Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 8
Physical therapy for individuals with amyotrophic lateral sclerosis: current insights
Authors Dal Bello-Haas V ![]()
Received 21 July 2017
Accepted for publication 11 December 2017
Published 16 July 2018 Volume 2018:8 Pages 45—54
DOI https://doi.org/10.2147/DNND.S146949
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Müller
Vanina Dal Bello-Haas
Physiotherapy Program, School of Rehabilitation Science, McMaster University, Hamilton, ON, Canada
Abstract: Amyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative, and inevitably fatal disease. There is no cure for ALS and life expectancy is typically 2–5 years after symptom onset. Despite the lack of a cure and the rapidly progressive nature of the disease, ALS is considered a “treatable disease” and rehabilitation is integral to optimal, comprehensive care. In addition to the other health care professions making up the health care team, physical therapy provides a critical role in the overall management in individuals with ALS. Physical therapy that is tailored to the individual’s needs and goals and focused on addressing symptoms and maximizing function and participation enables people with ALS to live their lives to the fullest and with quality. The purpose of this paper is to review some of the recent ALS research findings that have implications for physical therapy practice.
Keywords: amyotrophic lateral sclerosis, physical therapy, disease stage, falls, exercise, pain, cognition
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative, and inevitably fatal disease associated with loss of upper and lower motor neurons.1 There is no cure for ALS, life expectancy is typically 2–5 years after symptom onset, and the only approved drug, riluzole, has a modest effect on survival.2 The clinical management of people with ALS (PALS) is complex and requires a comprehensive and multidisciplinary approach. There is an increasing interest in a rehabilitation approach toward PALS, as illustrated in Figure 1, with an increasing number of articles published related to rehabilitation for PALS or specific elements of rehabilitation, such as exercise and falls. The sharp increase in the number of articles since the year 2000 indicates the ever-expanding interest related to these themes.
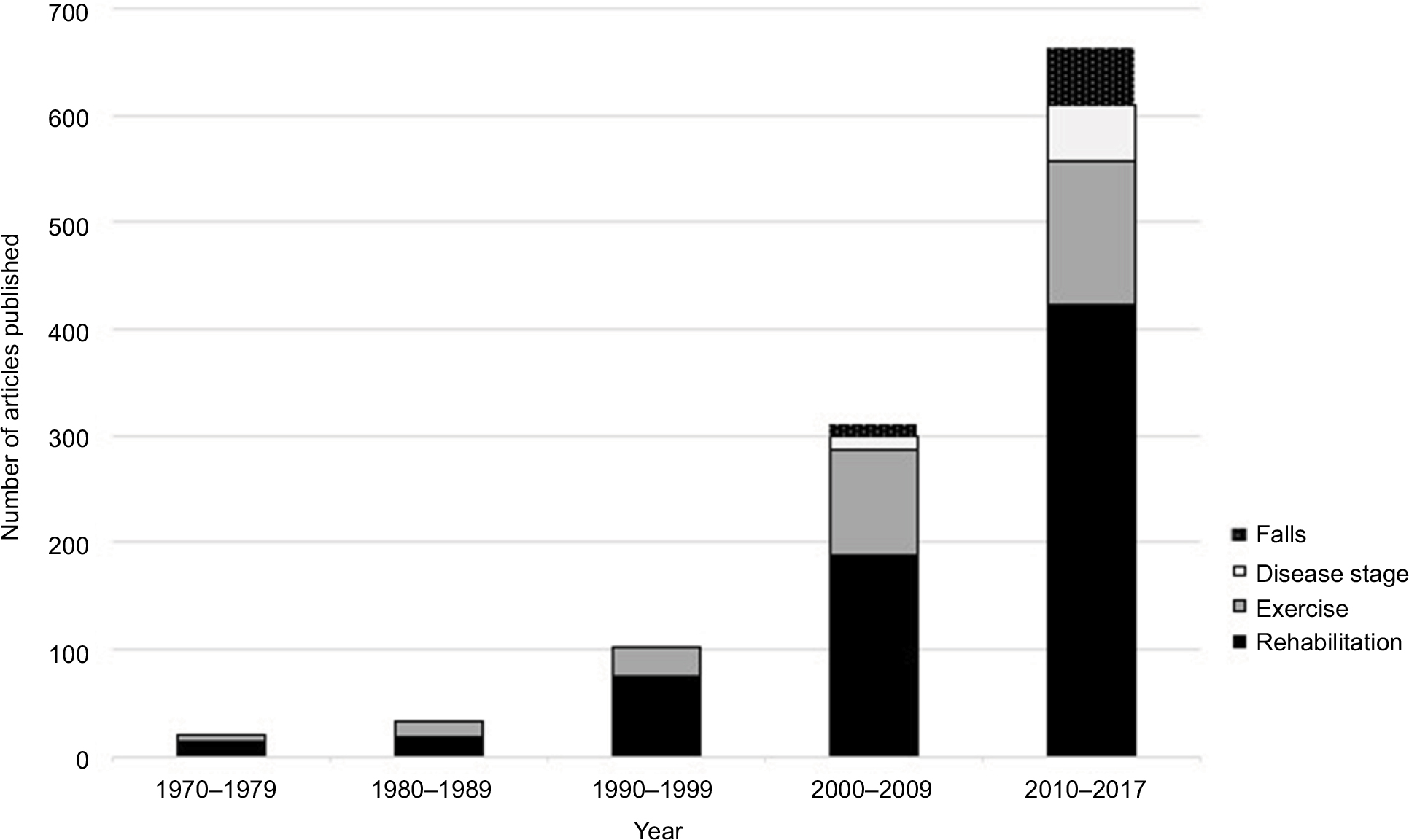 | Figure 1 PubMed search results, 1970 to 2017: Amyotrophic lateral sclerosis and rehabilitation, amyotrophic lateral sclerosis and disease stage, amyotrophic lateral sclerosis and falls, and amyotrophic lateral sclerosis and exercise. |
The Latin root of rehabilitation, rehabilitare – “to provide again with means” – exemplifies the optimal approach to care of PALS; as does active living, whereby healthy, optimal, and productive living is paramount irrespective of disease state. Effective management of PALS is complex and requires a comprehensive and multidisciplinary approach. Physical therapy is an integral component of the ALS multidisciplinary team and is well grounded in rehabilitation and active living concepts. Despite the lack of a cure and rapidly progressive nature of the disease, physical therapy that is tailored to the individual’s needs and goals, and focused on addressing the symptoms and maximizing the function and participation enables PALS to live their lives to the fullest and with quality. The purpose of this