Back to Journals » Drug Design, Development and Therapy » Volume 8
Pilot Phase II study of mazindol in children with attention deficit/hyperactivity disorder
Authors Konofal E, Zhao W ![]() , Laouénan C, Lecendreux M, Kaguelidou F, Benadjaoud L, Mentré F
, Laouénan C, Lecendreux M, Kaguelidou F, Benadjaoud L, Mentré F ![]() , Jacqz-Aigrain E
, Jacqz-Aigrain E
Received 4 April 2014
Accepted for publication 4 June 2014
Published 11 December 2014 Volume 2014:8 Pages 2321—2332
DOI https://doi.org/10.2147/DDDT.S65495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Eric Konofal,1,2 Wei Zhao,3–5 Cédric Laouénan,5–7 Michel Lecendreux,1,2 Florentia Kaguelidou,3–5 Lila Benadjaoud,4 France Mentré,5–7 Evelyne Jacqz-Aigrain3–5
1Pediatric Sleep Disorders Center, 2Child and Adolescent Psychiatric Department, 3Department of Pediatric Pharmacology and Pharmacogenetics, Hôpital Robert Debré, Assistance Publique – Hôpitaux de Paris (APHP), 4Clinical Investigation Center, Institut National de la Santé et de la Recherche Médicale (INSERM), 5Université Paris Diderot, Sorbonne Paris Cité, Paris, 6INSERM, Infection, Antimicrobiens, Modélisation, Evolution (IAME), UMR1137, Paris, 7Department of Biostatistiques, Hôpital Bichat, APHP, Paris, France
Objective: Mazindol has been proposed as a potential treatment of children with attention deficit/hyperactivity disorder (ADHD). The purpose of this pilot study was to assess its pharmacokinetics, short-term efficacy, and safety.
Subjects and methods: A total of 24 children (aged 9–12 years) with ADHD (according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, text-revision criteria) received a daily dose of 1 mg for 7 days and were followed for 3 additional weeks. Pharmacokinetic samples were collected after the first administration. ADHD symptoms were assessed using the ADHD Rating Scale (RS)-IV, Conners’ Parent Rating Scale – Revised: Long (CPRS-R:L) at screening, baseline, and the end of the study. The Clinical Global Impression – Severity (CGI-S) scale was assessed at baseline, and the CGI – Improvement (CGI-I) scale was assessed at subsequent visits.
Results: Twenty-one subjects (aged 10±1 years) were analyzed. Pharmacokinetic data were described by a one-compartment model with first-order absorption, elimination, and lag time. The typical apparent clearance and apparent volume of distribution were 27.9 L/h and 234 L, and increased with fat-free mass and age, respectively. The mean change in score in ADHD RS-IV after 1 week of mazindol was −24.1 (P<0.0001), greater than a 90% improvement from baseline. Reduction of CPRS-R:L and CGI-S scores were −52.1 (P<0.0001) and −2.5 (P<0.01), respectively. Adverse events were mild to moderate, decreased appetite and upper abdominal pain being the most common.
Conclusion: This preliminary study shows that mazindol might be an effective, well-tolerated, and long-acting (more than 8 hours) agent for the treatment of ADHD in children.
Keywords: pharmacokinetics, efficacy, safety, mazindol, ADHD, pediatrics
Introduction
Attention deficit/hyperactivity disorder (ADHD) is a common neurobehavioral disorder of childhood, affecting 3%–12% of school-age children1–4 and characterized by developmentally inappropriate symptoms of inattention, hyperactivity, and impulsivity.5,6 Additionally, children present with an objectively abnormal daytime sleepiness.7,8
Current medications with the most evidence of efficacy in ADHD are stimulants.9 However, some critical issues make their use controversial. Stimulants have a recognized abuse potential, and are under regulatory control. The length of action is short, and in immediate-release formulations is effective for only 3–4 hours (for methylphenidate) or 4–6 hours (for amphetamine). In controlled-release formulations, the length of action has been extended to 12 hours for both medications. The stimulants have side effects, mostly sleep and appetite disturbances, and potential detrimental effects on growth.10 Deleterious cardiovascular effects, including arrhythmias, hypertension, and cardiomyopathy, were initially reported, primarily in patients with associated risk factors,11–13 but this was not confirmed by long-term studies.14
Here, we explore an alternative treatment strategy by using mazindol, a known catecholaminergic agonist with a greater consistency of drug delivery and a lower potential for abuse. Mazindol is a known imidazoisoindole derivative, not related to amphetamines or metabolized to an amphetamine-like compound, but it acts by blocking the dopamine and norepinephrine reuptake similarly to amphetamine.15–17 Mazindol is metabolized primarily by hepatic conjugation, and eliminated through the kidney.18 In order to investigate the potential new therapeutic use of mazindol in children with ADHD, we designed a pilot Phase II study to assess its pharmacokinetics, short-term efficacy during daytime, and safety of immediate-release mazindol in children treated with 1 mg/day for 7 days.
Subjects and methods
This open-label study (NCT00508677) was conducted from March 2008 to April 2009 at Robert Debré Hospital. Written informed consent was obtained from both parents/guardians of each child. It was reviewed and approved by the Ethical Committee (Comité de Protection des Personnes, Ile-de-France II, France), and was conducted in accordance with guidelines from the Declaration of Helsinki and the International Conference on Harmonisation guidelines for good clinical practice.
Prior to study initiation and during each visit, children and their parents met with the investigators and the clinical staff of the Clinical Investigation Center. The study included 1) a screening visit (visit 1), 2) a 10-day drug washout of all ADHD treatments (methylphenidate, other psychostimulants, and wake-promoting agents), 3) a baseline visit (visit 2), 4) followed by 7 days of treatment with a single daily dose of 1 mg mazindol per day for 7 days (visit 3 at day 7), 5) and 3 weeks’ follow-up (visit 4 between day 7 and day 28) for a total of 40 days.
Subjects
The screening visit (visit 1, within one week before inclusion) included clinical examination and psychiatric evaluation for initial selection of participants. The day of inclusion (visit 2): inclusion criteria were impairing symptoms of ADHD that were severe enough to require treatment change, and ADHD Rating Scale (RS)-IV (parent-reported and investigator-rated) severity score of ≥31 at baseline. In all cases, children were low responders to methylphenidate for at least 6 months before eligibility. Inclusion criteria also included normal physical examination, and normal cardiac function (blood pressure [BP], pulse rate [PR], and electrocardiography [ECG] were to be within the normal range according to the physician/investigator or cardiologist).
