis
Systematic analysis was performed with use of Cytoscape version 2.8.1 (Institute for Systems Biology, Seattle, WA, USA). Gene Ontology terms of PKN1, TGFβ, and EGF were uploaded to generate a network, including neighbors of the uploaded proteins. Databases were explored by Cytoscape plug-in MiMI (National Center for Integrative Biomedical Informatics, Ann Arbor, MI, USA).
Statistical analysis
Statistical significance of observed differences was evaluated using the Mann–Whitney U test among unpaired groups and among multiple groups by the Kruskal–Wallis test followed by Dunn’s multiple comparison test. Analyses were conducted using Prism® 6 software (GraphPad Software, Inc., La Jolla, CA, USA) and P<0.05 was considered significant.
Results
Generation of transiently transfected HEC-1-A cells
PKN1 has been identified as a protein deregulated in endometrial cancer.17 To study the impact of PKN1 on cells, WT, KN, and CA PKN1 was transiently expressed in HEC-1-A endometrial cancer cells. The expression of PKN1 was controlled by immunoblotting with anti-PKN1 antibody (Figure 1A). Expression of endogenous PKN1 was observed in six out of seven tested cell lines (Figure S1). Enhanced expression of PKN1 constructs allowed the impact of PKN1 on cell physiology to be accentuated (Figure 1A). Transiently transfected cells were then used in the tests described below. Systemic analysis of potential connections between PKN1, TGFβ, and EGF showed involvement of a number of potent regulators of cell proliferation and cytoskeleton rearrangement (Figure 1B). This prompted the exploration of whether PKN1 plays a role in modulation of TGFβ- and EGF-dependent regulation of cell proliferation, migration, and invasiveness.
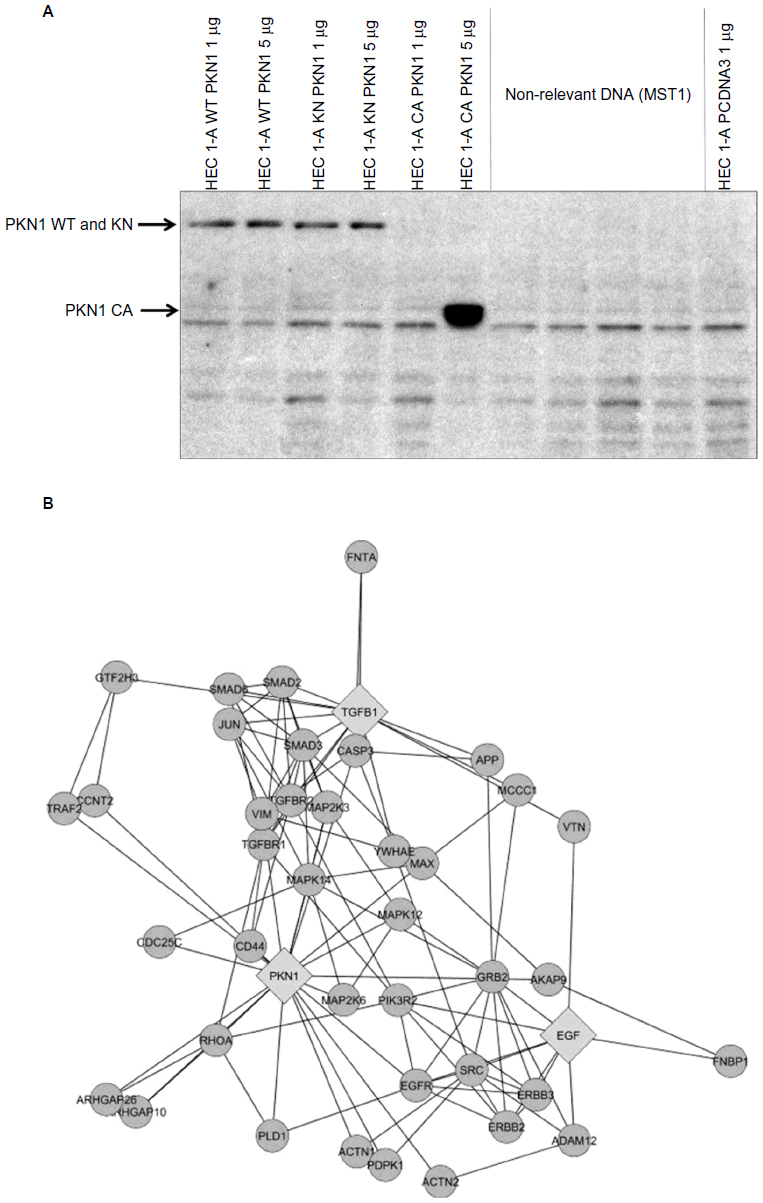 | Figure 1 PKN1 is a potential component of TGFβ and EGF crosstalk. |