paper is to review some of the recent research findings that have implications for physical therapy for PALS in general, and related to physical therapy evaluation and treatment for PALS specifically. Although not a systematic review, the research evidence published within the past 5 years was systematically searched using the following databases (CINAHL, Cochrane, Web of Science, PubMed, OVID) to address the specific objectives of this paper: 1) to describe important and relevant recent research breakthroughs and implications for physical therapy practice; and, 2) to describe the most recently published ALS research related to cognitive, behavioral, and psychological impairments, respiratory impairments, falls, pain, and exercise.
ALS: more than just a motor disease?
There is increasing evidence that ALS should be regarded as a multisystem health condition rather than solely a motor disease. Clinical, genetic, and postmortem studies have demonstrated that there is considerable variability in the phenotypic expression of ALS.3–7 Neuropathological and imaging findings have confirmed that ALS includes various non-motor (extrapyramidal) areas.3–5 Regions beyond the motor system affected by ALS include the autonomic nervous system, the basal ganglia, and the cerebellar, frontotemporal, oculomotor, and sensory systems.8,9 More recent ALS research has greatly enhanced the knowledge and understanding of the genetics and molecular mechanisms of the disease. Over 20 chromosomal regions and a number of identified genes have been linked to ALS. A substantial proportion of familial ALS (FALS) has now been traced to an expansion of the intrinsic hexanucleotide repeat sequence in C9orf72. The recent C9orf72 breakthrough reinforces the concept that ALS is comprised of multiple, complex pathogenic pathways.10 This body of research highlights the need for the physical therapist to not only focus on assessment of the motor system, but rather, the physical therapist should include evaluation of multiple systems within the subjective and objective assessments, for example, asking questions about sensory changes, testing for sensory system integrity, screening for cognitive impairments, screening for extrapyramidal signs and symptoms.
Classification of ALS – time for a change?
It has been recently suggested that currently used ALS classification systems, such as the El Escorial criteria and the International Classification of Diseases, are not consistently applied and omit features that are important in the clinical management of PALS, such as rate of progression, genetics, or functional effect.11 Al-Chalabi et al approach to ALS classification is based on the latest research findings and combines current classification systems with the highly variable presentation of ALS.11 The authors propose that a more comprehensive classification system would improve communication with and among health professionals, and patient care and research processes, and would assist with indications of prognosis and treatment needs.11
The proposed classification system incorporates 4 mandatory components. These are: 1) stage of disease; 2) phenotype; 3) diagnosis; 4) El Escorial category, as well as diagnostic modifiers and optional terms can be added as needed11 (Figure 2). A more fulsome “picture” of the individual with ALS is provided by the proposed classification system components.