Exclusion criteria were significant abnormalities revealed by physical examination, vital signs, or laboratory analysis (hematology, biochemistry including low ferritin levels), and treatment with anticonvulsants, antihistamines, or other medications that might affect the central nervous system (including psychoanaleptics or wake-promoting agents). Children with other psychiatric conditions – mood disorder, bipolar disorder, obsessive–compulsive disorder, pervasive developmental disorders (including Asperger’s syndrome), sleep disorders including narcolepsy, hypersomnia (according to International Classification of Sleep Disorders criteria), tic disorders, chronic diseases (including asthma), current or history of eating disorder (eg, bulimia, anorexia nervosa), mental retardation (intelligence quotient <80), hyperthyroidism or iron deficiency (serum ferritin level <15 μg/L), seizures, glaucoma, familial hypertension, or heart pathologies – were excluded for this study.
Drug administration
The senior research pharmacist prepared blister MEMS® (Medication Event Monitoring System) for each subject, containing seven tablets of 1 mg mazindol. Drug dosage was selected in agreement with the Agence National de Securité des Madicaments et des Produits de Santé.
The first tablet of mazindol was administered in the Clinical Investigation Center on day 1 (visit 2), and the treatment was then taken every day at home for 6 days at 7 am under parental supervision. The time of drug intake was recorded by MEMS.19 All additional medications were recorded by parents in a notebook and reported to investigators within 24 hours. Children were asked to refrain from food intake, beverages containing alcohol or caffeine/xanthine, and smoking during the study.
Efficacy
The primary outcome was the change in mean ADHD RS-IV total score from baseline to treatment end and after treatment with mazindol. The ADHD RS-IV contains 18 items in two subscales: inattention and hyperactivity–impulsivity. Each item is scored between 0 (never or rarely) and 3 (very often). The ADHD RS-IV was completed at baseline and then on a weekly basis for all subsequent study visits.
Secondary efficacy assessments included the Clinical Global Impression (CGI) scale and the Conners’ Parent Rating Scale – Revised: Long, (CPRS-R:L); 80-items.20,21 The CGI was completed on a weekly basis by clinicians with the CGI – Severity (CGI-S) scale at baseline and the CGI – Improvement (CGI-I) scale thereafter at visits 3 and 4. The clinician-rated CGI-S allowed for a global evaluation of a subject’s severity of illness at baseline, using a 7-point scale ranging from 1 (normal) to 7 (severely ill subjects). The CGI-I was used to assess patient improvement, also using a 7-point scale (1, great improvement, to 7, greatest severity). For the purpose of analysis, CGI-S scores were divided into two categories: improved (1, greatest improvement, or 2, fair improvement) versus not improved (all other categories). The CPRS-R:L scale was completed on a weekly basis by the parents.
Tolerability and safety
Medical history was obtained at screening. Physical examination and clinical laboratory testing (8 mL samples for hematological testing, serum chemistry analysis, and urinalysis) were conducted at screening and at each visit. Vital signs at rest (respiratory rate, BP, PR, and oral temperature) were measured at each visit immediately before (baseline) and at 1.5, 4, 8, 12, and 24 hours after drug administration. BP was measured using a standard-cuff sphygmomanometer (manual or automated was acceptable). All ECGs were analyzed by a board-certified pediatric cardiologist. ECG parameters (heart rate, PR, QRS, QT, and QTc intervals using Bazett’s formula [QTc-B]) were measured at screening and at each visit. Adverse events (AEs) were recorded throughout the study in the parents’ notebook and/or spontaneous reporting to the investigators during the planned visits.
Pharmacokinetics
The pharmacokinetic profile of mazindol was determined after optimization of the sampling times using the Fedorov–Wynn algorithm.22 Nine blood samples (1 mL each) were obtained at times of 0.5, 2, 4, 6 and 12 hours (day 1 [visit 2]), after administration of the first tablet, and at times of 12, 24, 36, and 48 hours, after administration of the last tablet (day 7 [visit 3]).Blood samples (2 mL) were drawn in heparin lithium tubes, immediately centrifuged, and kept frozen until analysis.
Mazindol plasma concentrations were measured using high-performance liquid chromatography with mass spectrometry detection. The calibration curve ranged from 0.5 to 10 ng/mL. The interday precision (coefficient of variation) of quality-control samples was less than 7.0% and 6.9%, respectively. The lower limit of quantification was 0.5 ng/mL.
Statistical analyses
Efficacy data
Data are summarized as frequencies and percentages for categorical variables. Quantitative variables are presented as mean values ± standard deviation. Statistical analyses were carried out in children with ADHD RS-IV results at day 0 and day 7. It was planned to include 24 patients in order to obtain at least 20 patients test-worthy for statistical analysis, to show a difference of 11 in ADHD RS-IV mean score after 1 week of mazindol using the nonparametric Wilcoxon paired test with type I error 5% and power 80%.
All comparisons between scores (ADHD RS-IV, CGIs, CPRS-R:L) were performed using nonparametric Wilcoxon paired tests. More specifically, a comparison was performed between baseline (day 0) and end of treatment (day 7), and then between end of treatment and last visit (visit 4). All tests were two-sided at 0.05 significance level. All analyses were performed with SAS version 9.1 (SAS Institute, Cary, NC, USA).
Pharmacokinetic analysis
Pharmacokinetic analysis was carried out using the nonlinear mixed-effect modeling program NONMEM VI (version 2.0; Icon Development Solutions, San Antonio, TX, USA). The Laplacian estimation method with interaction option was used to estimate pharmacokinetic parameters and their variability. The M3 method was used to handle below-the-limit-of-quantification (BLQ) concentrations.23
Several one- or two-compartment open models with first-order elimination were compared, each with various models to describe the absorption phase. Interindividual variability of the pharmacokinetic parameters was estimated using an exponential model. A residual variability (additive, proportional, exponential, or mixed) model was selected according to improvement of objective function value (OFV) and visual inspection of routine diagnostic plots.
The effects of age, hemoglobin, hematocrit, alanine aminotransferase, aspartate aminotransferase, and serum creatinine were investigated as potential variables influencing pharmacokinetic parameters. The effect of size was investigated as a biological covariate. Four measures of size were tested and compared24–26: total body weight (TBW), lean body weight (LBW), free fat mass (FFM), and normal fat mass (NFM). The following equations were used for estimating LBW, FFM, and NFM:
LBWmale = 1.10 × TBW −0.0128 × body mass index (BMI) × TBW, |
LBWfemale = 1.07 × TBW −0.0148 × BMI × TBW, |
where BMI is expressed as BMI = TBW/height (m)2;
FFM = WHSmax × height (m)2 × (TBW/[WHS50 × height {m}2 + TBW]), |
for boys, WHSmax was 42.92 kg·m–2 and WHS50 was 30.93 kg·m–2, |
for girls, WHSmax was 37.99 kg·m–2 and WHS50 was 35.98 kg·m–2, |
NFM = FFM + Ffat × (TBW - FFM), |
where Ffat is a special coefficient for mazindol, which was estimated by the model and WHSmax is the weight height squared maximum.
