")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
PIF1 Promotes Autophagy to Inhibit Chronic Hypoxia Induced Apoptosis of Pulmonary Artery Endothelial Cells
Authors Zhao Y, Wu J, Guan S, Xue T, Wei X, Cao D, Kong P, Zhang X
Received 13 February 2023
Accepted for publication 14 June 2023
Published 26 June 2023 Volume 2023:18 Pages 1319—1332
DOI https://doi.org/10.2147/COPD.S406453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Yujing Zhao,1– 4 Juan Wu,2– 4 Shuai Guan,5 Ting Xue,2– 4 Xiaolei Wei,1– 4 Dawei Cao,2– 4 Pengzhou Kong,6 Xinri Zhang2– 4
1Department of the First Clinical Medicine, Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China; 2Department of NHC Key Laboratory of Pneumoconiosis, The First Hospital of Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China; 3Department of Shanxi Key Laboratory of Respiratory Diseases, The First Hospital of Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China; 4Department of Pulmonary and Critical Care Medicine, The First Hospital of Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China; 5Department of Pediatric Infectious Diseases, The First People’s Hospital of Datong, Datong, Shanxi, People’s Republic of China; 6Department of Translational Medicine Research Center, Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China
Correspondence: Pengzhou Kong, Department of Translational Medicine Research Center, Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China, Email [email protected] Xinri Zhang, Department of Pulmonary and Critical Care Medicine, NHC Key Laboratory of Pneumoconiosis; Shanxi Key Laboratory of Respiratory Diseases, The First Hospital of Shanxi Medical University, Taiyuan, Shanxi, People’s Republic of China, Email [email protected]
Purpose: Pulmonary artery hypertension (PAH) is a common complication of chronic obstructive pulmonary disease and obstructive sleep apnea/hypopnea syndrome worldwide. Pulmonary vascular alterations associated with PAH have multifactorial causes, in which endothelial cells play an important role. Autophagy is closely related to endothelial cell injury and the development of PAH. PIF1 is a multifunctional helicase crucial for cell survival. The present study investigated the effect of PIF1 on autophagy and apoptosis in human pulmonary artery endothelial cells (HPAECs) under chronic hypoxia stress.
Methods: Chronic hypoxia Gene expression profiling chip-assays identified the PIF1 gene as differentially expressed, which was verified by RT-qPCR analysis. Electron microscopy, immunofluorescence, and Western blotting were used to analyze autophagy and the expression of LC3 and P62. Apoptosis was analyzed using flow cytometry.
Results: Our study found that chronic hypoxia induces autophagy in HPAECs, and apoptosis was exacerbated by inhibiting autophagy. Levels of the DNA helicase PIF1 were increased in HPAECs after chronic hypoxia. PIF1 knockdown inhibited autophagy and promoted the apoptosis of HPAECs under chronic hypoxia stress.
Conclusion: Based on these findings, we conclude that PIF1 inhibits the apoptosis of HPAECs by accelerating the autophagy pathway. Therefore, PIF1 plays a crucial role in HPAEC dysfunction in chronic hypoxia-induced PAH and may be a potential target for the treatment of PAH.
Keywords: PIF1, autophagy, HPAEC, PAH, chronic hypoxia
Introduction
Pulmonary artery hypertension (PAH) is a common complication of chronic obstructive pulmonary disease (COPD) and obstructive sleep apnea/hypopnea syndrome (OSAHS) worldwide. PAH predominantly affects patients with severe COPD and predicts a poor prognosis. PAH occurs due to occlusion, blockage, and remodeling of the pulmonary vasculature. Long-term home oxygen therapy is the most effective treatment to slow or reverse PAH progression in COPD patients.1 OSAHS is characterized by intermittent hypoxia during sleep, and it predisposes to PAH and right heart dysfunction.2,3 Approximately 66% of patients with moderate-to-severe COPD have concomitant OSAHS, known as overlap syndrome.4 The prognosis of overlap syndrome patients is worse than that of patients with OSAHS or COPD alone, predominantly due to more-frequent complications of PAH. PAH is associated with an increased hospitalization rate, high risk of acute exacerbation and mortality, and diminished quality of life. Most vasodilators are not useful for the treatment of PAH because of their potentially harmful effects on gas exchange. Furthermore, inhibiting pulmonary vasoconstriction is ineffective for long‑term treatment.5 Understanding the pathophysiological mechanism underlying hypoxia-induced PAH may aid in the development of novel drugs and therapeutic strategies for PAH.
Pulmonary vascular alterations in PAH have multifactorial causes, in which endothelial cells play a vital role.6,7 Studies have reported apoptosis of pulmonary artery endothelial cells and vascular dysfunction in the early stages of PAH.8 Subsequently, the loss of endothelial integrity causes uncontrolled growth of the vascular adventitia and smooth muscle media, leading to occlusion of pulmonary small vessels.7 Endothelial dysfunction, manifesting as enhanced susceptibility to apoptogenic stimuli, is a critical initiating factor in PAH.9
Autophagy is a catabolic process that eliminates proteins and damaged organelles via lysosomal degradation. Several recent studies have suggested that dysregulation of autophagy is involved in the development of PAH.10,11 Autophagy also regulates the apoptosis, proliferation, and migration of vascular endothelial cells and tube formation,7,12 but the role of autophagy in PAH remains unclear.
PIF1 is a multifunctional helicase and DNA processing enzyme, it encodes a DNA-dependent adenosine triphosphate metabolic enzyme. PIF1 also plays a role in maintaining genome stability, and several studies have reported that PIF1 plays roles in regulating autophagy and cell survival as well.13–16 However, whether PIF1 plays a crucial role in PAH remains unclear. The potential role of PIF1 regulating HPAEC autophagy and survival also remains unclear. OSAHS and COPD are characterized by chronic hypoxia that ultimately leads to PAH. In the present study, we investigated the role of PIF1 in chronic hypoxia-induced PAH by evaluating autophagy and apoptosis in HPAECs subjected to chronic hypoxia stress.
Materials and Methods
Cell Lines and Culture
HPAECs (BLUEBIO Co. Shanghai, China) were cultured at 37°C with 5% CO2 in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (BI) and 100 U/mL of penicillin and streptomycin (Gibco). The cells were assigned to normal and chronic hypoxia (CH) groups. Cells between passages 3 and 9 were used for the experiments.
Establishment of Chronic Hypoxia Model
Cells in the CH group were cultured in a hypoxic incubator for 24 h at 37°C under continuous hypoxic conditions with 5% CO2 and 10% O2. The 1% hypoxic condition was repeated every 30 min. The autophagy inhibitor 3-MA (10 mM, HY-19312, MCE) and chloroquine (40 µM, HY-17589A, MCE) were used to study the relationship between autophagy and apoptosis. Rapamycin (100 nM, HY-10219, MCE) was used to activate autophagy. The experiments were independently repeated three times, with three well replicates for each experiment.
shRNA-Mediated Silencing of Gene Expression
Human shRNA-PIF-1 lentivirus (PIF-1 shRNA, 5ʹ-GCCACAGCTTTCTGAGGAA-3ʹ) and negative control lentivirus (NC shRNA) were obtained from Shanghai GENEPHARMA Co., Ltd. (Shanghai, China). The lentiviruses were produced using HEK-293T cells according to the manufacturer’s instructions. HPAECs were incubated with lentiviral particles, and at 72 h after infection, the cells were screened for stable expression of lentiviral constructs using puromycin (25 mg, P8230; Beijing Solarbio Science and Technology Co., Ltd., Beijing, China).