 | Figure 2 Proposed classification system for ALS. Notes: The classification system begins at the left of the figure and proceeds through each block toward the right, as noted by the arrow, providing a “picture” of the individual with ALS. Reprinted from The Lancet Neurology; 15(11); Al-Chalabi A, Hardiman O, Kiernan MC, Chi.A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system; 1182–1194; Copyright © 2016 Elsevier Ltd; with permission from Elsevier.11 Abbreviations: ALS, amyotrophic lateral sclerosis; FTD, frontotemporal dementia; LMN, lower motor neuron; PLS, primary lateral sclerosis; PMA, progressive muscle atrophy; UMN, upper motor neuron. |
The stage of disease refers to a measure of disease progression, with stage 1 being early disease and stage 4 late-stage disease.11 Phenotypic descriptors include site of onset, for example, limb onset versus bulbar onset, age of onset, as well as others.11 Diagnosis refers to the spectrum of motor neuron diseases and the El Escorial category is included as a component of the larger classification system.11 Diagnostic modifiers may include familial (family history) and other features, for example, frontotemporal dementia (FTD); and optional components include specific ALS mutation or cause.11
The key to effective physical therapy examination and management, and the improvement and standardization of examination procedures and interventions is in understanding phenotype in an effort to ensure that clinical manifestations are comprehensively and appropriately identified and managed. The proposed classification system is much more robust in terms of describing the PALS and informing health care professionals of “what to expect” upon initial interaction. For example, the physical therapist who is to complete a subjective and objective evaluation, and develop an appropriate management plan for an individual with ALS whose diagnosis is stage 3 limb onset UMN-predominant FALS secondary to C9orf72 mutation, fulfilling El Escorial criteria for possible ALS11 versus whose diagnosis is possible ALS will have a much clearer initial picture of the clinical presentation and potential impairments, activity limitations and participation restrictions, potential focus of treatment, prognosis as it relates to the plan of care, and critical considerations for the overall approach and decision-making, for example, later disease stage, and cognition impairment(s).
A stage approach for physical therapy evaluation and treatment
Because of the variable clinical phenotypes of ALS, the highly variable presentation of PALS, the great variability in prognosis, and the typically rapid, progressive, and deteriorating nature of ALS, physical therapists are constantly challenged when developing the most appropriate intervention plan for PALS.12 The physical therapist requires a solid understanding of the nature and course of the disease, and needs to consider future problems in addition to current status.12 In order to make appropriate and effective decisions, the nature and significance of the interrelationships among impairments, activity limitations, and participation restrictions need to be determined.12 In addition, decision-making involves determining which impairments, activity limitations, and participation restrictions: 1) can be restored; 2) require compensatory strategies or interventions; 3) require referral to different health care professional(s); and 4) cannot be affected by physical therapy interventions at all.12
In 2002, Dal Bello-Haas proposed a 3-stage (early, middle, and late) framework to assist with decision-making as it relates to physical therapy examination and management for PALS.12 In the early stage of ALS, individuals are independent with mobility, function, and activities of daily living despite muscle weakness in specific muscle groups, and activity limitations and participation restrictions are not present or minimal.12 During the middle stage of ALS, PALS have an increasing number and severity of impairments and activity limitations due to disease progression and participation restrictions are common.12 Progressive decline in function and mobility results in the need for compensatory interventions and dependence on others.12 In the late stage of ALS, the PALS become totally dependent for mobility and function due to severe weakness of axial and extremity muscles.12 Dysarthria, dysphagia, and respiratory issues are most common in the late stage, although these problems may be present at any stage; and other impairments, such as cognitive and psychosocial, may also be present. Physical therapists should develop appropriate management plans, for example, preventative, compensatory, and restorative interventions based on the stage of the disease, as well as other factors, which include patient factors such as goals, psychosocial factors, social and financial resources; disease course and progression; and lifespan time of disease onset.12 Other interventions, including patient, family, and caregiver education and training, psychological support, and referral to other health care professionals, also comprise the overall management plan across the ALS disease continuum.12
A “stage” approach is increasingly being adopted when describing rehabilitation in general and physical therapy more specifically, for example, 2014 Irish Guidelines for the Physiotherapy Management of Motor Neuron Disease13 and Majumdar et al.14 In addition, the “stage” approach is increasingly being recognized as providing useful information with regard to disease progression, and for improving patient management, resource allocation, research classification(s), and clinical trial design, for example, Balendra et al15, Chio et al16, Roche et al.17 Thinking about which stage of the disease a PALS is in (early, middle, or late) is useful for guiding the physical therapist’s decision-making, focusing physical therapy management plans, and developing more patient-specific goals.
Amyotrophic symptom management: is enough being done?