For covariate analysis, the systematic covariate-analysis process and likelihood-ratio test was used to test the effect of each variable. The selection of variables was determined using a forward and backward selection process. During forward selection, a covariate was selected only if a significant decrease in OFV from the basic model was obtained (reduction >3.84, P<0.05). Then, all the significant variables were included simultaneously in a “full” model. The importance of each variable was then reevaluated by backward selection. Each variable was independently removed from the full model to confirm its relevance. An increase in OFV of more than 6.635 (P<0.01) was required for confirmation. The resulting model was called the “final” population pharmacokinetic model, and included all significant variables.
The stability and performance of the final model were assessed using a nonparametric bootstrap with 1,000 times resampling and replacement. The values of estimated parameters from the bootstrap procedure were compared with those from the original data set. The entire procedure was performed using Perl-speaks-NONMEN (PsN).27 The final model was also assessed by a visual predictive check (VPC) and normalized prediction distribution error (NPDE).28 A total of 1,000 data sets were simulated using the final population model parameters. For VPC, the plot was processed by Xpose (version 4.3.0).29 For NPDE, the plots were processed by the NPDE R package (version 1.2).30
Results
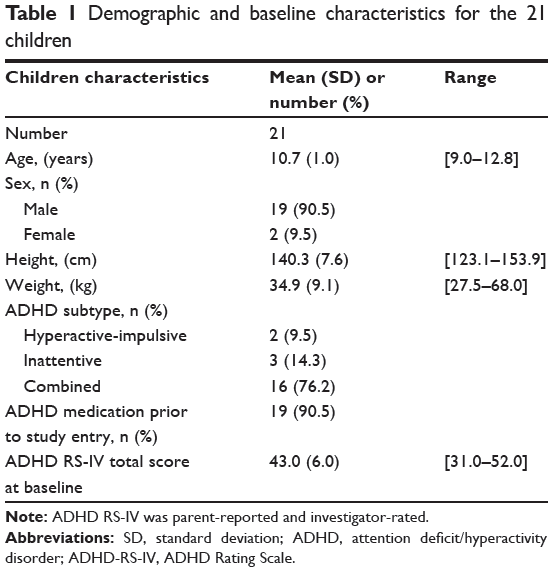
A total of 24 children aged 9.0 to 12.8 years who met Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, text-revision criteria for a primary diagnosis of ADHD combined subtype (predominantly hyperactive–impulsive subtype or inattentive subtype) were included in this study. Among them, three were excluded prior to mazindol first administration, for the following reasons: inclusion-criteria violation (n=1), chronic asthmatiform bronchitis (n=1), and appendicitis (n=1). The demographic and baseline characteristics for the 21 participating children are presented in Table 1. All were low responders to methylphenidate for at least 6 months prior to screening, with an ADHD severity score of ≥28 at baseline. Among them, 19 had a full mazindol pharmacokinetic profile, while two had incomplete mazindol pharmacokinetic profiles.
|
Table 1 Demographic and baseline characteristics for the 21 children |
Evaluation of efficacy
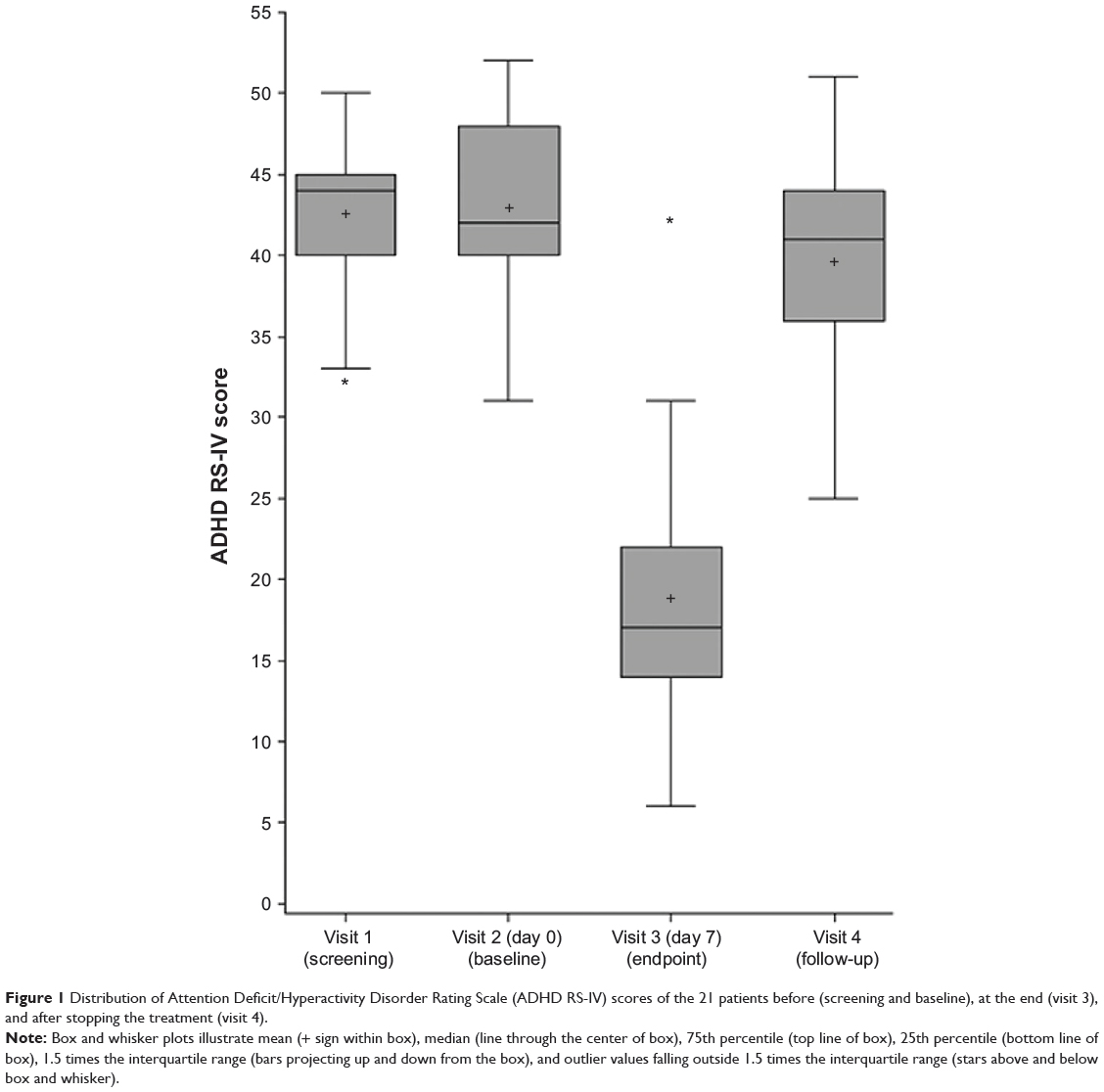
The change in ADHD RS-IV total score from baseline after 1 week of mazindol was −24.1 (95% confidence interval [CI] −28.9 to −18.29, P<0.0001), with >90% improvement from baseline. The mean ADHD RS-IV total score at baseline was 43.0±6.0, 18.9±8.4 after 1 week of mazindol, and 39.7±2.0 after mazindol discontinuation at visit 4 (last week of follow-up) (P<0.0001; Figure 1).