PKN1 reduced proliferation of HEC-1-A cells
It was observed that the treatment of cells with TGFβ1 and EGF significantly reduced the proliferation rate of the control parental empty vector-transfected and the WT PKN1-transfected HEC-1-A cells (Figure 2). Transfection of HEC-1-A cells with KN PKN1 and CA PKN1 resulted in a reduced rate of proliferation of the nontreated cells. It was observed that expression of KN PKN1 countered the effects of TGFβ1 and EGF. An unexpected effect was that CA PKN1 countered the effects of EGF and combined TGFβ1 and EGF treatments. This indicates that the truncation of PKN1 has a similar impact on PKN1 contribution as the KN construct. It also indicates that the kinase activity itself is not sufficient to mimic WT PKN1, and the intactness of PKN1 is required for the full functional capacity of PKN1. Thus, PKN1 is required for inhibition of cell proliferation by combined action of TGFβ1 and EGF, as impairment of PKN1 functions by blocking its kinase activity or by truncation preventing the inhibitory effect by TGFβ1 and EGF.
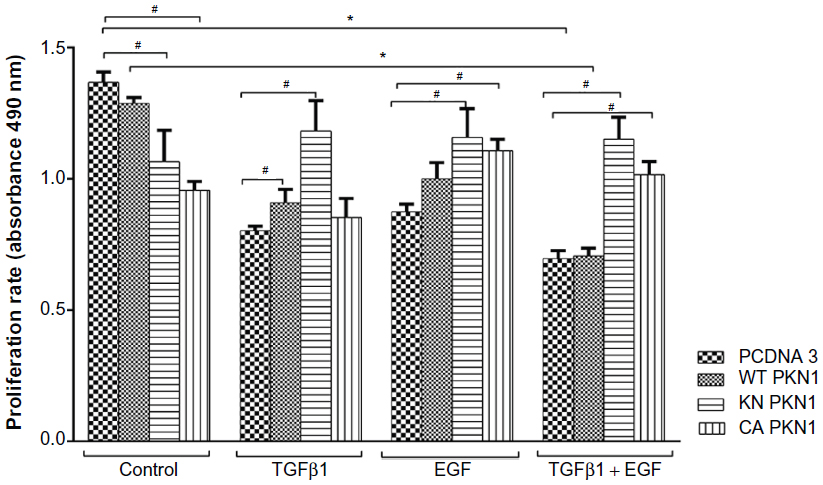 | Figure 2 Impact of PKN1 on TGFβ1- and EGF-dependent regulation of cell proliferation. |
PKN1 did not affect apoptosis
To explore whether PKN1 may affect cell death, an apoptosis assay was performed. It was observed that double treatment of the empty vector-transfected cells with TGFβ1 and EGF reduced cell apoptosis (Figure 3). Transfection of WT, KN, and CA PKN1 had only a marginal effect on cell death. The only significant, although weak, effect was the reversal of the combined treatment of TGFβ1 and EGF. There were no apoptotic cells or apoptotic bodies upon visual inspection of transfected and treated cells (Figure S2). Thus, strong effects of PKN1 on cell death were not observed.