A recent paper provides insight into the types of symptoms being reported by individuals with ALS and the current state of symptom management in a particular sample of PALS.18 Data related to symptom presence and severity, degree to which symptoms were problematic, and symptom management were collected.18 In order of decreasing prevalence, PALS reported the symptoms as follows: fatigue (90%), muscle stiffness (84%), muscle cramps (74%), shortness of breath (66%), difficulty sleeping (60%), pain (59%), anxiety (55%), depression (52%), increased saliva (52%), constipation (51%), pseudobulbar affect (38%), loss of appetite (37%), and weight loss (29%).18 Fatigue was not only the most frequently reported symptom, but was also the symptom least frequently treated.18 Other symptoms that had low treatment prevalence included: dyspnea (49%), pain (44%), muscle stiffness (39%), sialorrhea (32%), pseudobulbar affect (29%), and weight loss (22%)18 Many of these symptoms can be addressed by appropriate physical therapy interventions, for example, fatigue (energy conservation), muscle stiffness (stretching exercises), muscle cramps (stretching exercises), and pain (intervention dependent on source of pain). The Nicholson et al paper highlights that more can and should be done for the overall management of PALS, and reinforces the fact that the physical therapist should be diligent in asking the PALS about commonly reported signs and symptoms during the subjective assessment to develop effective management plans, including referral to other health care professionals for problems outside the physical therapy scope of practice.18
Similar to Nicholson et al’s paper, the 2013 ALS Quality Measures report also identified gaps in care and has specified several recommendations, including: 1) a multidisciplinary care plan is needed for each individual with ALS, in order to optimize health care delivery (measure #1); 2) ALS symptomatic therapy treatments should be offered (measure #4); 3) ALS non-invasive ventilation treatment for respiratory insufficiency should be provided (measure #6); and 4) ALS nutritional support via enteral feeding should be offered (measure #8).19 Additional measures have been reported and full reports, as data supplements, are available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3863352/bin/supp_81_24_2136__index.html.
Cognitive, behavioral, and psychological impairments
Cognitive, behavioral, and psychological impairments have important clinical implications for physical therapists related to communication, legal, and ethical issues, for example, consent, decision-making, and treatment adherence. As described previously, although once thought to be a purely motor disorder, it is increasingly being recognized that ALS is comprised of extra-motor signs and symptoms. Cognitive impairments are common and may be present in up to 50% of individuals with ALS;20,21 and up to 15% of PALS who have cognitive impairment and meet the Neary criteria22 for FTD, and have ALS–FTD.20,21 ALS and FTD are known to share distinct clinical, neuropathological, and genetic features, and it is now recognized that they are part of a continuum in which “pure” ALS, with no cognitive involvement, and “pure” FTD, with no motor involvement, form ends of a disease spectrum.23,24 The 2011 discovery of a mutation in the C9orf72 gene unambiguously linked ALS and FTD.23 The C9orf72 mutation is known to be responsible for about 40% of FALS, 25% of familial FTD, and up to 80% of ALS–FTD cases.23,24
ALS–FTD is characterized by cognitive decline, executive functioning impairments, difficulties with planning, organization and concept abstraction, and personality and behavior changes.24 Individuals with ALS, without FTD, may also have a variety of cognitive impairments, such as generalized impairments in intellectual function, as well as difficulties with verbal fluency, language comprehension, memory, and abstract reasoning.24 The presence of cognitive impairment(s) is associated with reduced survival, more rapid disease progression, decreased functional ability, higher rates of treatment non-adherence, and increased caregiver distress and burden.25,26
Because of the prevalence of cognitive impairments and FTD, and the impact on goal setting and physical therapy examination and interventions, it is critical for the physical therapist to screen for and consider these impairments when developing and implementing management plans. The utility of incorporating screening instruments in a busy ALS clinic has been documented,27,28 and it is recommended that PALS be screened at least once annually for cognitive impairment, for example, FTD screening, and cognitive and behavioral impairment screening.19
Two ALS-specific screening tools that can be administered by non-neuropsychologists exist. The ALS Cognitive Behavioral Screen assesses executive function via 2 components. These are 1) cognitive, which evaluates attention, concentration, working memory, fluency, and tracking and 2) behavioral, which is completed by a family member or caregiver and examines changes in empathy, personality, judgment, language, and insight.27 It has been reported that the cognitive section differentiated patients with cognitive impairments with 71% specificity and 85% sensitivity, and that the behavioral section predicted ALS–FTD with 80% sensitivity and 88% specificity.27 The Edinburgh Cognitive and Behavioral ALS Screen (ECAS) is an additional screening tool that incorporates short cognitive tests that have been shown to be sensitive to cognitive impairment in PALS. A brief Behavioral and Psychosis interview is completed with family or caregivers, and executive functions, memory, language, visuospatial skills, and social cognition are assessed. The ECAS takes about 15 min to administer and can be found at: https://www.era.lib.ed.ac.uk/handle/1842/6592.
Anxiety and depression can greatly affect the quality of life of an individual with ALS and the patient’s family, as well as the ability to cope with and adapt to the progressive changes and associated physical and losses. Prevalence of anxiety and depression in PALS is relatively high.18 The ALS Depression Inventory 12 (ADI-12) is the only ALS-specific depression screening instrument.29 The ADI-12 excludes statements addressing activities that depend on an intact motor system, but has not undergone extensive or rigorous cross-cultural methodology or psychometric testing. There is no ALS-specific anxiety screening measure; however, the Hospital Anxiety and Depression Scale,30 and the State-Trait Anxiety Inventory31 have been used in clinical studies. Similar to screening for cognitive and behavioral issues, the physical therapist should also screen PALS for depression and anxiety, and make referrals to appropriate health care professionals as needed.
