Back to Journals » Drug Design, Development and Therapy » Volume 8
Phyllostachys edulis extract induces apoptosis signaling in osteosarcoma cells, associated with AMPK activation
Authors Chou C, Cheng Y, Tsai C
Received 13 June 2014
Accepted for publication 15 July 2014
Published 24 September 2014 Volume 2014:8 Pages 1577—1584
DOI https://doi.org/10.2147/DDDT.S69342
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Chi-Wen Chou,1,2 Ya-Wen Cheng,3 Chung-Hung Tsai1
1Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan; 2Department of Orthopedics Surgery, National Taiwan University Hospital Yun-Lin Branch, Yun-Lin, Taiwan; 3Graduate Institute of Cancer Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan
Objective: Bamboo is distributed worldwide, and its different parts are used as foods or as a traditional herb. Recently, antitumoral effects of bamboo extracts on several tumors have been increasingly reported; however, antitumoral activity of bamboo extracts on osteosarcoma remains unclear. In the present study, we investigated effects of an aqueous Phyllostachys edulis leaf extract (PEE) on osteosarcoma cells and the underlying mechanism of inhibition.
Methods: The growth of human osteosarcoma cell lines 143B and MG-63 and lung fibroblast MRC-5 cells was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Apoptosis was demonstrated using TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay and flow cytometric analysis. Phosphorylation and protein levels were determined by immunoblotting.
Results: After treatment with PEE, viability of 143B and MG-63 cells was dose-dependently reduced to 36.3%±1.6% of control values, which were similar to AICAR (5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside) treatments. In parallel, ratios of apoptotic cells and cells in the sub-G1 phase were significantly increased. Further investigation showed that PEE treatments led to activation of caspase cascades and changes of apoptotic mediators Bcl2, Bax, and p53. Consistently, our results revealed that PEE activated adenosine monophosphate-activated protein kinase (AMPK) signaling, and the AMPK activation was associated with the induction of apoptotic signaling.
Conclusion: Our results indicated that PEE suppressed the growth of 143B and MG-63 cells but moderately affected MRC-5 cells. PEE-induced apoptosis may attribute to AMPK activation and the following activation of apoptotic signaling cascades. These findings revealed that PEE possesses antitumoral activity on human osteosarcoma cells by manipulating AMPK signaling, suggesting that PEE alone or combined with regular antitumor drugs may be beneficial as osteosarcoma treatments.
Keywords: bamboo, antitumor, AMPK, Bcl2, Bax
Introduction
Osteosarcoma is a primary bone tumor which is believed to be derived from malignant mesenchymal stem cells.1 The tumors mostly occur in the metaphyses of long bones, particularly the distal femur, the proximal tibia, and the proximal humerus.2 Although the application of neoadjuvant chemotherapy coupled with local control has ameliorated the outcomes of osteosarcoma patients in the past decades, the 5-year survival rate remains approximately 65%, and the rates after recurrence or metastasis are dramatically lowered to 30%.3,4 In addition, osteosarcoma patients with metastasis to other tissues such as lung usually decease in 6–12 months.5 Therefore, the mining of the therapeutic compounds and/or investigation of molecular mechanisms to suppress tumor progression is urgent and critical to develop effective osteosarcoma treatment. Use of herbal medicine has gained increased interest as an alternative for treating neoplastic disorders.
There are approximately 1,050 different species of bamboo in the world, from the tiny Sasa to the subtropical giants. Bamboo is widely distributed, and several edible parts constitute a range of important nutrients. Leaves of bamboo have been utilized in traditional Chinese medicine for treating fever and inflammation for centuries. Bamboo leaf extracts have been demonstrated to possess different biological and pharmacological activities including antioxidant activity and anti-inflammatory activity.6,7 Recently, antitumor effects of bamboo extracts on different cancers such as tongue squamous carcinoma and colon cancer have been increasingly investigated.8–11 However, the functional components of antitumor activity and the underlying mechanisms remain unclear and need further investigation. Moreover, the therapeutic effects of herbal remedies need to be justified in different age groups of patients.12–14
Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is an important sensor for cellular energy level in all eukaryotic cells. AMPK is activated under conditions of low intracellular ATP (adenosine triphosphate) attributing to nutrient deprivation and hypoxia.15 Previous studies have shown that AMPK governs important cellular physiology including cell growth, cell proliferation, and autophagy.16,17 Activation of AMPK has been reported to be involved in suppressed growth of different carcinoma cells such as HepG2 (liver cancer) and SW620 (colorectal cancer).18,19 Recent studies have found that genetic manipulation of the AMPK upstream activator LKB1 is crucial for hepatoma development,20 and activated AMPK inhibits hepatoma growth by destabilizing p53 in a SIRT1-dependent manner.21 These findings suggest that AMPK may be considered as a potential target for treating carcinomas.