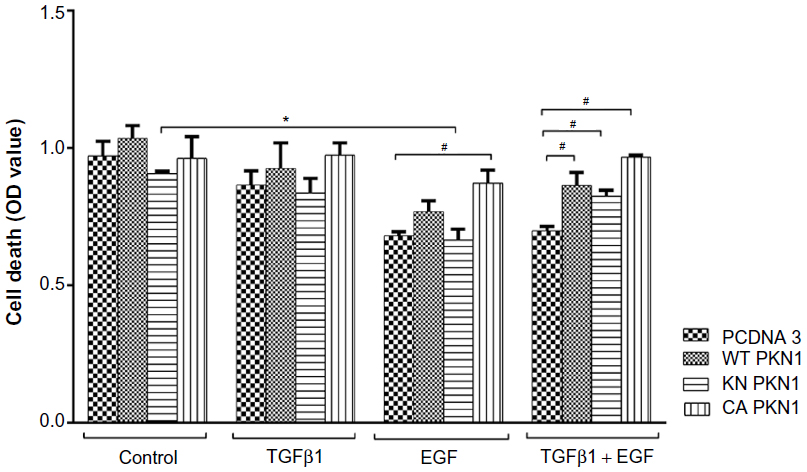 | Figure 3 PKN1 has a marginal effect on HEC-1-A cell death. |
PKN1 modulates migration of cells stimulated by TGFβ1 and EGF
As the systemic analysis indicated that PKN1 may affect migration of cells, wound healing18 and membrane migration19 assays were performed (Figures 4 and 5). Wound healing assays explore migration capacities of cells that are under contact inhibition of proliferation. Membrane migration assays explore proliferating cells in a sparse culture. The molecular mechanisms triggering cell migration in these two tests may differ due to unequal conditions of tested cells. Despite differences, the tests may allow complementation of assessment of migration. The wound healing assay showed that PKN1 constructs prevented TGFβ1-induced closure (Figure 4). However, expression of the PKN1 constructs strongly promoted wound closure when cells were treated with both TGFβ1 and EGF (Figure 4).
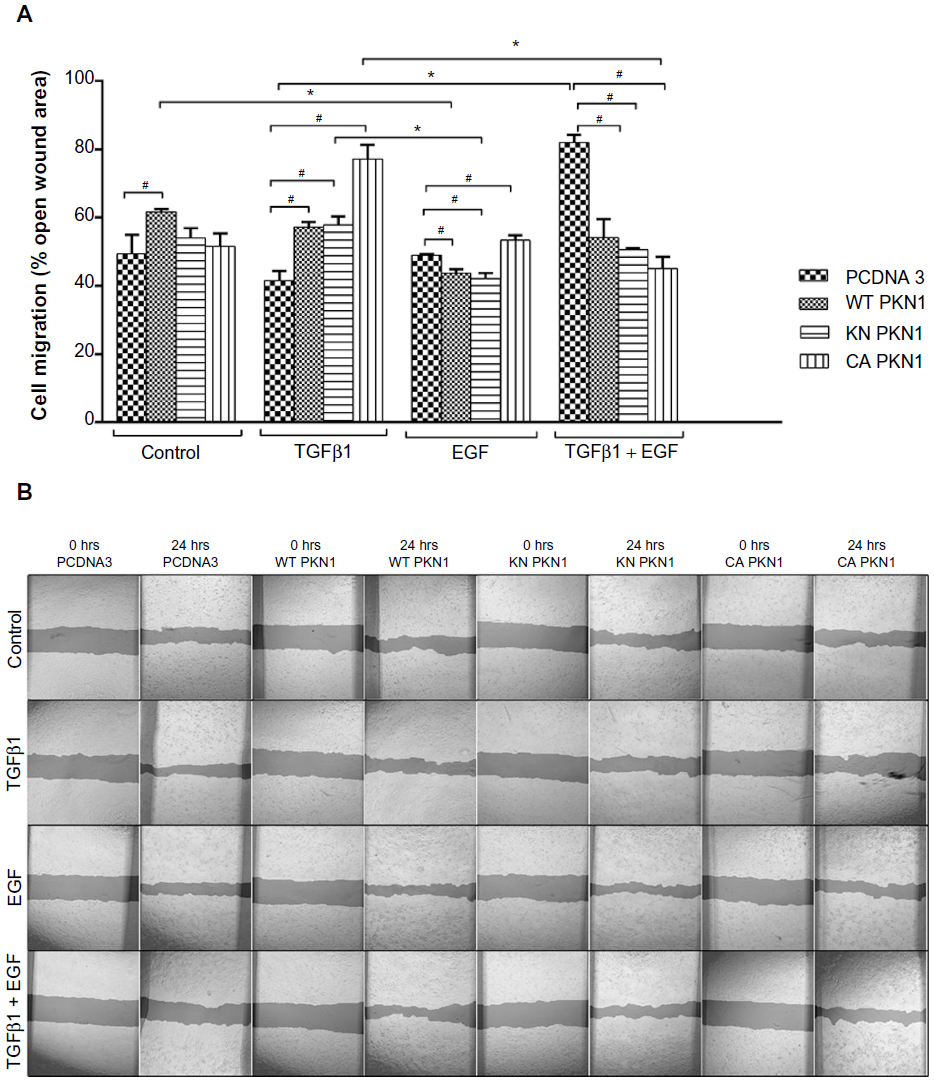 | Figure 4 Effect of PKN1 on cell migration in a wound healing assay. |
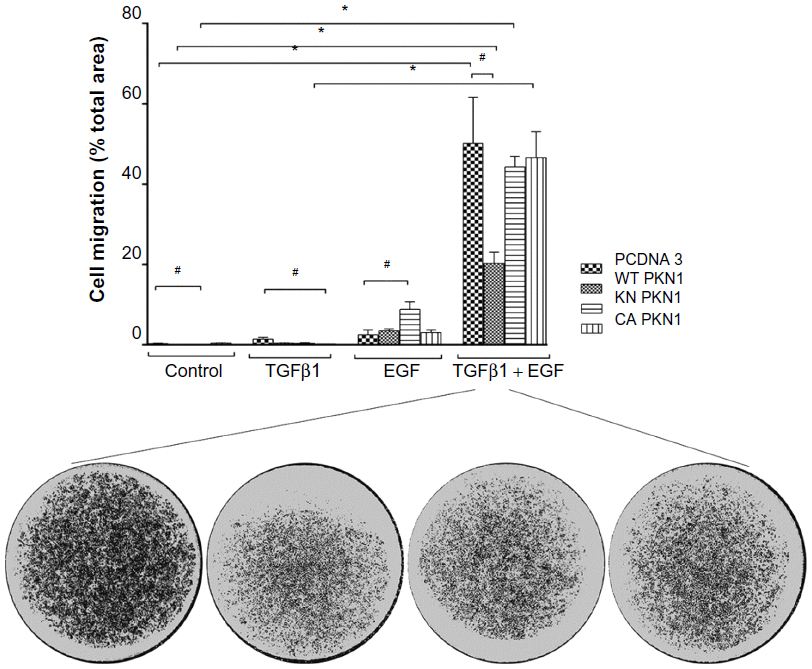 | Figure 5 PKN1 suppressed migration of cells stimulated by TGFβ1 and EGF. |
The membrane migration assay showed that the treatment with both TGFβ1 and EGF promoted cell migration (Figure 5). Single treatments and transfections with PKN1 constructs did not have significant effects. The difference in response pattern between wound healing and membrane migration is that impairment of PKN1 activity in the KN and CA mutants did not mimic the WT construct effect in the membrane migration assay as compared to the wound healing tests (Figures 4 and 5). Despite this difference, these results show that PKN1 may indeed counteract the action of combined TGFβ1 and EGF treatment.
TGFβ1- and EGF-dependent regulation of cell invasiveness is modulated by PKN1
Invasiveness of cells into a collagen matrix is one of the key mechanisms in the development of metastasis. Therefore, we explored whether PKN1 constructs may affect TGFβ1- and EGF-dependent regulation of cell invasiveness (Figure 6). It was observed that double treatment of HEC-1-A cells with TGFβ1 and EGF increased cell invasiveness in the cells transfected with KN PKN1, while expression of WT or CA PKN1 did not show such action. Expression of KN PKN1 significantly increased the invasiveness of cells. This stimulatory effect was strong. Thus, PKN1 did modulate the invasiveness of cells, with the intracellular mechanisms apparently different from mechanisms of regulation of cell migration.