|
Figure 1 Distribution of Attention Deficit/Hyperactivity Disorder Rating Scale (ADHD RS-IV) scores of the 21 patients before (screening and baseline), at the end (visit 3), and after stopping the treatment (visit 4). |
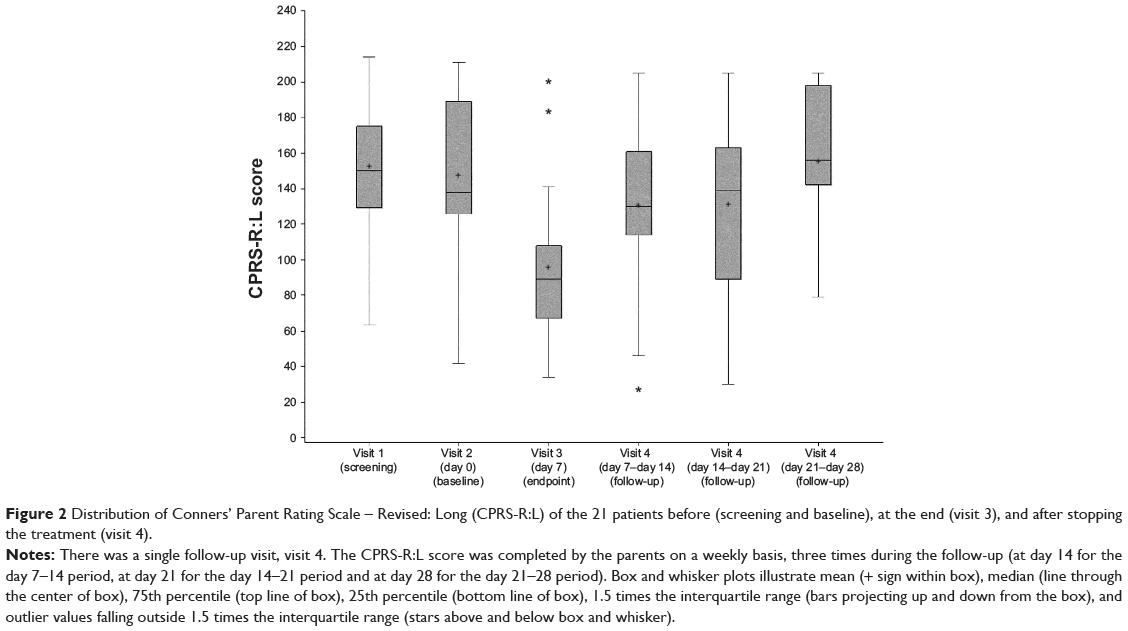
The mean CPRS-R:L total score at baseline (147.9±42.7) significantly decreased (95.8±40.2), with a change after one week of −52.1 (95% CI −70.9 to −34.9, P<0.0001) (Figure 2). The CPRS-R:L score increased to 155.6±45.7 at the last week of follow-up, thus indicating significant alteration in the level of symptomatology of ADHD after mazindol withdrawal (Figure 2).
|
Figure 2 Distribution of Conners’ Parent Rating Scale – Revised: Long (CPRS-R:L) of the 21 patients before (screening and baseline), at the end (visit 3), and after stopping the treatment (visit 4). |
At baseline, the mean CGI-S score in subjects was 5.6±1.2. The mean change in CGI-S score after 1 week of mazindol was −2.5 (95% CI −3.2 to −1.9, P<0.01) (Figure 3). Only 9.5% of children (n=2) had a severity of illness of 5 after this week of treatment, and none of them had a severity of illness greater than 5. Most of them (57.1%, n=12) had a CGI-S score smaller than 4, in accordance with a clinical improvement in ADHD symptoms.

|
Figure 3 Distribution of Clinical Global Impression – Severity (CGI-S) score of the 21 patients before (screening and baseline), at the end (visit 3), and after stopping the treatment (visit 4). |
Based on a CGI-I rating, 18 of 21 (85%) children were “improved” (Table 2). There was strong (or important) improvement in symptoms, based on a CGI-I rating. Eighteen of 21 children (85.7%) were “improved”, while only 2 (9.5%) were “worse” at the last study visit (Table 2). The proportion of children rated as “much” or “very much” improved on the CGI-I after only 1 week of mazindol was 57.1%.

|
Table 2 Clinical Global Impression – Improvement scores after 1 week of mazindol (1 mg/day) in the 21 patients |
Safety and tolerability
Forty-six AEs were reported in 19 patients. AEs were mild (45.7%), moderate (34.8%), or severe (19.6%), but did not require treatment discontinuation. They included decreased appetite (37.0%), drowsiness (17.4%), intestinal distension (8.7%), and upper abdominal pain (6.5%). Decrease of appetite was reported as “severe” in four (22%) patients: three during day 1 and one (5.3%) at day 7. Mean weight at baseline was 35.2±8.9 kg, 34.7±8.7 kg at the end of treatment, and 35.5±9.1 kg at visit 4. No insomnia was reported. Blood pressure (systolic and diastolic), HR and ECG parameters remained unchanged between baseline, day 7, and the end of the study. Changes in laboratory values were not significant.