The present study was aimed to investigate antitumoral effects of an aqueous Phyllostachys edulis leaf extract (PEE) on human osteosarcoma cells and the underlying mechanism, with emphasis on AMPK signaling.
Materials and methods
Preparation of PEE
The young leaves of bamboo P. edulis were collected and washed with distilled water. Five grams of the leaves were homogenized with 20 mL of distilled water. The homogenate was heated at 95°C for 30 minutes to inactivated proteins, and then filtered through gauze. After centrifugation at 15,000× g for 10 minutes, the supernatant was passed through an ultrafiltration membrane (1 kDa cutoff), lyophilized (PEE), and then stored at −70°C for subsequent experiments.
Cell culture and experimental treatments
Human osteosarcoma cell lines 143B and MG-63 and human non-tumor lung fibroblast MRC-5 cells were purchased from American Type Culture Collection (ATCC; Rockville, MD, USA) and maintained in Dulbecco’s modified Eagle medium (DMEM; Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco BRL) and 100 μg/mL penicillin/streptomycin (Gibco BRL, Basel, Switzerland) at 37°C in a humidified atmosphere containing 5% CO2. Cells were seeded at a density of 1×105 cells/mL and cultured for 24 hours to reach 80% confluence. For experiments, 80% confluent cells were treated with serial concentrations of PEE (w/v) in serum-free DMEM for 24 or 48 hours. After the treatments, the cells were washed with phosphate-buffered saline (PBS) and collected for subsequent assays.
Cell viability assay
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described.22 After 24 or 48 hours incubation with PEE or 5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside (AICAR; Sigma-Aldrich, St Louis, MO, USA) at serial concentrations, culture medium was aspirated, and the cells were washed with PBS followed by incubation with MTT (0.5 mg/mL) at 37°C for 4 hours. After incubation, the supernatant was removed, and the cells were given with isopropanol to dissolve formazan. Concentration of formazan was determined by measuring the absorbance at 570 nm using SpectraMAX 360 pc microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Cellular apoptosis assay
The apoptotic cells were detected using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) system according to the manufacturer’s instructions (Promega, Madison, WI, USA). For nuclei staining, 4′,6-diamidino-2-phenylindole (DAPI) was used. Signal detection was performed using fluorescence microscopy (LSM 710; Carl-Zeiss, Oberkochen, Germany). The apoptotic index was presented as average percentages of positive nuclei including apoptotic bodies per total number of nuclei from five random observation fields at 200× magnitudes. Three independent experiments were performed for statistical analysis.
Flow cytometric analysis for cell cycle distribution
Cells were synchronized at G0 phase by serum starvation for 16 hours and subsequently incubated in fresh serum-containing medium to allow cell-cycle progression. At designated time points, cells released from G0 arrest were analyzed using a flow cytometer (FACSCalibur™; BD Biosciences, San Jose, CA, USA). At the end of treatment, cells were harvested, fixed with 1 mL of ice-cold 70% ethanol, and centrifuged at 400× g for 5 minutes at room temperature. Cell pellets were stained with l mL of 20 μg/mL propidium iodide for 15 minutes, protected from light. The resulting samples were analyzed using FACS Calibur system (BD Biosciences) incorporating with CellQuest software (version 5.1).
Immunoblotting
After treatments, cells were washed with PBS twice and then lysed with lysis buffer (50 mM Tris-HCl, pH 7.5, 1% Nonidet P-40; Sigma-Aldrich) supplemented with protease inhibitor cocktail (Roche, Mannheim, Germany). The cell lysates were centrifuged at 20,000× g for 15 minutes, and then the resulting supernatants were collected for protein quantitation using BCA method (Pierce Biotechnology, Rockford, IL, USA) and the subsequent analyses. Of each sample, 20 μg protein was subjected to a 12.5% SDS (sodium dodecyl sulfate)-polyacrylamide gel followed by a transfer onto a nitrocellulose membrane (Millipore, Bedford, MA, USA). The blotted membrane was blocked with 5% w/v skimmed milk, incubated with primary antibodies (1/1,000 dilution) for 2 hours, flowed by an incubation with peroxidase-conjugated secondary antibodies (1/2,000 dilution). Primary antibodies against caspase-3, caspase-8, caspase-9, Bax, Bcl-2, phosphorylated AMPK at threonine 172, AMPK, and poly(ADP-ribose) polymerase (PARP) were purchased from Cell Signaling Technologies (Beverly, MA, USA). Signal development was performed using ECL (electrochemiluminescence) reagent (Millipore).