Flow Cytometry
Apoptosis of HPAECs subjected to chronic hypoxia was assessed using a Annexin V-FITC/PI Cell Apoptosis kit (G1511, Servicebio, Wuhan, China). The effect of PIF-1 on the apoptosis of HPAECs was evaluated using an Annexin V-PE/7-AAD Cell Apoptosis kit (G1512, Servicebio, Wuhan, China). Fluorescence signals were measured using an fc500/NAVIOS flow cytometer (Beckman Coulter Inc., Brea, CA, USA) and the data were analyzed using Kaluza software (Beckman Coulter Inc.).
MTT Assay
Cell proliferation was analyzed using an MTT Cell Proliferation Assay kit (Sigma-Aldrich, St. Louis, MO, USA). HPAECs were plated in 96-well plates at a density of 6×103 cells per well and cultured overnight. The cells were then incubated in a humidified incubator at 37°C and exposed to no or chronic hypoxia treatment. After 24 h, 10μL of MTT was added to each well, and the plates were incubated at 37°C for an additional 4 h. Next, 100μL of dimethyl sulfoxide was added to each well, and the plates were placed on a shaker for 10 min. Finally, the absorbance of each well was measured at 490 nm. The experiments were repeated three times.
Wound Healing Assay
Monolayers of HPAECs in 6-well plates were scratched using a sterile pipette tip, washed with phosphate-buffered saline (PBS) and then subjected to no or chronic hypoxia treatment. Images of the areas were acquired at the initiation of the experiment and again after 24 h of chronic hypoxia. The migration area (%) was calculated as follows: (A0 – A24)/A0 × 100, where A0 and A24 represent the area of the wound at 0 and 24 h, respectively. The experiments were performed three times.
RT-qPCR
Total RNA was extracted from HPAECs using TRIzol reagent (Sigma-Aldrich, Shanghai, China). The RNA was reverse-transcribed into cDNA using Hiscript QRT Supermix (Vazyme, Nanjing, China). The expression of candidate genes was examined using RT-qPCR with AceQ qPCR SYBR Green Master Mix (Vazyme, Nanjing, China) and an Applied Biosystems StepOnePlus Real-Time PCR System (ThermoFisher Scientific, Waltham, MA, USA). The expression levels of target genes were normalized to that of GAPDH and expressed as ΔCt. Data were analyzed using the 2−ΔΔCt method. The sequences of human mRNA primers (Sangon Biotech, Shanghai, China) were as follows: GAPDH (forward TGACTTCAACAGCGACACCCA; reverse CACCCTGTTGCTGTAGCCAAA) and PIF1 (forward GGCAGGTGTTCAGATGAGGT; reverse AGGAGCTGGCTAACAGGACA). Finally, RT-qPCR was used to analyze the expression of PIF-1 in HPAECs.
Western Blotting
HPAECs were dissolved in RIPA lysis buffer (Cell Signaling Technology, Danvers, MA, USA). Protease inhibitors were added, and the lysate was centrifuged at 12,000 rpm for 15 min at 4°C. The protein concentration was determined using the bicinchoninic acid protein assay. Proteins were separated and an adequate concentration was added for SDS-PAGE, followed by electrotransfer onto polyvinylidene difluoride membranes. The membranes were blocked using 5% bovine serum albumin with 0.1% Tween 80, incubated with primary antibodies overnight at 4°C, and then washed three times with TBST. The membranes were probed with antibodies against LC3 (ab192890; 1:2000; Abcam) as an autophagy marker protein. An increase in the LC3II/LC3I ratio is indicative of autophagy activation. The membranes were also probed with antibodies against P62 (ab109012; 1:10,000; Abcam), PIF1 (sc-48,377; 1:100; SANTA CRUZ), and β-actin (ab8226; 1:1000; Abcam). Finally, the membranes were incubated with anti-rabbit (ab216773; 1:10,000; Abcam) or anti-mouse (ab216772; 1:10,000; Abcam) secondary antibodies for 1 h. Protein bands of interest were detected using an Amersham Imager 600 (Cytiva, Marlborough, MA, USA) and quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence
Slides of human HPAECs were fixed with 4% polyformaldehyde at room temperature for 30 min, washed three times with PBS, treated with 3% H2O2 for 5 min to block endogenous peroxidases, and washed three times with PBS. The cells were then permeabilized by treatment with 0.5% Triton X-100 for 30 min, washed three times with PBS, and treated with 5% goat serum for 15 min. The slides were then incubated with anti-LC3 (ab192890; 1:100; Abcam) antibody overnight at 4°C and washed three times with PBS. Finally, the slides were incubated with fluorescein-conjugated secondary antibodies (Ab6717, 1:500; Abcam) for 60 min at room temperature. A laser confocal microscope (Leica, Bensheim, Germany) was used to observe and photograph the slides.
Transmission Electron Microscopy
Cells were prefixed with 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4) and postfixed with 1% osmium tetroxide buffer. After dehydration in a gradient series of ethanol, the cells were permeabilized and embedded with acetone and 812 Embedding Medium (SPI Supplies, West Chester, PA, USA). Ultrathin sections (60 to 80 nm thick) were stained with uranyl acetate and lead citrate, and examined under a transmission electron microscope (HT7700; Hitachi, Tokyo, Japan).
JC-1 Staining
HPAECs were collected, precipitated, washed with PBS, added to cell culture medium, and stained with JC-1 staining working solution (C2006; Beyotime Biotechnology Inc., Shanghai, China). The cells were incubated at 37°C for 20 min, and the supernatant was removed by aspiration. The pellet was washed two times with JC-1 staining buffer (1×), mixed with 2 mL of cell culture medium, and analyzed by flow cytometry.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 8.0 software (GraphPad Software Inc., San Diego, CA, USA). All data passed normality and equal variance tests using the Shapiro–Wilk test and F-test, respectively. Results are expressed as the mean ± standard error of the mean (SEM). The Student’s t-test was used to compare differences between two groups, whereas one-way or two-way analysis of variance followed by Tukey’s test and sidak’s multiple comparison test was performed to compare differences between multiple groups. Differences were considered statistically significant at P<0.05.
Results
Chronic Hypoxia Increased the Apoptosis and Migration of HPAECs
HPAEC dysfunction is an important factor in PAH. To elucidate the effect of chronic hypoxia on HPAECs, flow cytometry was performed to characterize the effect of chronic hypoxia on the apoptosis of HPAECs. Compared to the control group, the apoptosis ratio of HPAECs was significantly increased after chronic hypoxia treatment for 24 h (Figure 1A and D). The mitochondrial membrane potential changes in the early stage of apoptosis. JC-1 assay results showed that the mitochondrial membrane potential decreased after chronic hypoxia treatment for 24 h, as compared to the control group (Figure 1B and E). Therefore, chronic hypoxia induced the apoptosis of HPAECs.