Falls
Falls in PALS are common, with fall rates reported to be 46% in an ALS clinic.32 Falls and their implications in PALS are garnering much more attention. Falls in PALS have serious consequences, resulting in increased morbidity and mortality.33 Fall-related head trauma has been found to be significantly higher in PALS compared with a control group the year before and after diagnosis, and Gil et al found that 1.7% of ambulatory PALS experienced fall-related deaths.33 An interesting 2014 study found that 37% of ambulatory individuals with relatively normal clinical balance and mobility test findings (eg, Dynamic Gait Index, Berg Balance Scale Score, Timed Up and Go, Stair-Climbing Test, 25-Feet Walk Test) had decreased ability to use vestibular input and required increased reliance on visual input for postural orientation to sustain equilibrium, as detected via sensory organization testing (EquiTest, Neurocom).34 The authors suggested that peripheral and central pathologic abnormalities or ALS-related cerebellum pathology may have contributed to the vestibular deficits.34 The involvement of the cerebellum in the pathologic abnormalities of ALS is increasingly recognized,35 lending further support that ALS is a multisystem, rather than purely a motor disease. The physical therapist should ask PALS about their recent falls at each clinical visit and further evaluate for the presence of known risk factors,19 and appropriate physical therapy interventions and management plans should be implemented in PALS to minimize their risk of falling, for example, through prescription of assistive devices or orthotics, and balance training as appropriate.
Pain
ALS has been typically considered a “painless” disease. However, many PALS report pain even in early stages of the disease,36,37 and as illustrated in Figure 3, there are multiple and various sources of pain in PALS. Depending on the cause of pain, physical therapy interventions may include modalities, range of motion (ROM) exercises, passive stretching, joint mobilizations, and education about proper joint support and protection. Although ALS does not involve the pain pathways primarily, musculoskeletal impairments, immobility, loss of ROM, decreased support from weakened muscles, positioning difficulty, dependent edema, and acute injuries (sprains, strains, and falls) can all cause pain. Spasticity and cramps, especially if severe, and pre-existing conditions can also cause pain in PALS.
 | Figure 3 Sources of pain and interventions in people with ALS. Notes: Adapted with permission from The UK Motor Neurone Disease Networking Group. A pathway for the management of pain in motor neurone disease; 2001. Available from: https://parkinsonsacademy.co/resources/care-pathways/. Copyright © 2001 The UK Motor Neurone Disease Networking Group.55 Abbreviations: ALS, amyotrophic lateral sclerosis; ROM, range of motion; TENS, transcutaneous electrical nerve stimulation. |
Common sites of pain include the low back, the neck, and the shoulder region.38
Pain in PALS has been poorly studied and is often poorly managed.39 Physical therapists should regularly assess for pain, as this may exacerbate depression and fatigue, both of which have been associated with decreased quality of life in individuals with ALS.36,37 The 2014 Irish Guidelines for the Physiotherapy Management of Motor Neuron Disease13 and the National Institute for Health and Care Excellence clinical guidelines on motor neuron disease,40 updated in 2016, succinctly summarize possible causes of pain in PALS and potential intervention strategies (Figure 3), and Chiò et al’s review paper41 summarizes the current understanding of pain in PALS.
Exercise for PALS
The effects of exercise in PALS are not well understood, and the role of resistance and aerobic exercise, and fitness in general, for those with ALS does continue to be controversial. The role of exercise in ALS has been the subject of much animal and human research. Many PALS ask about exercise, and exercise may have been central to the individual’s pre-ALS life. It is becoming recognized that exercise plays a very important role in health and wellbeing for many populations, including those with other neurodegenerative diseases such as Parkinson’s disease, Huntington’s disease, and multiple sclerosis. Physical therapists, with their education and training, are considered experts in movement and exercise, and thus, are the ideal health care professionals to prescribe exercise for PALS.42
Despite the lack of research evidence, some clinicians continue to discourage any formal resistance or aerobic exercise programs for fear of overuse weakness or “what’s the point” thinking and recommend no exercise other than everyday activities. The evidence related to the benefits and risks of exercise in PALS is limited. However, the view that exercise, when prescribed appropriately, may be physiologically and psychologically beneficial for PALS, especially when implemented in the earlier stages of the disease, is being increasing adopted, for example, Paganoni et al43, Plowman et al.44
A 2013 update to a Cochrane review45 titled Therapeutic Exercise for People with Amyotrophic Lateral Sclerosis or Motor Neuron Disease found no new studies. This review is in the process of being updated again. Two exercise trials have been recently completed: Muscle Training of Patients with Amyotrophic Lateral Sclerosis (NCT01504009, Sweden) and Trial of Resistance and Endurance Exercise in Amyotrophic Lateral Sclerosis (NCT01521728 USA), and the results are pending. Until the research findings regarding the role of exercise in individuals with ALS is more definitive, integrating standard principles of exercise prescription, for example, American College of Sports Medicine,46 with physical therapy examination findings, the rehabilitation framework,12 and a thorough understanding of the nature and course of ALS is the best practice. See Dal Bello-Haas and Krivickas47 or Paganoni et al43 for practical recommendations.