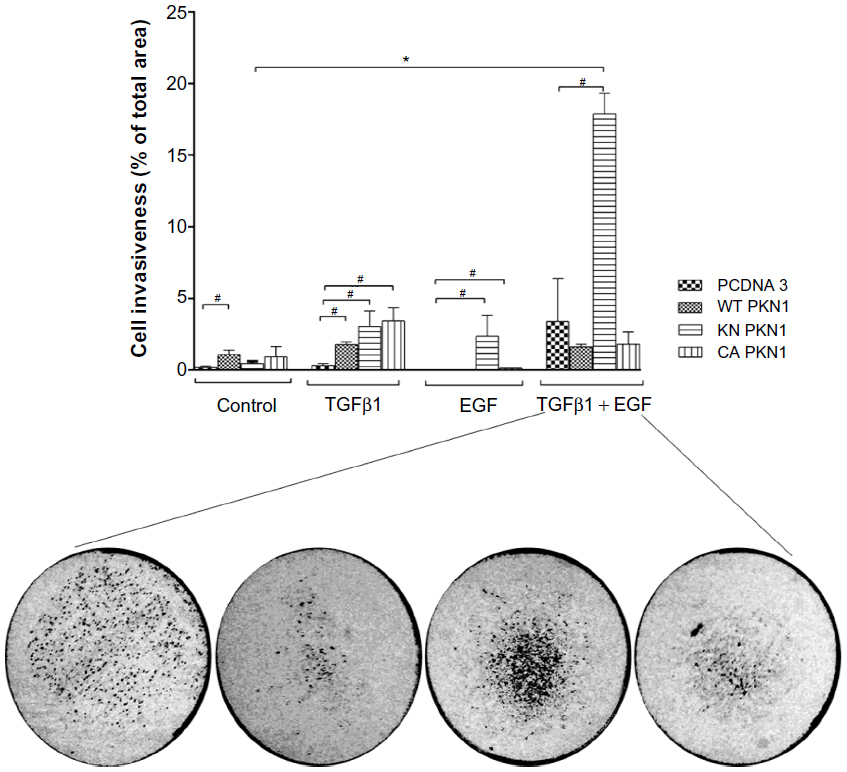 | Figure 6 TGFβ1- and EGF-dependent regulation of cell invasiveness is modulated by PKN1. |
PKN1 expression in modulation of phosphorylation of Smad2, Erk1/2, and FAK, and expression of E-cadherin and vimentin
To explore the molecular mechanisms of PKN1 involvement in TGFβ and EGF signaling, the expression and phosphorylation of Smad2, Erk1/2, FAK, E-cadherin, and vimentin were studied. Phosphorylation of Smad2 and Erk1/2 reflect the activation of signaling downstream of TGFβ and EGF. It was observed that expression of WT, KN, and CA PKN1 decreased the intensity of Smad2 phosphorylation upon treatment with TGFβ1. The effect was more significant in CA PKN1-transfected cells. At the same time, phosphorylation of Smad2 upon treatment with combined TGFβ1 and EGF and single EGF decreased in KN and CA PKN1-transfected cells, while there was no change in WT PKN1-transfected cells (Figure 7A). It was observed that expression of WT, KN, and CA PKN1 decreased the intensity of Erk1/2 phosphorylation upon treatment with TGFβ1 (Figure 7B). It was observed that EGF-dependent Erk1/2 phosphorylation increased in WT, KN, and CA PKN1-transfected cells but not in the vector-transfected cells, which indicates that the cells were responsive to EGF under condition of the enhanced level of PKN1. The effect was most pronounced in KN PKN1-transfected cells. Double treatment of cells with TGFβ1 and EGF led to no significant effects on intensity of Erk1/2 phosphorylation in WT and KN PKN1-transfected cells, while it caused a significant decrease in HEC-1-A CA PKN1 cells.