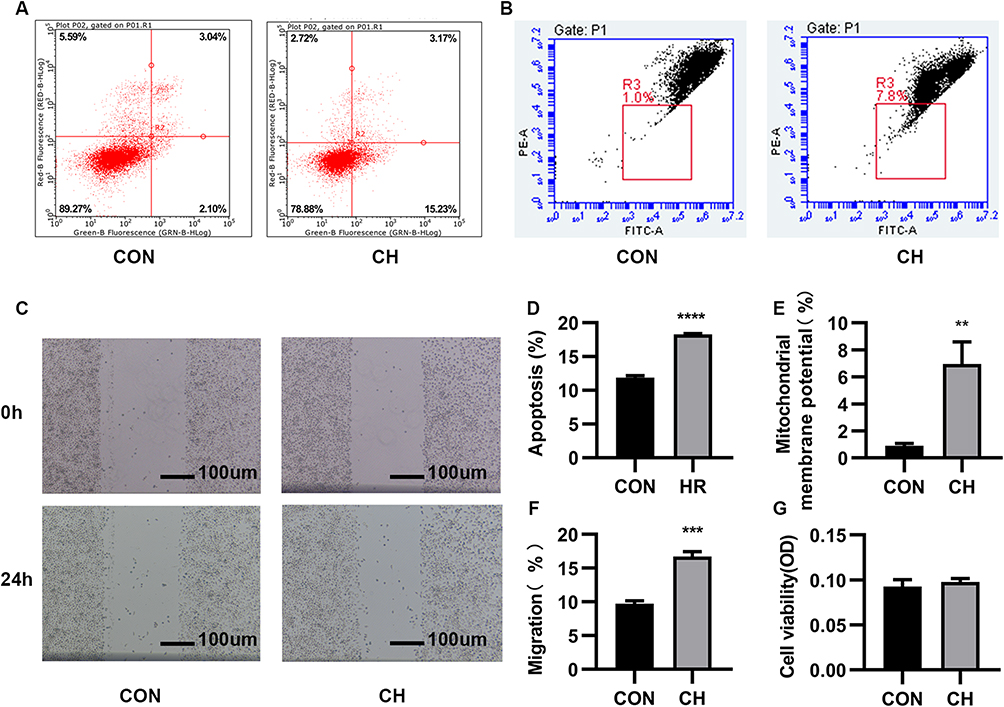 |
Figure 1 Chronic hypoxia (CH) promotes the apoptosis and migration of human pulmonary artery endothelial cells (HPAECs). (A). The apoptosis of CH-induced HPAECs was measured using flow cytometry. (B). JC-1 was used to determine the mitochondrial membrane potential. (C). Scratch-wound assay to assess cell migration. (D). Quantification (percentage) of apoptotic cells (****P<0.0001). (E). Quantification of mitochondrial membrane potential (**P<0.01). (F). Quantification of cell migration (***P<0.001). (G). MTT assay to assess the proliferative capacity of HPAECs. Abbreviation: CON, Control. |
The scratch-wound assay was used to examine the migration of cells. Chronic hypoxia increased the migration of HPAECs (Figure 1C and F). The MTT assay was used to evaluate the viability of HPAECs. No significant difference in optical density values was observed between the chronic hypoxia treatment and control groups (Figure 1G). Chronic hypoxia altered the function of HPAECs. Our results demonstrate that chronic hypoxia causes endothelial dysfunction and increases the migration and apoptosis of HPAECs, which becomes the basis of PAH.
Autophagy is Activated in HPAECs Subjected to Chronic Hypoxia
Autophagy can be activated by hypoxia and plays an important role in the occurrence of PAH. Immunofluorescence images showed that the number of LC3 cells was increased after chronic hypoxia treatment compared to the control group (Figure 2A). Western blotting and semi-quantitative analyses were performed to evaluate the expression of autophagy-related proteins. The expression of LC3-II/LC3-I was increased, and that of P62 decreased under chronic hypoxia conditions (Figure 2B–D). Transmission electron microscopy showed that autophagosome formation was increased after chronic hypoxia treatment for 24 h compared to the control group (Figure 2E). These results demonstrate that chronic hypoxia activates autophagy, which plays an important role in the development of PAH.
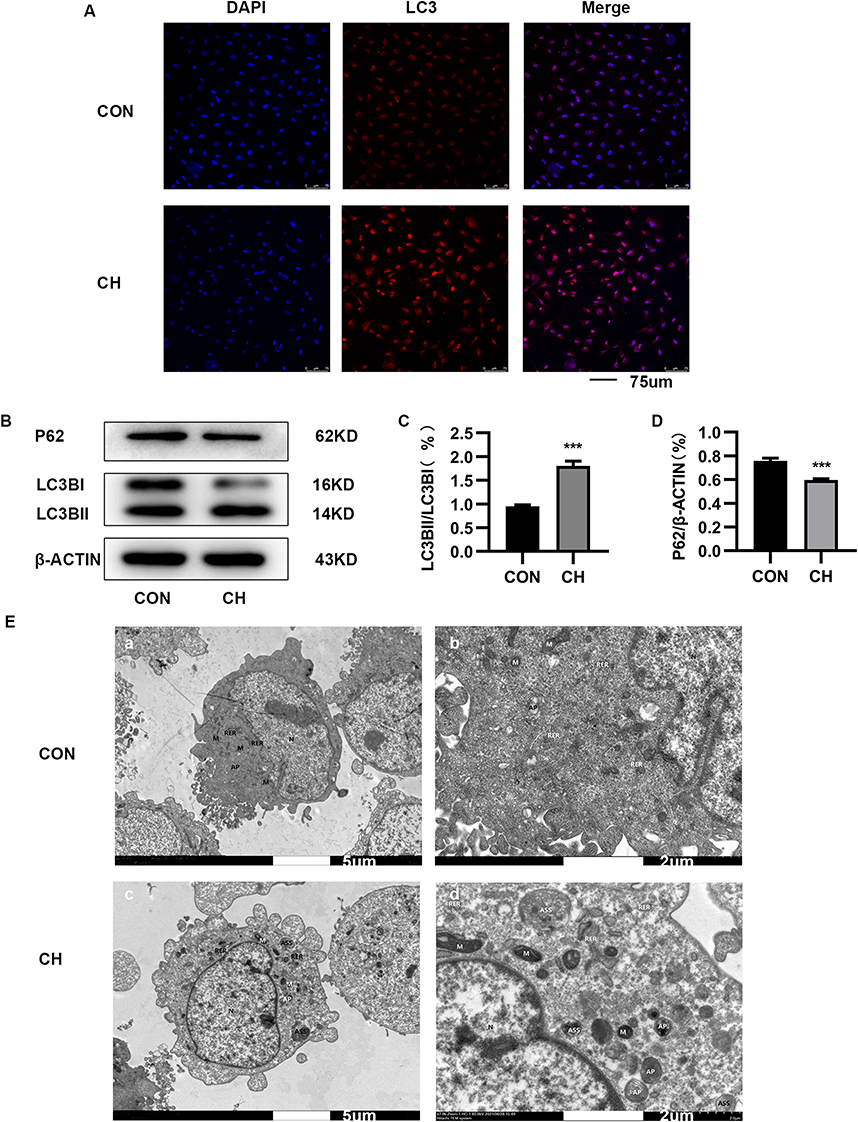 |
Figure 2 Autophagy is activated in HPAECs subjected to CH. (A). Immunofluorescence staining was used to examine the relative fluorescence intensity and levels of LC3 in HPAECs. (B). Expression of LC3 and P62 proteins in HPAECs as determined by Western blotting. (C). Semi-quantification of LC3-II/LC3-I protein expression (***P<0.001). (D). Semi-quantification of P62 protein expression (***P<0.001). E. Electron micrographs showing the formation of autophagic vacuoles in HPAECs. a. Electron microscopy of HPAECs in control group (×2.0k). b. Electron microscopy of HPAECs in control group (×7.0k). c. Electron microscopy of chronic hypoxia-induced HPAECs (×2.0k). d. Electron microscopy of chronic hypoxia-induced HPAECs (×7.0k). Abbreviations: AP, autophagosome; ASS, autolysosome; RER, rough endoplasmic reticulum; M, mitochondria; N, nucleus. |
Autophagy Inhibitors Promoted Apoptosis Under Chronic Hypoxia Conditions
Cells in the control and chronic hypoxia groups were treated with the autophagy inhibitors 3-MA and chloroquine to study the relationship between autophagy and apoptosis. Flow cytometry was performed to detect apoptotic HPAECs. The apoptosis ratio of HPAECs was increased in the chronic hypoxia + 3-MA group compared to the chronic hypoxia group without 3-MA treatment (Figure 3A and B). Western blotting showed that the expression of LC3-II/LC3-I proteins was downregulated, whereas that of P62 was upregulated after 3-MA treatment (Figure 3C–E). Apoptosis of HPAECs was further increased after treatment with the autophagy inhibitor 3-MA under chronic hypoxia conditions. However, apoptosis increased after autophagy was inhibited by treatment with chloroquine (Figure S1A–D). These results thus demonstrate that the activation of autophagy suppresses the apoptosis of HPAECs under chronic hypoxia conditions.