ROM and stretching exercises are typically accepted modes of exercise for PALS. Resistance exercises of unaffected muscles (and possibly affected muscles with strength of at least grade 3 or above) using a low-to-moderate load and intensity, and aerobic activities, such as swimming, walking, and stationary cycling (mode dependent on safety of PALS), at sub-maximal levels (eg, between 50% and 65% of heart rate reserve) may be safe and effective in achieving therapeutic goals.47 Aerobic and resistance exercises are more appropriate for PALS in the early or early-middle stage of ALS and those with more slowly progressive disease, and should be prescribed as soon as possible after diagnosis. When designing any exercise program for PALS, the physical therapists must continuously consider the balance between underwork (“doing too little”) and overwork (“doing too much”), and make adjustments based on a PALS’ response to exercise and other disease-specific factors, for example, onset of respiratory impairments. The physical therapist needs to carefully monitor and adjust the exercise mode and intensity according to disease progression, in order to prevent excessive fatigue and potential overwork damage. while at the same time, poting optimal use of intact muscle groups. Physical therapists should educate PALS not to carry out any activities to the point of extreme fatigue; and PALS should keep track of signs and symptoms of overuse (eg, inability to perform daily activities following exercise due to exhaustion or pain; a reduction in maximum muscle force that gradually recovers; increased or excessive muscle cramping, soreness, fatigue, or fasciculations) via an exercise log that the physical therapist can review. In addition, PALS may be advised to exercise for several brief sessions throughout the day versus completing aerobic or resistance exercises for a sustained period of time, with sufficient rest periods in between exercise sessions. If signs or symptoms of overuse occur, PALS should be advised to stop the exercises and the physical therapist should re-assess.
Respiratory impairments
Respiratory impairments in PALS are related to decreased respiratory muscle strength. Early signs and symptoms of respiratory muscle weakness are varied and may include shortness of breath, orthopnea, sleep disturbances, poor concentration, confusion, daytime sleepiness, morning headaches, and fatigue. In addition, expiratory muscle weakness leads to a decreased ability to cough and clear secretions. As the disease progresses, almost all PALS will eventually require some intervention to address respiratory-related issues. Physical therapists should ask PALS about symptoms of respiratory insufficiency as a part of the subjective assessment at each clinical visit; and vital capacity, maximum inspiratory pressure, sniff nasal pressure, or peak cough expiratory flow testing should be conducted at least every 3 months.19,40
Cough augmentation, lung volume recruitment training, and airway clearance techniques, such as breath stacking, assisted breath stacking, and manually-assisted cough are common physical therapy interventions for individuals with weak expiratory muscles and secretion retention. While there are no randomized control trials of cough augmentation techniques in PALS, there is evidence that suggests that cough augmentation techniques and devices can assist with secretion clearance,48,49 and that regular breath stacking may be beneficial for individuals with early respiratory impairments who do not require non-invasive ventilation.49–52 A recent systematic review found that specific physical therapy interventions (eg, inspiratory muscle training, lung volume recruitment training, and manually assisted cough) were effective in improving respiratory outcome measures and increasing survival, and should be included in the overall management of PALS.53 Mechanical insufflation–exsufflation (MI-E) is beneficial when manually assisted coughing or breath stacking are no longer effective,40,49 and a systematic review of the effects of MI-E and the breath-stacking technique for reducing morbidity and mortality, and enhancing quality of life in PALS is currently underway.54
Conclusion
Despite advances and improvements in care and the recognition of the importance of multidisciplinary care for PALS, the disease progression for those with ALS remains relatively unchanged, as does the availability of disease-modifying agents. It is increasingly being recognized that rehabilitation, in general, and physical therapy, in particular, can greatly enhance function, participation, and quality of life for those with ALS. The current state of the evidence indicates that current practice guidelines for physical therapy management heavily relies on expert opinion and consensus, although physical therapy research is being to emerge. More evidence is needed to substantiate specific physical therapy interventions for individuals with ALS.
Acknowledgments
Special thanks to Humna Malik and Briana Virag for their assistance with this manuscript.
Disclosure
The author reports no conflicts of interest in this work.