Pharmacokinetics
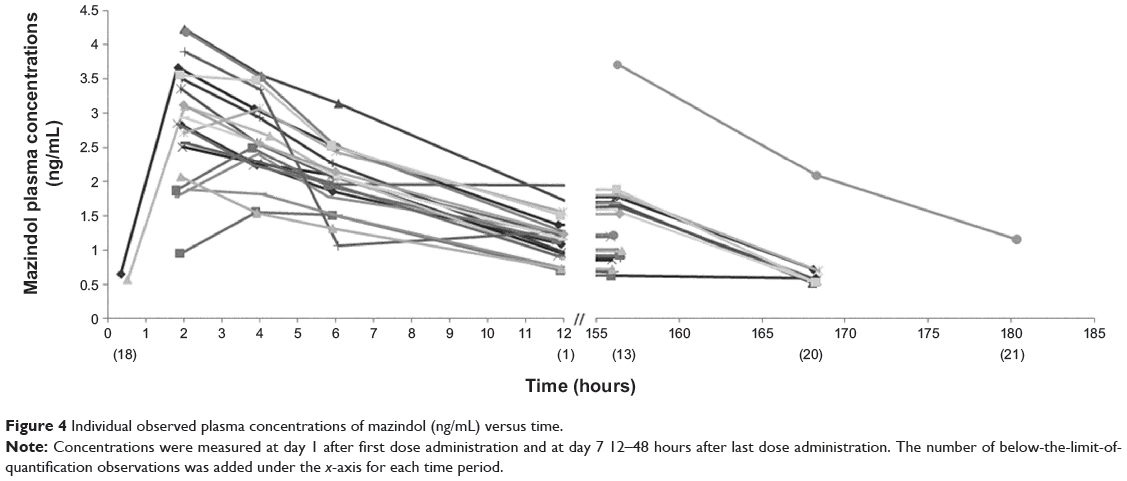
Nineteen children (90%) had a full mazindol pharmacokinetic profile, and two had incomplete pharmacokinetic profiles (for one child, pharmacokinetic analysis was not performed after the first dose, and the sample at 12 hours after the first dose was missing for the other child). Therefore, 183 plasma mazindol concentrations were available for population modeling (73 concentrations BLQ). Mazindol plasma concentrations at a steady state (ie, 24 on day 7) ranged from <0.5 to 2.09 ng/mL. The maximum concentration (Cmax) measured after the first dose was 2.99±0.73 ng/mL (range 1.56–4.23 ng/mL). The MEMS was used to record the entire mazindol dosing history, and was used in population pharmacokinetic analysis. All included children showed good adherence to the protocol, including MEMS records and mazindol doses. The maximum delay of administration time between any of the two doses in the same patient varied between 3 and 101 minutes (average 30 minutes). Individual concentrations are plotted in Figure 4. Data were adequately fitted by a one-compartment model with lag time and first-order absorption and elimination. Inclusion of lag time improved the fitting, and the value of 0.3 hour was fixed. Interindividual variability was estimated for apparent volume of distribution (V/F) and apparent oral clearance (CL/F). Residual variability was best described by a constant variance model.
|
Figure 4 Individual observed plasma concentrations of mazindol (ng/mL) versus time. |
The influence of four different measures of size was compared. The use of FFM for CL/F and V/F showed greater improvement in OFV than TBW or LBW. The use of allometric three-quarter fixed-power scaling and the use of NFM did not improve the model.
The addition of age to both CL/F and V/F significantly decreased the OFV in the forward selection step. FFM and age were highly related (s=0.706, P<0.001, Spearman’s test), and after the backward selection, the influence of FFM but not age was retained for CL/F. However, V/F was better related to age than FFM. No other potential covariates were found to be significant.
Therefore, CL/F and V/F were expressed with the following equations:
CL/Fi = CL/Fref × (FFMi/FFMref)θ1, |
V/Fi = V/Fref × (agei/ageref)θ2. |
where i stands for the ith individual and ref for a reference population.
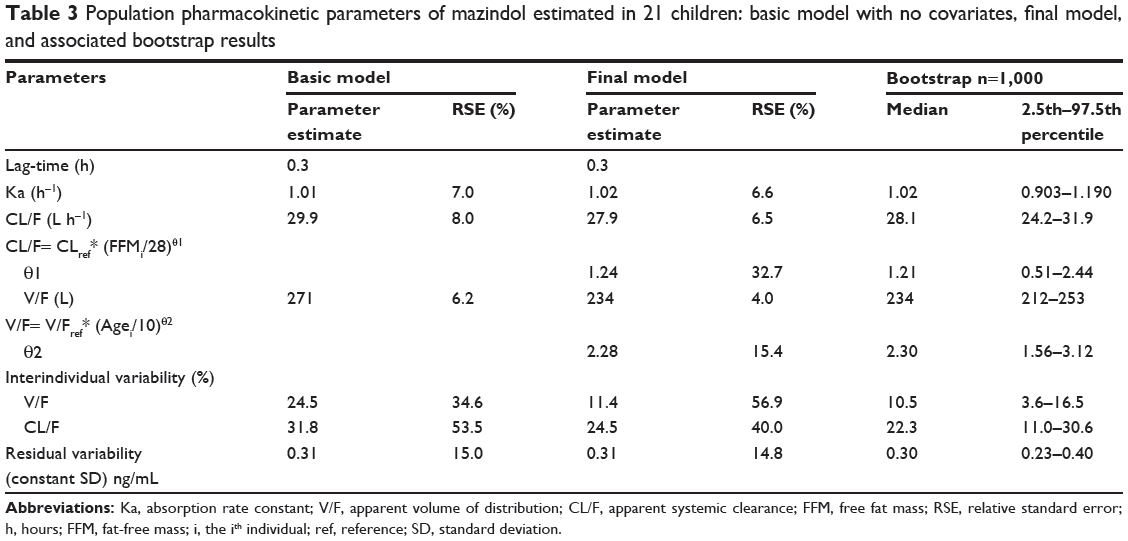
CL/Fref and V/Fref were estimated to be 27.9 L/hour and 234 L, respectively. FFMref and ageref were the median values of the present population, which were 28 kg and 10 years, respectively. The exponents were estimated to be 1.24 for CL/F and 2.28 for V/F. The interindividual variability of both V/F and CL/F decreased from 24.5% to 11.4% and 31.8% to 24.5%, respectively, when including FFM in CL/F and age in V/F.
Routine diagnostic plots, including observed versus individual prediction, observed versus population prediction and weighted residuals versus time, indicate goodness of fit (Figure 5). The ε-shrinkage was 6%. The percentage of BLQ concentrations of mazindol simulated by the model was 35%, which corresponds to an observed value of 40%. This showed good agreement between model prediction using the M3 method and observation for BLQ data.