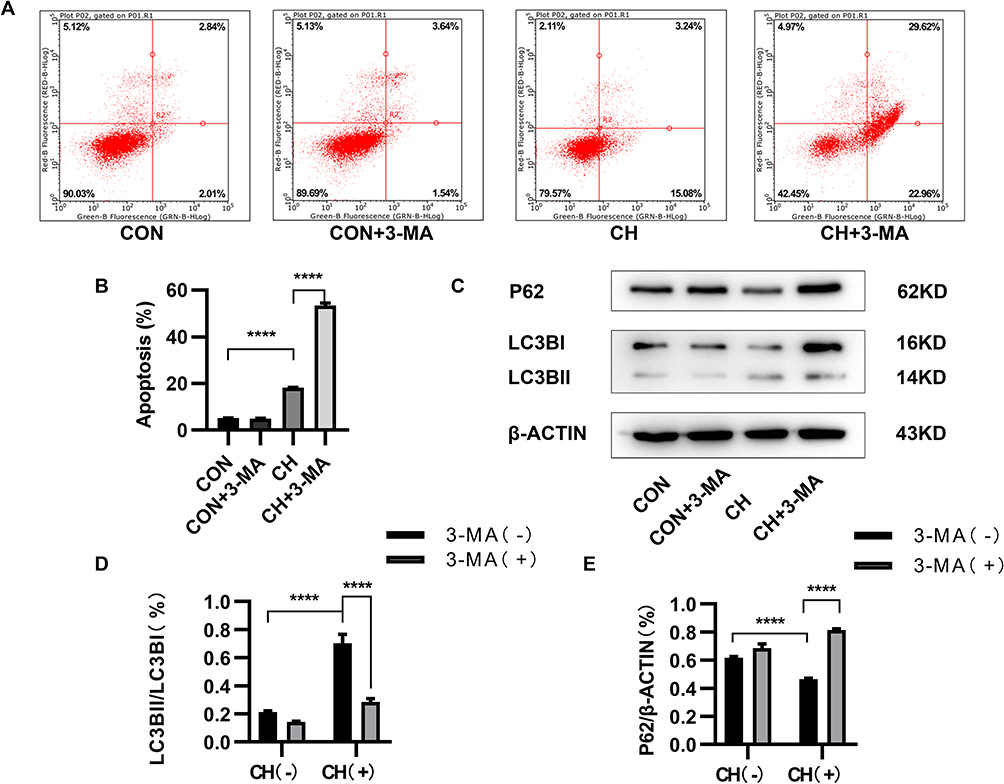 |
Figure 3 3-Methyladenine (3-MA) inhibits autophagy and promotes apoptosis. (A and B) Apoptosis of HPAECs subjected to CH after inhibition of autophagy using 3-MA, as measured using flow cytometry. (C) Expression of LC3 and P62 proteins in HPAECs, as determined by Western blotting. (D and E) Quantification of the expression of LC3B and P62 in HPAECs (****P<0.0001). |
Identification of Differentially Expressed Genes (DEGs) in HPAECs Under Chronic Hypoxia Conditions
We performed gene expression profiling for the chronic hypoxia and control groups. We screened important target genes associated with chronic hypoxia by identifying DEGs between the chronic hypoxia and control groups. In total, 3743 DEGs were identified between the chronic hypoxia and control groups. Among these genes, the expression of 1767 was upregulated (fold change ≥1.3, false discovery rate [FDR] < 0.05), and that of 1976 was downregulated (fold change ≤1.3, FDR < 0.05). A volcano plot and heatmap of gene expression are presented in Figure 4A and B. We performed a cluster analysis of the top 15 upregulated and top 5 downregulated genes. The 20 hub genes are shown in Figure 4C. These results indicate a clear change in the expression of PIF1.