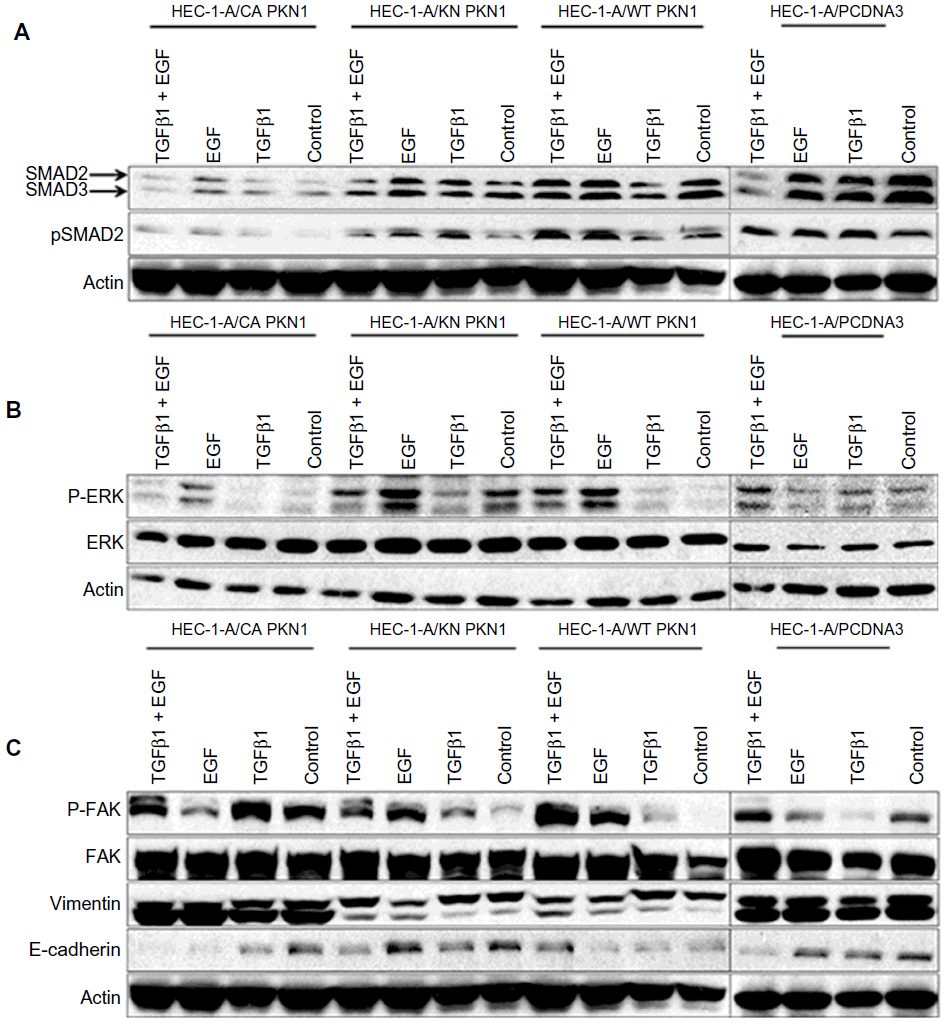 | Figure 7 Impact of PKN1 on the expression and phosphorylation of intracellular proteins (A) SMAD2, (B) Erk1/2, and (C) FAK, E-cadherin, and vimentin. |
The expression and activity of FAK, E-cadherin, and vimentin may reflect molecular mechanisms involved in regulation of cell migration and invasiveness. It was observed that expression of WT PKN1 increased TGFβ1- and EGF-dependent phosphorylation of FAK while KN PKN1 expression decreased this effect (Figure 7C).
It was observed that expression of WT and CA PKN1 predominantly inhibited E-cadherin expression upon single and double treatment with TGFβ1 and EGF (Figure 7C). KN PKN1 did not have such inhibitory effect. Vimentin expression in tested conditions was not modulated by treatments with TGFβ1 and/or EGF (Figure 7C). The only observed effect was of WT and KN PKN1, which inhibited levels of vimentin irrespective of treatments. The observations of changes in E-cadherin, vimentin, Smad2, and Erk1/2 expression and phosphorylation indicate that these proteins are indeed affected by PKN1. These observations indicate that further studies of the tested proteins (Figure 7) in the context of PKN1’s role in proliferation, migration, and invasiveness are justified to gain insights into underlying molecular mechanisms.
Discussion
Unveiling the complexity of intracellular signaling crosstalk results in the identification of more and more components and interactions. Systems biology tools allow the extraction of potential interactions between proteins and genes, which otherwise may not be detected. The search for the mechanisms of crosstalk between TGFβ and EGF indicates that PKN1 may be a convergence point for these two potent regulatory pathways.17 Here, it was described that PKN1 is a modulator of the crosstalk between TGFβ and EGF in the regulation of cell proliferation, migration, and invasiveness. The modulating effects of PKN1 were dependent on its kinase activity. The description of cellular responses shows involvement of PKN1 in TGFβ and EGF signaling (Figures 2–6). Exact molecular mechanisms of this crosstalk would require a further dedicated study. The data (Figure 7) indicate that Smad2 and Erk1/2 may be involved in the crosstalk. Smad2 is a direct target of TGFβ receptor type I and Erk1/2 is a convergence target of many different regulators of cell proliferation. It has been shown that Smad2 can be phosphorylated by Erk1/2, and that phosphorylation may inhibit nuclear localization of Smad2.20 Focal adhesion may be regulated by multiple factors, and the results show that PKN1 may interfere with TGFβ- and EGF-dependent phosphorylation of FAK (Figure 7). The effects on FAK phosphorylation are in line with PKN1 modulation of cell invasion and migration. The expression of vimentin and E-cadherin upon explored conditions (Figure 7) is also in line with cellular responses (Figures 4–6). It has to be noted that the observed correlations indicate involvement of other intracellular components, as types and amplitude of changes were not fully overlapping with cellular responses. Systemic analysis and a number of reports by others showed that the number of molecules involved in the network signaling by TGFβ and EGF may account for more than 100.21,22 These molecules are engaged on different stages of signaling cascades, and may include various intracellular processes, eg, gene transcription, protein synthesis, protein activities, localization, and metabolic processes. These intracellular processes then converge on regulators of cellular responses. This report includes PKN1 in this network of combined signaling by TGFβ and EGF, which leads to regulation of cell migration, invasiveness, and proliferation, and sets the direction for further exploration of intracellular mechanisms.