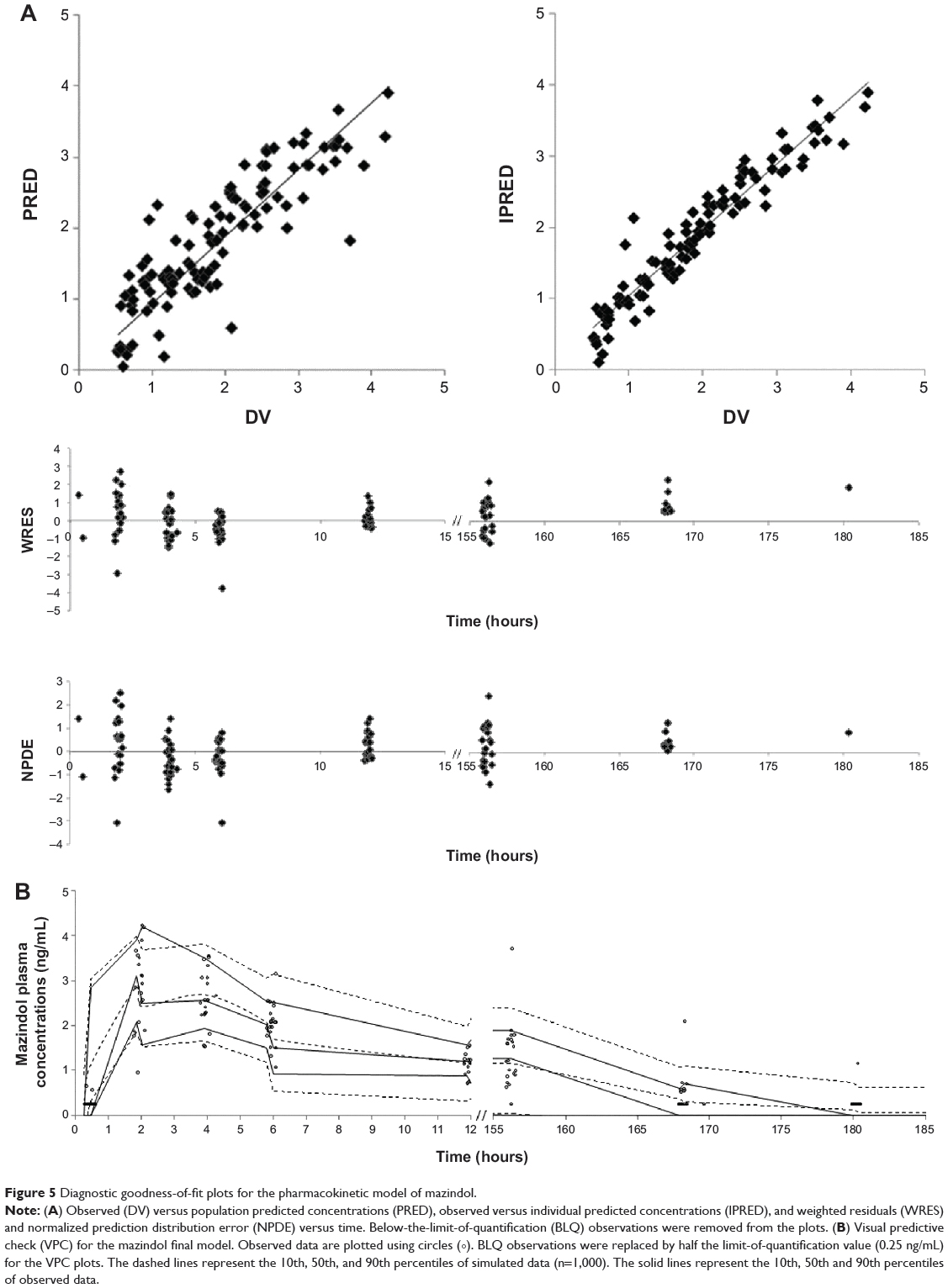
|
Figure 5 Diagnostic goodness-of-fit plots for the pharmacokinetic model of mazindol. |
The mean parameter estimates resulting from the bootstrap procedure agreed closely with their respective values from the final population model, indicating that the estimates for the population pharmacokinetic parameters in the final model were accurate and that the model was stable. The results of 1,000 bootstrap replicates are summarized in Table 3. The VPC is presented in Figure 5. The prediction interval was obtained by simulating 1,000 data sets with the final model. The median concentration was well predicted by the final model. The slight misfit was observed 6 hours post-dose for high (95th percentile) and low (5th percentile) concentrations because of the limited number of samples and high variability. The NPDE is presented in Figure 5. Plots of NPDE versus time and NPDE versus population prediction showed a symmetric distribution around zero, except for the late times of the last dose. This problem was caused by omitting the NPDE of BLQ data. The final population pharmacokinetic parameters are shown in Table 3.
|
Table 3 Population pharmacokinetic parameters of mazindol estimated in 21 children: basic model with no covariates, final model, and associated bootstrap results |
Relationship between exposure, efficacy, and safety
Twenty children were included, as one child was excluded due to missing concentrations at day 1. From population estimates, individual parameters were obtained by specification of the post hoc option in NONMEM. Then, for each patient, area under the curve (AUC)0–∞ and estimated Cmax after the first dose of 1 mg were derived using the model. Individual mean (standard deviation) Cmax and AUC at day 1 were 2.99 (0.73) ng/mL and 32.91 (7.73) hours × ng/mL.
There was no significant correlation between efficacy (ΔADHD RS-IV) and Cmax (P=0.33) or AUC (P=0.94). Only one child had a negative change in ADHD RS-IV score and was classified as nonresponder, but he had relatively high AUC and Cmax values (47.07 hours × ng/mL and 4.23 ng/mL, respectively). Cmax or AUC were not significantly different in children with or without decreased appetite or somnolence.
Discussion
The present pilot study was conducted to evaluate the efficacy of mazindol (1 mg per day for 7 days) in children with ADHD. We observed a significant reduction in ADHD RS-IV and CPRS-R:L total scores, concordant with the clinician-rated CGI-I scale. Pharmacokinetic analysis showed that a one-compartment model with lag time and first-order absorption and elimination was adequate for data modeling. It also showed that FFM and age were significant covariates contributing to mazindol pharmacokinetic variability in children. Although frequent, adverse drug effects did not require drug interruption. No relation between pharmacokinetics and response was evidenced.
Mazindol is an imidazoisoindole derivative unrelated to amphetamine.15,16 It is not metabolized to an amphetamine-like compound, but acts by blocking dopamine and norepinephrine reuptake similarly to amphetamine.17 Mazindol has previously been marketed under the brand names Teronac® (Europe) and Sanorex® (Canada), and used in short-term treatment of exogenous obesity, in combination with a regimen of weight reduction in patients with such risk factors as hypertension, diabetes, or hyperlipidemia.31–34 Mazindol (1–6 mg/day) has also been studied in the treatment of excessive daytime sleepiness (eg, narcolepsy),35–39 and in the present study, the low dose of 1 mg per day for 7 days was selected based on these previous data.