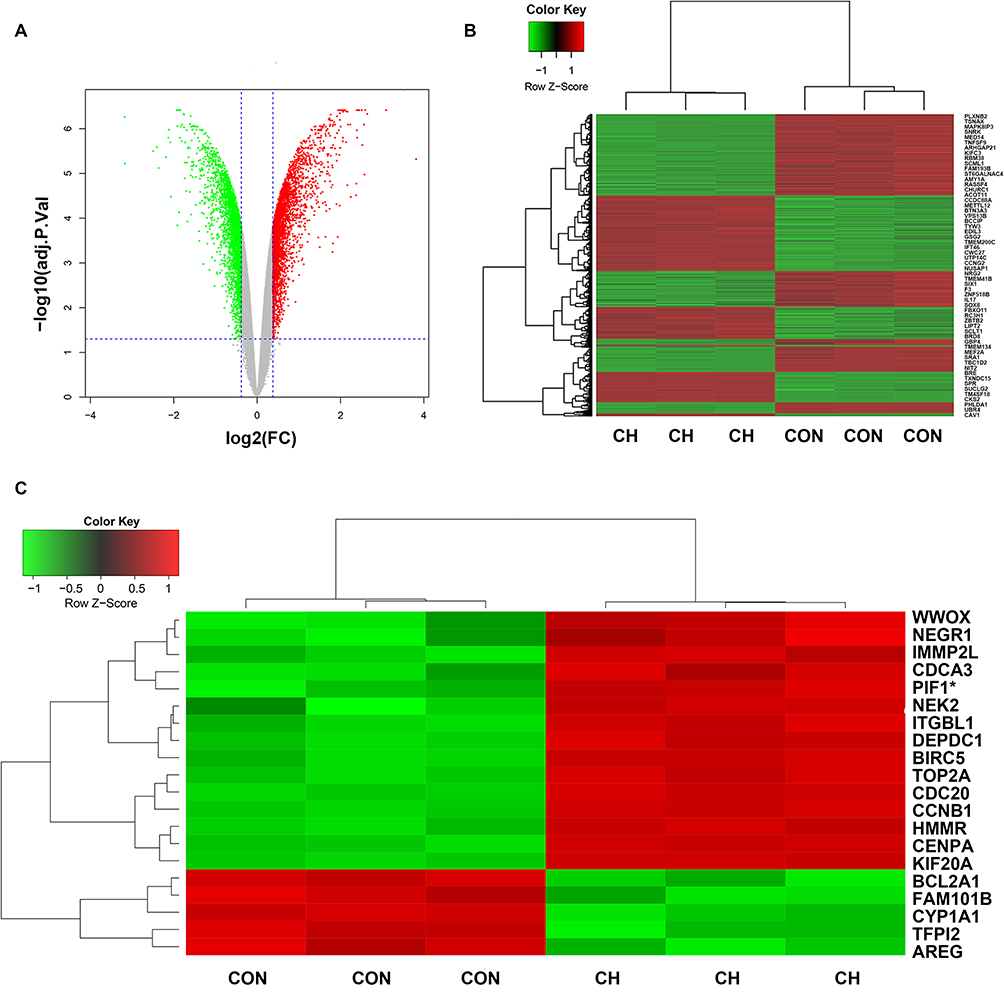 |
Figure 4 Identification of differentially expressed genes (DEGs) in the CH and CON groups using gene expression profiling chips. (A) Volcano plot of CH and CON groups. (B) Heatmap of DEG expression. (C) Heatmap of expression of 20 hub genes. PIF1* (the gene we studied). |
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analyses of DEGs
To investigate the functions of the 3743 DEGs, we used Cluster Profiler software (p. adjust <0.05, q. values <0.05) to perform GO and KEGG analyses. The 10 most significant molecular functions, biological processes, and cellular components obtained from GO enrichment analysis were used to construct a histogram (Figure 5A). The histogram showed that the DEGs were mainly involved in the biological processes of chromosome segregation, organelle fission, nuclear division, cell cycle G2/M phase transition, regulation of chromosome organization, and mitotic nuclear division. The DEGs were mainly located in the cell components of chromosomal region, spindle, cell-substrate junction, focal adhesion, chromosome, and centromeric region. The molecular functions of the DEGs mainly included ATPase activity, cell adhesion molecule binding, ubiquitin-like protein ligase binding, ubiquitin-like protein transferase activity, and nucleoside-triphosphatase regulator activity.
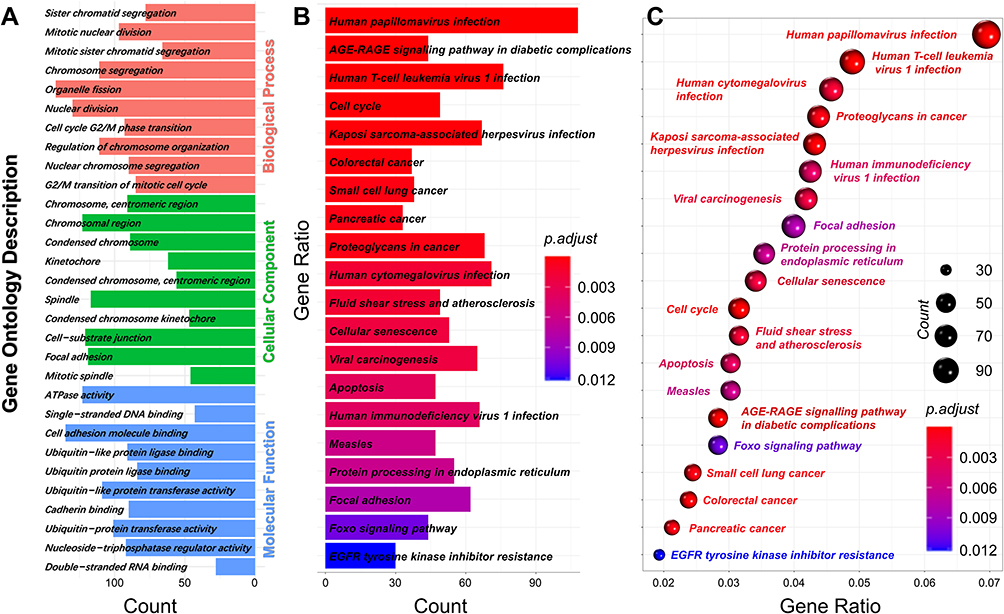 |
Figure 5 Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of DEGs. (A) GO term enrichment analysis for biological process, molecular function, and cellular component. (B and C) KEGG pathway analysis. Node size represents the gene ratio; node color indicates P-value. |
KEGG analysis was performed to investigate the pathways of the 3743 DEGs. The top 10 KEGG terms of the DEGs are shown in Figure 5B and C. The DEGs were primarily enriched in human papillomavirus infection, human T-cell leukemia virus 1 infection, cell cycle, and apoptosis. In the previous experiment, we confirmed that chronic hypoxia induces apoptosis, cell migration, and autophagy. Among the top 20 genes, PIF1 was the most significantly elevated, and this gene regulates the apoptosis and migration of tumor cells.14,16 Moreover, PIF1 overexpression in neurons was shown to improve autophagy.15 Based on these results, we investigated the relationship between PIF1 and autophagy, as well as the effects of PIF1 on HPAEC apoptosis, migration, and proliferation.
Increased PIF-1 Expression Promotes Autophagy of HPAECs Under Chronic Hypoxia Conditions
Western blotting and RT-qPCR were performed to evaluate PIF1 expression and determine the effects of PIF1 knockdown (Figure 6A–F). PIF1 expression was markedly increased in the chronic hypoxia group compared to the control group. The effect on autophagy was investigated by inhibiting PIF1 expression. Compared to the control group under chronic hypoxia conditions and the chronic hypoxia group, LC3-II/LC3I protein expression was downregulated and P62 protein expression upregulated in the CH-shPIF1 group (Figure 6G, H and J–L). To confirm the reduction in autophagic flux after PIF1 knockdown, we evaluated changes in autophagic flux after manipulating the process using specific modulators. Leupeptin suppresses lysosomal degradation, leading to the accumulation of autolysosomes.17 LC3-II accumulated after leupeptin treatment in PIF1-knockdown HPAECs (Figure 6I and M). Collectively, these results indicated that PIF1 promotes autophagic flux. Transmission electron microscopy showed decreased autophagosome formation in the CH-shPIF1 group compared to the control group under chronic hypoxia conditions (Figure 6N), indicating that autophagy was inhibited by PIF1 knockdown. Thus, PIF1 may be involved in regulating autophagy in HPAECs under chronic hypoxia conditions.