Acknowledgments
The authors are grateful to Dr Hideyuki Mukai for his gift of the PKN1 constructs. They are also grateful to Oves Minnesfond for support and encouragement. This work is supported in part by grants from the Radiumhemmet research funds (#121202), the Swedish Cancer Society, the Swedish Research Council, the Swedish Institute, INTAS, Erasmus KI-UWM, and STINT to SS, and grants from the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and the Karolinska Institute and Swedish Labour Market Insurance (AFA) to MM.
Disclosure
The authors report no conflicts of interest in this work.
References
Ong CC, Jubb AM, Haverty PM, et al. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2011;108(17):7177–7182. | |
Galgano MT, Conaway M, Spencer AM, Paschal BM, Frierson HF Jr. PRK1 distribution in normal tissues and carcinomas: overexpression and activation in ovarian serous carcinoma. Hum Pathol. 2009;40(10):1434–1440. | |
Carter JH, Douglass LE, Deddens JA, et al. Pak-1 expression increases with progression of colorectal carcinomas to metastasis. Clin Cancer Res. 2004;10(10):3448–3456. | |
Metzger E, Müller JM, Ferrari S, Buettner R, Schüle R. A novel inducible transactivation domain in the androgen receptor: implications for PRK in prostate cancer. EMBO J. 2003;22(2):270–280. | |
Adam L, Vadlamudi R, Mandal M, Chernoff J, Kumar R. Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J Biol Chem. 2000;275(16):12041–12050. | |
Liu F, Li X, Wang C, et al. Downregulation of p21-activated kinase-1 inhibits the growth of gastric cancer cells involving cyclin B1. Int J Cancer. 2009;125(11):2511–2259. | |
Takahashi M, Mukai H, Toshimori M, Miyamoto M, Ono Y. Proteolytic activation of PKN by caspase-3 or related protease during apoptosis. Proc Natl Acad Sci U S A. 1998;95(20):11566–11571. | |
Vadlamudi RK, Adam L, Wang RA, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275(46):36238–36244. | |