Pharmacokinetic analysis showed that a one-compartment model with lag time and first-order absorption and elimination was adequate for data modeling. The estimated CL/F was comparable to that reported in healthy adults volunteers, as Kim et al reported that an oral dose of 2 mg mazindol resulted in a mean AUC of 72.68 hours·ng·mL–1 in 24 healthy male volunteers, and a calculated mean CL/F of 27.5 L/hour.40 Among the four parameters tested to express size, FFM gave the best results, which might reflect the different impact of the nonfat and fat components of weight on the mazindol hepatic metabolic rate.24 Such expression of size might therefore be significant in children in the presence of wide variations in body-fat composition, as in our population FFM ranged from 22.9 to 47.2 kg/m2. In the final model, FFM was incorporated into CL/F with an exponential relationship, estimated by the model, and not fixed by allometric scaling. Age was also a significant covariate affecting volume of distribution, which increased exponentially with age. This result should be interpreted carefully, because of the narrow age range of our children and the significant correlation between FFM and age. A larger data set over a wilder age range may give a better understanding of the changes in mazindol disposition during childhood. However, this variable should probably be considered in future studies.
After 1 week of mazindol administration, all children were clinically responders to mazindol. A significant reduction in the ADHD composite score was observed both by investigators and parents. In addition, the CPRS-R:L score indicates significant change in the level of symptomatology of ADHD after mazindol withdrawal.
No correlation was evident between pharmacokinetics and response to mazindol. As previously reported, clinical evaluation of the response to psychostimulants is a challenge in patients with ADHD.41,42 On the other hand, systemic exposure may not reflect brain levels. In addition, genetic factors may play an important role in interindividual variability in response to treatment. The impact of polymorphisms at the dopamine-transporter gene on methylphenidate and atomoxetine response has been reported.43,44 However, such pharmacogenetic investigations would require a large number of patients.
Previous long-term studies have shown that neither tolerance nor dependence develop during treatment. In addition, no drug-related adverse effects have been reported in trials on narcoleptic patients, making mazindol a second-line treatment for narcolepsy with a positive effect on deficits of alertness and wakefulness.45 In our study, although AEs occurred in most patients, they were mild or moderate, and none of them required treatment discontinuation. Decreased appetite, already reported with mazindol,39 is consistent with a pharmacological effect of psychostimulants.11 Similarly, cardiac function and ECG measurements remained unchanged.41,46
Our study has the limitations of a pilot study, as it was open-label and the sample size was small. The study was not designed to evaluate long-term efficacy and safety, but previous data have shown that treatment of children with Duchenne muscular dystrophy up to 12 months with 2 mg mazindol daily is safe, effective, and without major AEs.47 However, the positive results are important, as all children were clinically responders to mazindol. The efficacy of mazindol needs to be confirmed in a randomized controlled trial.
Conclusion
The present pilot study of mazindol administered at the daily dose of 1 mg per day for 7 days confirms that mazindol, positioned as an interesting potent catecholaminergic reuptake inhibitor, might have a positive impact on ADHD symptoms in children. The pharmacokinetics of mazindol were evaluated for the first time in children, and pharmacokinetic population parameters can be used for dosage optimization. Our preliminary results warrant further investigations to confirm the significant improvement in ADHD RS-IV total score in children with ADHD.
Acknowledgments
This study was supported by a grant from Assistance Publique – Hôpitaux de Paris (APHP). Genopharm helped to support this study and provided the tablets of mazindol. The authors wish to thank the nurses, technicians, and statisticians involved in this study. The authors also wish to thank Ms Delphine Prieur (assistant) and Dr Florence Tubach (URC Bichat), who contributed to the study design and methodology, and MedEdGlobal Solutions France. Preliminary results of this study were presented at the 56th Annual Meeting of the American Academy of Child and Adolescent Psychiatry, October 27–November 1, Honolulu, HI, and at NeuroTalk, June 25–28, 2010, Singapore.
Author contributions
EK, ML, and FK participated in patient enrollment. CL, FM, WZ, and EJA collected data and performed analysis. LB performed protocol development and outcome. EK and EJA prepared the manuscript, which was critically reviewed by the other authors. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. All authors approved the final version of the manuscript, which is being submitted for publication.
Disclosure
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). EK has served on advisory boards for Shire Pharmaceuticals and Vifor. He has consulted for Shire Pharmaceuticals. He has served as a medical writer for Remidica. He has served on the speaker’s bureau of UCB and served as a principal investigator in clinical trials supported by Eli Lilly and Janssen-Cilag, and has been a coinvestigator in studies sponsored by GlaxoSmithKline, Cephalon, Eli Lilly, and Shire Pharmaceuticals. He serves as a consultant for Shire Pharmaceuticals, BLK Pharma, and Vifor. EK is the inventor of the mazindol combination in the treatment of ADHD (WO/2007/116076). The other authors report no conflicts of interest in this work.