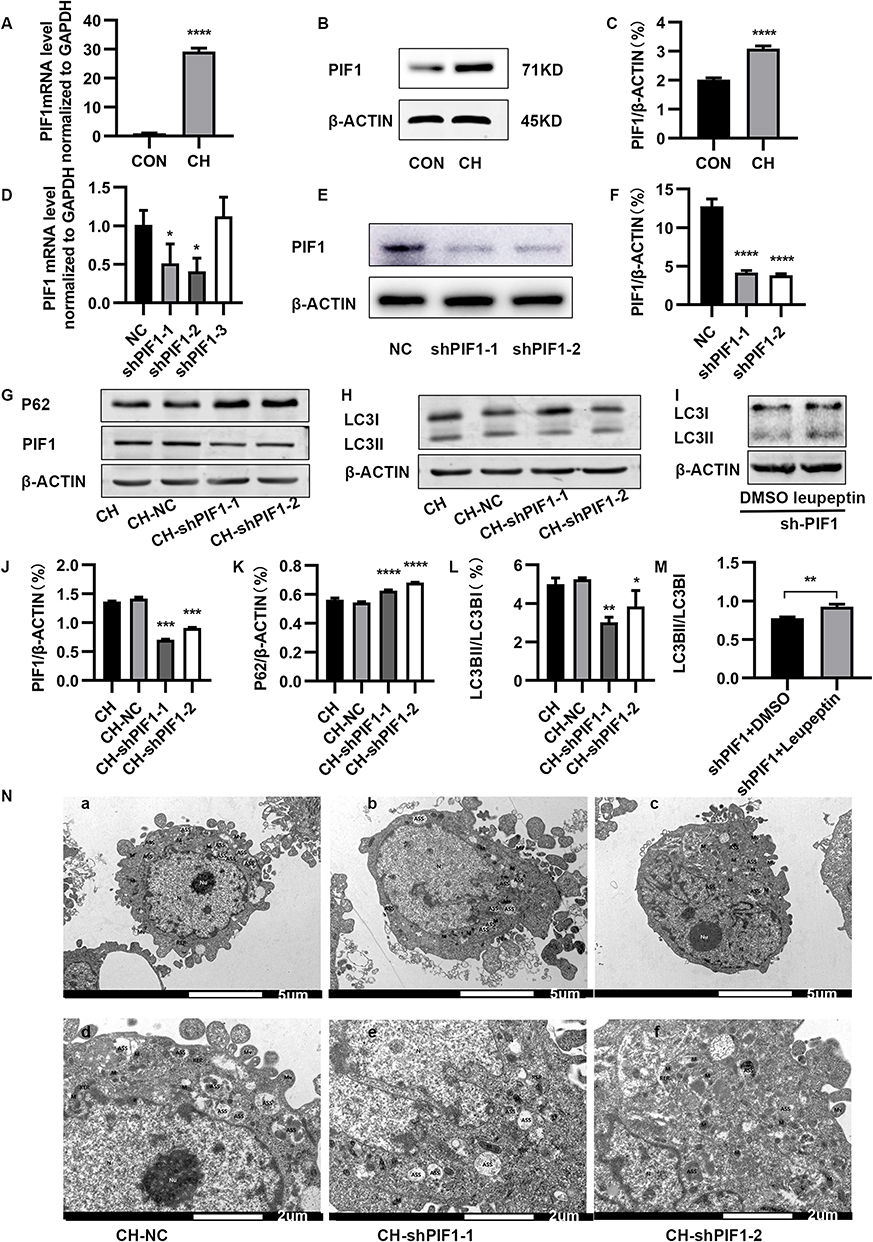 |
Figure 6 Effect of PIF1 knockdown on autophagy in HPAECs. (A) Expression of PIF1 mRNA (****P<0.0001). (B) Expression of PIF1 in HPAECs, as measured by Western blotting. (C) Quantification of the expression of PIF1 protein in HPAECs (****P<0.0001). (D–F) Validation of PIF1 knockdown efficiency (*P < 0.05, ****P<0.0001). (G) Expression of PIF1, and P62 in HPAECs, as measured by Western blotting. (H) Expression of LC3 protein in HPAECs, as measured by Western blotting. (I) HPAECs were transfected with shPIF1 and then, treated with 20 μg/mL of leupeptin for 24 h. Cell lysates were prepared for Western blotting analysis of LC3 and β-ACTIN. (J–M) Quantification of the expression of PIF1, P62, and LC3B in HPAECs (*P < 0.05, **P<0.01, ***P<0.001, ****P<0.0001). (N) Electron micrographs showing the formation of autophagic vacuoles in HPAECs. a. Electron microscopy of HPAEC in negative control (NC) group under CH conditions (×3.0k). b–c. Electron microscopy HPAEC subjected to CH after PIF1 knockdown (×3.0k). d. Electron microscopy of HPAEC in negative control group under CH conditions (×7.0k). e–f. Electron microscopy HPAEC subjected to CH after PIF1 knockdown (×7.0k). |
PIF1 Knockdown Inhibited Autophagy and Promoted Apoptosis in HPAECs Subjected to Chronic Hypoxia
Pulmonary vascular alterations in PAH have multifactorial causes, and endothelial cell dysfunction plays a critical role in this process. HPAEC apoptosis is an important contributor to the earliest changes during PAH. We therefore investigated whether PIF1 affects the apoptosis of HPAECs by transfecting the cells with shRNA-PIF1 lentivirus and exposing them to chronic hypoxia. Scratch-wound assays revealed that the migration of HPAECs was significantly inhibited by knockdown of PIF-1 (Figure 7A and B). In addition, the effects of PIF1 on HPAEC viability under chronic hypoxia conditions was examined using the MTT assay. No significant difference in optical density values was observed between the CH-shPIF-1 and control groups under chronic hypoxia conditions (Figure 7C). Flow cytometry demonstrated that the apoptosis ratio was higher in the CH-shPIF1 group than in the CH and CH-NC groups (Figure 7D and E). PIF-1 knockdown induced a significant increase in the apoptosis of HPAECs under chronic hypoxia conditions. These results showed that PIF1 regulates HPAEC apoptosis and migration. PIF1 affects the integrity and normal function of endothelium and plays an important role in the development of PAH.