Jakowlew SB. Transforming growth factor-β in cancer and metastasis. Cancer Metastasis Rev. 2006;25(3):435–457. | |
Dunfield LD, Nachtigal MW. Inhibition of the antiproliferative effect of TGFβ by EGF in primary human ovarian cancer cells. Oncogene. 2003;22(30):4745–4751. | |
Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. | |
Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77(6):400–410. | |
Kuramoto H, Hamano M, Imai M. HEC-1 cells. Hum Cell. 2002;15(2):81–95. | |
Mori Y, Mizuuchi H, Sato K, Okamura N, Kudo R. [The factors involved in invasive ability of endometrial carcinoma cells]. Nihon Sanka Fujinka Gakkai Zasshi. 1994;46(6):509–516. Japanese. | |
Gebäck T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46(4):265–274. | |
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. | |
Attarha S, Andersson S, Mints M, Souchelnytskyi S. Individualised proteome profiling of human endometrial tumours improves detection of new prognostic markers. Br J Cancer. 2013;109(3):704–713. | |
Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333. | |
Penno MB, Hart JC, Mousa SA, Bozarth JM. Rapid and quantitative in vitro measurement of cellular chemotaxis and invasion. Methods Cell Sci. 1997;19(3):189–195. | |
Kamato D, Burch ML, Piva TJ, et al. Transforming growth factor-β signalling: role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25(10):2017–2024. | |
Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial–mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25(1):76–84. | |
Ghosh S, Matsuoka Y, Asai Y, Hsin KY, Kitano H. Software for systems biology: from tools to integrated platforms. Nat Rev Genet. 2011;12(12):821–832. |
Supplementary material
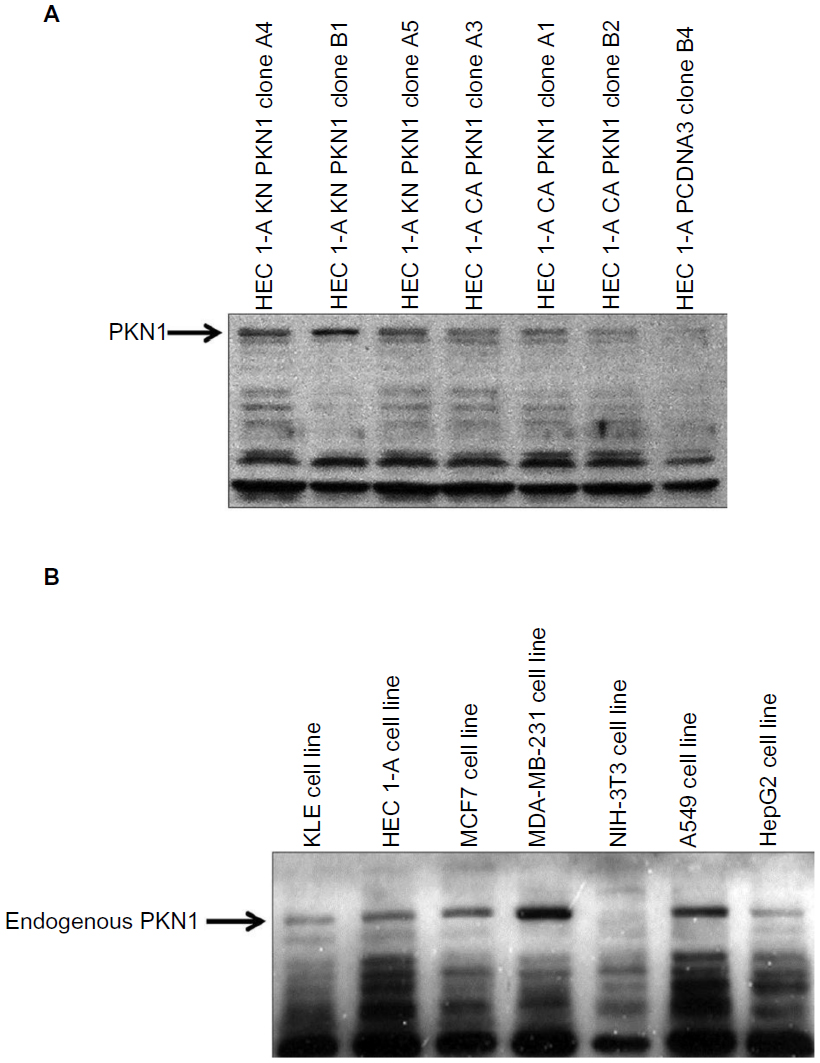 | Figure S1 Detection of endogenous PKN1. |
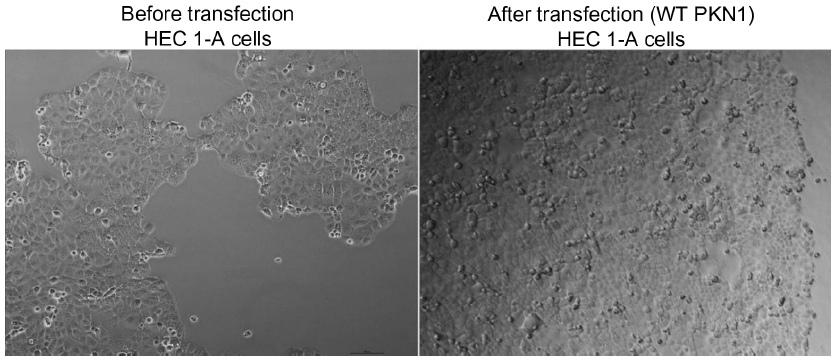 | Figure S2 Images of HEC1-A cells before and after transfection with WT PKN1. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.