References
Swash M. Clinical features and diagnosis of amyotrophic lateral sclerosis. In: Brown R, Jr, Meininger V, and Swash M, editors. Amyotrophic Lateral Sclerosis. London, UK: Martin Dunitz Ltd; 2000. | ||
Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND).Cochrane Database of Syst Rev. 2012;3:CD001447. | ||
Brettschneider J, Libon DJ, Toledo JB, et al. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012;123(3):395–407. | ||
Cistaro A, Valentini MC, Chiò A, et al. Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging. 2012;39(2):251–259. | ||
Prell T, Peschel T, Hartung V, et al. Diffusion tensor imaging patterns differ in bulbar and limb onset amyotrophic lateral sclerosis. Clin Neurol Neurosurg. 2013;115(8):1281–1287. | ||
Cardenas-Blanco A, Machts J, Acosta-Cabronero J, et al. Central white matter degeneration in bulbar- and limb-onset amyotrophic lateral sclerosis. J Neurol. 2014;261(10):1961–1967. | ||
Brettschneider J, Del Tredici K, Irwin DJ, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014;127(3):423–439. | ||
McCluskey L, Vandriel S, Elman L, et al. ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. J Neurol Sci. 2014;345(1–2):118–124. | ||
Brooks BR. ALS-Plus – where does it begin, where does it end? J Neurol Sci. 2014;345(1–2):1–2. | ||
Sabatelli M, Marangi G, Conte A, Tasca G, Zollino M, Lattante S. New ALS-related genes expand the spectrum paradigm of amyotrophic lateral sclerosis. Brain Pathol. 2016;26(2):266–275. | ||
Al-Chalabi A, Hardiman O, Kiernan MC, Chiò A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 2016;15(11):1182–1194. | ||
Dal Bello-Haas V. A framework for rehabilitation in degenerative diseases: planning care and maximizing quality of life. Neurol Report. 2002;26(3):115–129. | ||
MND Guideline Development Group. Irish Guidelines for the Physiotherapy Management of Motor Neuron Disease (MND). Available from: http://imnda.ie/wp-content/uploads/2014/09/MND-guidelines-on-Physiotherapy.pdf. Accessed December 6, 2017. | ||
Majumdar S, Wu J, Paganoni S. Rehabilitation in amyotrophic lateral sclerosis: why it matters. Muscle Nerve. 2014;50(1):4–13. | ||
Balendra R, Jones A, Jivraj N, et al; UK-MND LiCALS Study Group, Mito Target ALS Study Group. Use of clinical staging in amyotrophic lateral sclerosis for phase 3 clinical trials. J NeurolNeurosurg Psychiatry. 2015;86(1):45–49. | ||
Chiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86(1):38–44. | ||
Roche JC, Rojas-Garcia R, Scott KM, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135(Pt 3):847–852. | ||
Nicholson K, Murphy A, Mcdonnell E, et al. Improving symptom management for people with amyotrophic lateral sclerosis. Muscle Nerve. 2017;57(1):20–24. | ||
Miller RG, Brooks BR, Swain-Eng R, et al. Quality improvement in neurology: amyotrophic lateral sclerosis quality measures. Report of the quality measurement and reporting subcommittee of the American Academy of Neurology. Neurology. 2013;81(24):2136–2140. | ||
Phukan, J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. 2012;83(1):102–108. | ||
Montuschi A, Iazzolino B, Calvo A, et al. Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry. 2015;86(2):168–173. | ||
Neary D, Snowden JS, Gustfson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. | ||
Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–330. | ||
Beeldman E, Raaphorst J, Klein Twennaar M, de Visser Schmand BA, de Haan RJ. The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry. 2016;87(6):611–619. | ||
Olney RK, Murphy J, Forshew D, et al. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. 2005;65(11):1774–1777. | ||
Elamin M, Phukan J, Bede P, et al. Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology. 2011;76(14);1263–1269. | ||
Woolley SC, York MK, Moore DH, et al. Utility of the ALS cognitive behavioral screen. Neurodegen Dis Manage. 2011;1:473–479. | ||
Gordon PH, Wang Y, Doorish C, et al. A screening assessment of cognitive impairment in patients with ALS. Amyotroph Lateral Scler. 2007;8(6):362–365. | ||
Kübler A, Winter S, Kaiser J, Birbaumer N, Hautzinger M. Das ALS-Depressionsinventar (ADI). Z Klin Psychol Psychother (Gott). 2005;34(1):19–26. | ||
Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67(6):361–370. | ||
Spielberger CS, Gorsuch RL, Lushene RE. Manual for the State Trait Anxiety Inventory. Palo Alto, CA, USA: Consulting Psychologists Press; 1970. | ||
Dal Bello-Haas V, Andrews-Hinders D, Richer CB, et al. Development, analysis, refinement and utility of an interdisciplinary amyotrophic lateral sclerosis database. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(1):39–46. | ||