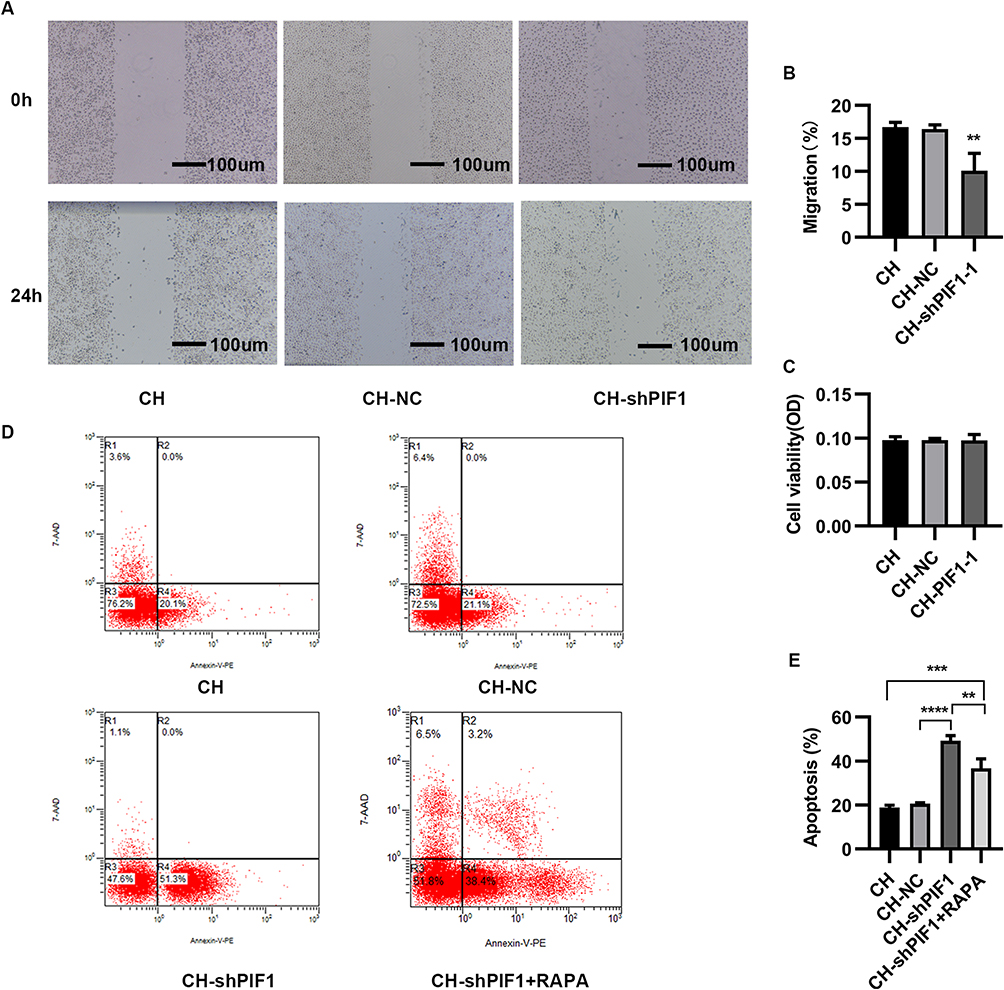 |
Figure 7 Migration, proliferation, and apoptosis of HPAECs subjected to CH after PIF1 knockdown. (A) Scratch-wound assay to assess migration of HPAECs subjected to CH after PIF1 knockdown. (B) Quantification of migration (**P<0.01). (C) Proliferation of HPAECs subjected to CH after PIF1 knockdown. (D) Apoptosis of HPAECs after PIF1 knockdown and of cells treated with rapamycin (RAPA) along with knockdown of PIF-1, as measured using flow cytometry. (E) Quantification (percentage) of apoptotic HPAECs (**P<0.01, ***P<0.001, ****P<0.0001). |
We also examined the effect of treating cells with the autophagy activator rapamycin along with knockdown of PIF1 and found that autophagy was activated (Figure S1E and F). Flow cytometry results showed that the increase in apoptosis was abrogated under these conditions (Figure 7D and E). These data suggested that PIF1 may inhibits the apoptosis of HPAEC by promoting autophagy under chronic hypoxia conditions.
Discussion
In the present study, we subjected HPAECs to chronic hypoxia to investigate the pathogenesis of PAH. PAH is caused by endothelial damage and the proliferation of vascular smooth muscle cells. The converging effects of hypoxia, inflammation, and oxidative and metabolic stresses play a key role in the development of PAH.6,11 Animal studies have demonstrated that the pathogenesis of PAH initially involves endothelial cell apoptosis, followed by the proliferation of apoptosis-resistant endothelial and smooth muscle cells.18 We subjected HPAECs to chronic hypoxia, and our data show that chronic hypoxia increases HPAEC apoptosis, migration, and autophagy. Inhibition of autophagy promoted the apoptosis of HPAECs. In HPAECs subjected to chronic hypoxia, 3743 DEGs were identified by gene expression profiling and bioinformatics analysis. Of these DEGs, 1767 were upregulated and 1976 downregulated (FC >1.3, FDR <0.05). GO analysis showed that the DEGs are involved in ATPase activity, cell adhesion, molecule binding, ubiquitin-like protein ligase binding, ubiquitin-like protein transferase activity, and nucleoside-triphosphatase regulator activity. KEGG pathway analysis showed that the 3743 DEGs were primarily enriched in human papillomavirus infection, human T-cell leukemia virus 1 infection, cell cycle, and apoptosis pathways. PIF-1 expression was markedly increased in HPAECs under chronic hypoxia, leading to promotion of autophagy and inhibition of apoptosis.
Autophagy is a homeostatic cellular process and imbalances in autophagy can cause several diseases.19 Autophagy is a highly regulated catabolic process related to the lysosomal degradation of cytosolic components and the regulation of dysfunctional organelles and misfolded proteins. Autophagy can be activated by stressors such as hypoxia, reactive oxygen species, inflammation, and DNA damage and plays an important role in several human diseases, including cancers and inflammatory diseases.20 Recent evidence has suggested that abnormal regulation of the autophagy pathway is involved in the occurrence of several lung diseases, including PAH.21,22 PAH, a major complication of COPD and OSAHS, is characterized by vascular remodeling, abnormal proliferation of vascular cells (such as endothelial cells), increased intimal thickness, and the formation of plexiform lesions. Animal studies have demonstrated initial EC apoptosis followed by the proliferation of apoptosis-resistant ECs.18 Accumulating evidence also suggests that autophagy plays a mixed role in regulating cell death and survival in response to various stimuli. Autophagy has been demonstrated in human lungs with PAH and protects against EC injury, which promotes vascular remodeling in PAH.23 While some studies have suggested that the activation of autophagy may have beneficial effects in PAH,24,25 other studies have reported contradictory results.12,26 Chloroquine-induced inhibition of the autophagy pathway was shown to prevent disease progression in a monocrotaline-induced PAH model.22 It is unclear whether autophagy activation in pulmonary vascular cells in PAH is beneficial or harmful in terms of pulmonary vascular lesion progression, and whether autophagy contributes to HPAEC survival or death under chronic hypoxia conditions.
In the present study, the protein levels of autophagy markers, autophagosomes, and autolysosome were significantly increased in HPAECs subjected to chronic hypoxia. In addition, chronic hypoxia affected the apoptosis of ECs in PAH. The apoptosis ratio of HPAECs was significantly increased after treatment with autophagy inhibitors, suggesting that apoptosis of HPAECs is related to autophagy in PAH. Chronic hypoxia may induce the autophagy of HPAECs, then affect the apoptotic function of HPAECs, thereby affecting the apoptotic function of HPAECs and leading to dysfunction of the pulmonary vascular endothelium. Chronic hypoxia caused a significant increase in PIF1 expression in HPAECs. PIF1 is a multifunctional helicase and DNA processing enzyme that inhibits apoptosis and promotes autophagy. The functions of human PIF1 are crucial for cell survival. Our study suggests that PIF1 induces autophagy and inhibits the apoptosis of HPAECs under chronic hypoxia conditions. In cells treated with the autophagy activator rapamycin along with knockdown of PIF1, the increase in apoptosis was inhibited. PIF1 may inhibit EC apoptosis by promoting autophagy, and our data indicate that PIF1 plays an important role in the occurrence and progression of PAH. However, we did not validate our results using an animal model of PAH. Future studies should explore the effects of autophagy and apoptosis, as well as the role of PIF1 in a chronic hypoxia-induced PAH animal model. PIF1 also plays a role in maintaining genome stability. The functions of human PIF-1 are crucial for cell survival during replication stress.27 In addition, PIF-1 regulates telomere length, replication, and G4-DNA, and is one of the most potent G4-DNA helicases, capable of unwinding G4-DNA stabilized by G4-DNA-interacting small molecules and mitochondria. Overexpression of the G4-DNA helicase PIF-1 in neurons improves autophagy.13,15,28 In addition, due to its significant role in cell apoptosis and proliferation, the role of PIF1 in cancer has been investigated.16 We found that PIF-1 interference inhibited autophagy and promoted the apoptosis of HPAECs under chronic hypoxia conditions, but PIF-1 interference did not affect the proliferation of HPAECs.