References
Faraone SV, Sergeant J, Gillberg C, Biederman J. The worldwide prevalence of ADHD: is it an American condition? World Psychiatry. 2003;2:104–113. | ||
Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164:942–948. | ||
Visser SN, Lesesne CA, Perou R. National estimates and factors associated with medication treatment for childhood attention deficit/hyperactivity disorder. Pediatrics. 2007;119:S99–S106. | ||
Lecendreux M, Konofal E, Faraone SV. Prevalence of attention deficit hyperactivity disorder and associated features among children in France. J Atten Disord. 2011;15:516–524. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Text revision. Washington: American Psychiatric Association; 2000. | ||
Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366:237–248. | ||
Lecendreux M, Konofal E, Bouvard M, Falissard B, Mouren-Simeoni MC. Sleep and alertness in children with ADHD. J Child Psychol Psychiatry. 2002;41:803–812. | ||
Golan N, Shahar E, Ravid S, Pillar G. Sleep disorders and daytime sleepiness in children with attention-deficit/hyperactive disorder. Sleep. 2004;27:261–266. | ||
Spencer TJ, Biederman J, Wilens TE, Faraone SV. Novel treatments for attention-deficit/hyperactivity disorder in children. J Clin Psychiatry. 2002;63:16–22. | ||
Swanson JM, Kinsbourne M, Nigg J, et al. Etiologic subtypes of attention deficit/and hyperactivity disorder: brain imaging, molecular genetic and environmental factors the dopamine hypothesis. Neuropsychol Rev. 2007;17:39–59. | ||
Findling RL, Biederman J, Wilens TE, et al. Short- and long-term cardiovascular effects of mixed amphetamine salts extended release in children. J Pediatr. 2005;147:348–354. | ||
Charach A, Figueroa M, Chen S, Ickowicz A, Schachar R. Stimulant treatment over 5 years: effects on growth. J Am Acad Child Adolesc Psychiatry. 2006;45:415–421. | ||
Faraone SV, Lecendreux M, Konofal E. Growth dysregulation and ADHD: an epidemiologic study of children in France. J Atten Disord. 2011;15:516–524. | ||
Vitiello B, Emslie G, Clarke G, et al. Long-term outcome of adolescent depression initially resistant to selective serotonin reuptake inhibitor treatment: a follow-up study of the TORDIA sample. J Clin Psychiatry. 2011;72:388–396. | ||
Gogerty JH, Penberthy C, Iorio LC, Trapold JH. Pharmacological analysis of a new anorexic substance: 5-hydroxy-5(4′-chlorophenyl)-2,3-dihydro-5H-imidazo-(2,1-a) isoindole (mazindol). Arch Int Pharmacodyn Ther. 1975;214:285–307. | ||
Gogerty JH, Trapold JH. Chemistry and pharmacology of mazindol. Triangle. 1976;15:25–36. | ||
Hadler AJ. Mazindol, a new non-amphetamine anorexigenic agent. J Clin Pharmacol New Drugs. 1972;12:453–458. | ||
Dugger HA, Madrid VO, Talbot KC, Coombs RA, Orwig BA. Biotransformation of mazindol. III. Comparison of metabolism in rat, dog, and man. Drug Metab Dispos. 1979;7:132–137. | ||
Staples M. Microchips and controlled-release drug reservoirs. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:400–417. | ||
Conners CK, Sitarenios G, Parker JD, Epstein JN. The revised Conners’ Parent Rating Scale (CPRS-R): factor structure, reliability, and criterion validity. J Abnorm Child Psychol. 1998;26:257–268. | ||
Conners CK. Conners’ Rating Scales – Revised: Technical Manual. Toronto: Multi-Health System; 2000. | ||
Retout S, Comets E, Samson A, Mentré F. Design in nonlinear mixed effects models: optimization using the Fedorov–Wynn algorithm and power of the Wald test for binary covariates. Stat Med. 2007;26:5162–5179. | ||
Bergstrand M, Karlsson MO. Handling data below the limit of quantification in mixed effect models. AAPS J. 2009;11:371–380. | ||
Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet. 2009;24:25–36. | ||
Green B, Duffull S. Caution when lean body weight is used as a size descriptor for obese subjects. Clin Pharmacol Ther. 2002;72:743–744. | ||
Duffull SB, Dooley MJ, Green B, Poole SG, Kirkpatrick CM. A standard weight descriptor for dose adjustment in the obese patient. Clin Pharmacokinet. 2004;43:1167–1178. | ||
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN) – a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75:85–94. | ||
Brendel K, Comets E, Laffont C, Laveille C, Mentré F. Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res. 2006;23:2036–2049. | ||
Jonsson EN, Karlsson MO. Xpose – an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58:51–64. | ||
Comets E, Brendel K, Mentré F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–166. | ||
de Felice EA, Chaykin LB, Cohen A. Double-blind clinical evaluation of mazindol, dextroamphetamine, and placebo in treatment of exogenous obesity. Curr Ther Res. 1973;15:358–366. | ||
Aeberli P, Eden P, Gogerty JH, Houlihan WJ, Penberthy C. 5-Aryl-2,3-dihydro-5H-imidazo[2,1-a] isoindols. A novel class of anorectic agents. J Med Chem. 1975;18:177–181. | ||
Inoue S. Clinical studies with mazindol. Obes Res. 1995;Suppl 4:549S–552S. | ||
Bray GA. A concise review on the therapeutics of obesity. Nutrition. 2000;16:953–960. | ||
Parkes, JD, Baraitser M, Marsden CD, Asselman P. Natural history, symptoms and treatment of the narcoleptic syndrome. Acta Neurol Scandinav. 1975;52:337–353. | ||
Parkes JD, Schachter M. Mazindol in the treatment of narcolepsy. Acta Neurol Scand. 1979;60:250–254. | ||
Vespignani H, Weber M, Atlas P, Barroche G. [Value of mazindol in Gélineau’s disease. A propos of 10 cases]. Rev Electroencephalogr Neurophysiol Clin. 1986;16:317–322. French. | ||
Iijima S, Sugita Y, Teshima Y, Hishikawa Y. Therapeutic effects of mazindol on narcolepsy. Sleep. 1986;9:265–268. | ||
Alvarez B, Dahlitz M, Grimshaw J, Parkes JD. Mazindol in long-term treatment of narcolepsy. Lancet. 1991;337:1293–1294. | ||
Kim SS, Lee HW, Lee KT. Validated method for determination of mazindol in human plasma by liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1011–1016. | ||
Wigal SB, Jun A, Wong AA, Stehli A, Steinberg-Epstein R, Lerner MA. Does prior exposure to stimulants in children with ADHD impact cardiovascular parameters from lisdexamfetamine dimesylate? Postgrad Med. 2010;122:27–34. | ||
Pichini S, Papaseit E, Joya X, et al. Pharmacokinetics and therapeutic drug monitoring of psychotropic drugs in pediatrics. Ther Drug Monit. 2009;31:283–318. | ||
Gilbert DL, Wang Z, Sallee FR, et al. Dopamine transporter genotype influences the physiological response to medication in ADHD. Brain. 2006;129:2038–2046. | ||
Joober R, Grizenko N, Sengupta S, et al. Dopamine transporter 3′-UTR VNTR genotype and ADHD: a pharmaco-behavioural genetic study with methylphenidate. Neuropsychopharmacology. 2007;32:1370–1376. | ||
Nittur N, Konofal E, Dauvilliers Y, et al. Mazindol in narcolepsy and idiopathic and symptomatic hypersomnia refractory to stimulants: a long-term chart review. Sleep Med. 2013;14:30–36. | ||
Stiefel G, Besag FM. Cardiovascular effects of methylphenidate, amphetamines and atomoxetine in the treatment of attention-deficit hyperactivity disorder. Drug Saf. 2010;33:821–842. | ||
Desnuelle C, Serratrice G, N’guyen VK, Richelme C, Pinsard N. [A therapeutic trial of mazindol versus placebo in Duchenne muscular dystrophy. A one-year follow-up study of 14 children]. Arch Fr Pediatr. 1989;46:759–765. French. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.