Gil J, Funalot B, Verschueren A, et al. Causes of death amongst French patients with amyotrophic lateral sclerosis: a prospective study. Eur J Neurol. 2008;15(11):1245–1251. | ||
Sanjak M, Hirsch MA, Bravver EK, Bockenek WL, Norton HJ, Brooks BR. Vestibular deficits leading to disequilibrium and falls in ambulatory amyotrophic lateral sclerosis. Arch Phys Med Rehabil. 2014;95(10):1933–1939. | ||
Prell T, Grosskreutz J. The involvement of the cerebellum in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(7–8):507–515. | ||
Pizzimenti A, Aragona M, Onesti E, Inghilleri M. Depression, pain and quality of life in patients with amyotrophic lateral sclerosis: a cross-sectional study. Funct Neurol. 2013;28(2):115–119. | ||
Rivera I, Ajroud-Driss S, Casey P, et al. Prevalence and characteristics of pain in early and late stages of ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(5–6):369–372. | ||
Ho DT, Ruthazer R, Russell JA. Shoulder pain in amyotrophic lateral sclerosis. J Clin Neuromuscul Dis. 2011;13(1):53–55. | ||
Wallace VC, Ellis CM, Burman R, Knights C, Shaw CE, Al-Chalabi A. The evaluation of pain in amyotrophic lateral sclerosis: a case controlled observational study. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):520–527. | ||
National Institute for Health and Care Excellence. Motor Neurone Disease: Assessment and Management. London, UK: Clinical Guideline NG42, NICE; 2016. | ||
Chiò A, Mora G, Lauria G. Pain in amyotrophic lateral sclerosis. Lancet Neurol. 2017;16(2):144–157. | ||
World Confederation for Physical Therapy. Physical therapists as exercise experts across the life span; 2017. Available from: http://www.wcpt.org/sites/wcpt.org/files/files/resources/policies/2017/PS_Physical_therapists_exercise_experts_lifespan_FINAL.pdf. Accessed June 30, 2017. | ||
Paganoni S, Chaficb IK, Nanettec J, Bedlack, R, Carter GT. Comprehensive rehabilitative care across the spectrum of amyotrophic lateral sclerosis.Neuro Rehabil. 2015;37(1):53–68. | ||
Plowman EK, Watts SA, Tabor L, et al. Impact of expiratory strength training in amyotrophic lateral sclerosis. Muscle Nerve. 2016;54(1):48–53. | ||
Dal Bello-Haas V, Florence JM. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst Rev. 2013;5:CD005229. | ||
Riebe D, Ehrman JK, Liguori G, Magal M. American College of Sports Medicine. ACSM’s Guidelines for Exercise Testing and Prescription. 10th ed. Philadelphia, PA: Wolters Kluwer; 2018. | ||
Dal Bello-Haas V, Krivickas L. Amyotrophic lateral sclerosis. In: Durstine JL, Moore GE, Painter PL, editors. ACSM’s Exercise Management for Persons with Chronic Diseases and Disabilities. 3rd ed. Champaign, IL: Human Kinetics; 2008:336–341. | ||
Cleary S, Misiaszek JE, Kalra S, Wheeler S, Johnston W. The effects of lung volume recruitment on coughing and pulmonary function in patients with ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(2):111–115. | ||
Rafiq MK, Bradburn M, Proctor AR, et al. A preliminary randomized trial of the mechanical insufflator-exsufflator versus breath-stacking technique in patients with amyotrophic lateral sclerosis. Amyotroph Lateral SclerFrontotemporal Degener. 2015;16(7–8):448–455. | ||
Molgat-Seon Y, Hannan LM, Dominelli PB, et al. Lung volume recruitment acutely increases respiratory system compliance in individuals with severe respiratory muscle weakness. ERJ Open Res. 2017;3(1):00135–02016. | ||
Toussaint M, Pernet K, Steens M, Haan J, Sheers N. Cough augmentation in subjects with Duchene Muscular Dystrophy: comparison of air stacking via resuscitator bag versus mechanical ventilation. Respir Care. 2015;61(1):61–67. | ||
Kaminska M, Browman F, Trojan DA, Genge A, Benedetti A, Petrof BJ. Feasibility of lung volume recruitment in early neuromuscular weakness: a comparison between amyotrophic lateral sclerosis, myotonic dystrophy, and postpolio syndrome. Phys Med Rehabil. 2015;7(7):677–684. | ||
Macpherson CE, Bassile CC. Pulmonary physical therapy techniques to enhance survival in amyotrophic lateral sclerosis: a systematic review. J Neurol Phys Ther. 2016;40(3):165–175. | ||
Rafiq MK, Bradburn M, Mustfa N, McDermott CJ, Annane D. Mechanical cough augmentation techniques in amyotrophic lateral sclerosis/motor neuron disease (Protocol). Cochrane Database Syst Rev. 2016;12(1):CD012482. | ||
The UK Motor Neurone Disease Networking Group. A pathway for the management of pain in motor neurone disease; 2001. Available from: https://parkinsonsacademy.co/resources/care-pathways/. Accessed April 23, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.