The present study examined the effects of PIF1 in HPAECs under chronic hypoxia conditions. PIF1 plays an important role in chronic hypoxia-induced pulmonary vascular endothelial dysfunction and inhibits EC apoptosis by regulating autophagy. These data indicate that PIF1 may inhibit the apoptosis of HPAECs by promoting autophagy in chronic hypoxia-induced PAH, thereby playing a crucial role in the occurrence and progression of PAH. Therefore, PIF1 is a potential target in the prevention and treatment of PAH.
Conclusion
PIF1 expression was increased in HPAECs under chronic hypoxia conditions, leading to activation of autophagy, inhibition of apoptosis, and promotion of HPAEC migration. PIF-1 plays a crucial role in HPAEC dysfunction in the early stages of chronic hypoxia-induced PAH, making it a potential treatment target for PAH.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study did not require approval from the Ethical and Informed Consent Committee because only involved cells.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the National Natural Science Foundation of China (no. 32200168), the Basic Research Project of Natural Science in Shanxi Province (no. 20210302124039), the Key Research and Development Program in Shanxi Province (no.201803D31093), and the Start-up Foundation for Doctoral Scientific Research of Shanxi Medical University (No. XD1905).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Blanco I, Tura-Ceide O, Peinado VI, Barberà JA. Updated perspectives on pulmonary hypertension in COPD. Int J Chron Obstruct Pulmon Dis. 2020;15:1315–1324. doi:10.2147/COPD.S211841
2. Bonsignore MR, Saaresranta T, Riha RL. Sex differences in obstructive sleep apnoea. Eur Respir Rev. 2019;28(154):190030. doi:10.1183/16000617.0030-2019
3. Adir Y, Humbert M, Chaouat A. Sleep-related breathing disorders and pulmonary hypertension. Eur Respir J. 2021;57(1):2002258. doi:10.1183/13993003.02258-2020
4. Soler X, Diaz-Piedra C, Ries AL. Pulmonary rehabilitation improves sleep quality in chronic lung disease. COPD. 2013;10(2):156–163. doi:10.3109/15412555.2012.729622
5. Shawon MSR, Perret JL, Senaratna CV, Lodge C, Hamilton GS, Dharmage SC. Current evidence on prevalence and clinical outcomes of co-morbid obstructive sleep apnea and chronic obstructive pulmonary disease: a systematic review. Sleep Med Rev. 2017;32:58–68. doi:10.1016/j.smrv.2016.02.007
6. Russomanno G, Jo KB, Abdul-Salam VB, et al. miR-150-PTPMT1-cardiolipin signaling in pulmonary arterial hypertension. Mol Ther Nucleic Acids. 2021;23:142–153. doi:10.1016/j.omtn.2020.10.042
7. Ou M, Li X, Cui S, Zhao S, Tu J. Emerging roles of let-7d in attenuating pulmonary arterial hypertension via suppression of pulmonary artery endothelial cell autophagy and endothelin synthesis through ATG16L1 downregulation. Int J Mol Med. 2020;46(1):83–96. doi:10.3892/ijmm.2020.4567
8. Feng W, Wang J, Yan X, et al. ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. 2021;54(6):e13048. doi:10.1111/cpr.13048
9. Pu X, Du L, Hu Y, Fan Y, Xu Q. Stem/Progenitor cells and pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2021;41(1):167–178. doi:10.1161/ATVBAHA.120.315052
10. Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy. 2018;14(2):221–232. doi:10.1080/15548627.2017.1389823
11. Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1):1801887. doi:10.1183/13993003.01887-2018
12. Kato F, Sakao S, Takeuchi T, et al. Endothelial cell-related autophagic pathways in Sugen/hypoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Lung Cell Mol Physiol. 2017;313(5):L899–L915. doi:10.1152/ajplung.00527.2016
13. Dai Y-X, Chen W-F, Liu -N-N, et al. Structural and functional studies of SF1B Pif1 from Thermus oshimai reveal dimerization-induced helicase inhibition. Nucleic Acids Res. 2021;49(7):4129–4143. doi:10.1093/nar/gkab188
14. Chen B, Hua Z, Gong B, et al. Downregulation of PIF1, a potential new target of MYCN, induces apoptosis and inhibits cell migration in neuroblastoma cells. Life Sci. 2020;256:117820. doi:10.1016/j.lfs.2020.117820
15. Moruno-Manchon JF, Lejault P, Wang Y, et al. Small-molecule G-quadruplex stabilizers reveal a novel pathway of autophagy regulation in neurons. Elife. 2020;9. doi:10.7554/eLife.52283
16. Wang J, Zhu X, Ying P, Zhu Y. PIF1 affects the proliferation and apoptosis of cervical cancer cells by influencing TERT. Cancer Manag Re. 2020;12:7827–7835. doi:10.2147/cmar.S265336
17. Pan M, Yin Y, Wang X, et al. Mice deficient in UXT exhibit retinitis pigmentosa-like features via aberrant autophagy activation. Autophagy. 2021;17(8):1873–1888. doi:10.1080/15548627.2020.1796015
18. Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. 2005;19(9):1178–1180. doi:10.1096/fj.04-3261fje
19. Suzuki Y, Aono Y, Akiyama N, et al. Involvement of autophagy in exacerbation of eosinophilic airway inflammation in a murine model of obese asthma. Autophagy. 2022;18(9):2216–2228. doi:10.1080/15548627.2022.2025571
20. Gomez-Puerto MC, van Zuijen I, Huang CJ, et al. Autophagy contributes to BMP type 2 receptor degradation and development of pulmonary arterial hypertension. J Pathol. 2019;249(3):356–367. doi:10.1002/path.5322
21. Liu X, Cao H, Li J, et al. Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. 2017;24(4):683–693. doi:10.1038/cdd.2017.1
22. Long L, Yang X, Southwood M, et al. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res. 2013;112(8):1159–1170. doi:10.1161/CIRCRESAHA.111.300483
23. Zhang D, Zhou J, Ye LC, et al. Autophagy maintains the integrity of endothelial barrier in LPS-induced lung injury. J Cell Physiol. 2018;233(1):688–698. doi:10.1002/jcp.25928
24. Wang K, Chen Y, Zhang P, Lin P, Xie N, Wu M. Protective features of autophagy in pulmonary infection and inflammatory diseases. Cells. 2019;8(2). doi:10.3390/cells8020123
25. Houssaini A, Abid S, Mouraret N, et al. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol. 2013;48(5):568–577. doi:10.1165/rcmb.2012-0429OC
26. McMurtry MS, Bonnet S, Michelakis ED, Bonnet S, Haromy A, Archer SL. Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamcyin-atorvastatin-simvastatin study. Am J Physiol Lung Cell Mol Physiol. 2007;293(4):L933–L940. doi:10.1152/ajplung.00310.2006
27. Dehghani-Tafti S, Levdikov V, Antson AA, Bax B, Sanders CM. Structural and functional analysis of the nucleotide and DNA binding activities of the human PIF1 helicase. Nucleic Acids Res. 2019;47(6):3208–3222. doi:10.1093/nar/gkz028
28. Decaux G. Long-term treatment of patients with inappropriate secretion of antidiuretic hormone by the vasopressin receptor antagonist conivaptan, urea, or furosemide. Am J Med. 2001;110(7):582–584. doi:10.1016/S0002-9343(01)00678-7
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.