Statistical analysis
The data were expressed as mean ± standard deviation of the three independent experiments. Differences between treated groups and control groups were statistically analyzed using Student’s t-test or Dunnett’s t-test. The differences were considered significant for P-values <0.05.
Results
PEE treatment inhibits cell viability of human osteosarcoma cells
Cytotoxicity of PEE was first investigated by using cell viability assay. Osteosarcoma 143B and MG-63 cells were treated with serial concentrations of PEE (50, 100, 200, and 400 μg/mL) for 24 and 48 hours, and subsequently measured for the cell viability. As shown in Figure 1, the viability of 143B and MG-63 cells exposed to PEE for both 24 and 48 hours declined in a dose-dependent fashion. In the 24-hour treatment groups, the cell viability was significantly decreased to 55.4%±2.1% (143B) and 56.1%±3.2% (MG-63) of control at concentration of 400 μg/mL PEE, respectively (P<0.05 as compared with control). For the 48-hour treatment, the cell viability was further lowered to 42.7%±2.2% and 35.2%±2.8% of control at a concentration of 400 μg/mL PEE, respectively (P<0.05 as compared with control). In parallel, cytotoxic effects of PEE on non-tumor lung fibroblast MRC-5 were also explored. Comparing with 143B and MG-63 cells, viability of MRC-5 cell was moderately affected by PEE treatment, and the cell viability was decreased to 83.5%±8.2% (24 hours) and 79.7%±2.8% (48 hours) of control at concentration of 400 μg/mL PEE, respectively (P<0.05 as compared with 143B).
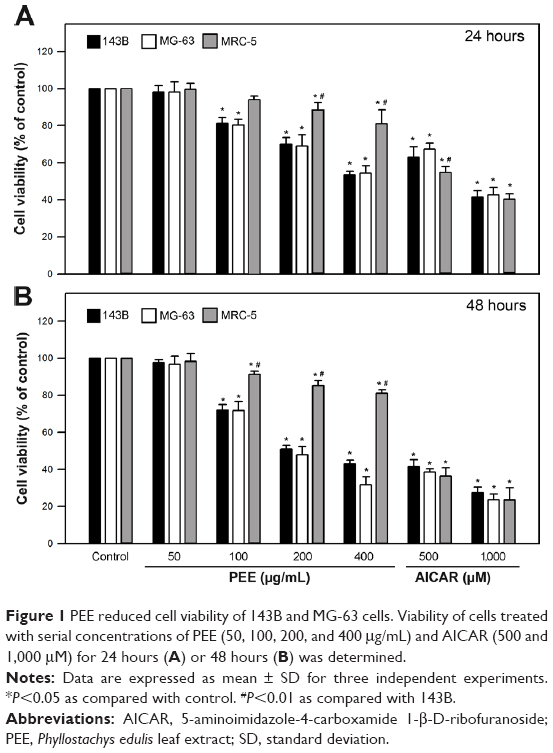 | Figure 1 PEE reduced cell viability of 143B and MG-63 cells. Viability of cells treated with serial concentrations of PEE (50, 100, 200, and 400 μg/mL) and AICAR (500 and 1,000 μM) for 24 hours (A) or 48 hours (B) was determined. |
Effects of an AMPK activator AICAR on viability of the osteosarcoma cells were also performed. As shown in Figure 1B, the cell viability was significantly decreased to 41.5%±3.1% (143B) and 38.4%±1.6% (MG-63) of control at concentration of 500 μM AICAR for 48 hours, respectively (P<0.05 as compared with control). Interestingly, the changes of viability between 143B and MRC-5 cells in response to AICAR (1,000 μM for 24 and 48 hours and 500 μM for 48 hours) were insignificant (P>0.05). Taken together, these findings showed that PEE treatment (200 and 400 μg/mL), similar to AICAR treatment, significantly suppressed the viability of 143B and MG-63 cells.
PEE induces apoptosis of 143B cells
To elucidate the cell death caused by PEE, cell cycle distribution of PEE-treated 143B cells was examined. As shown in Figure 2A, ratios of sub-G1 phase were increased in a dose-dependent manner in the cells with exposure to PEE treatments for 24 hours. The percentage of sub-G1 phase was increased to 32.5%±3.1% (400 μg/mL PEE, P<0.05 as compared with control). For AICAR treatment (500 μM), the percentage of sub-G1 phase was increased to 24.3%±2.3% (P<0.05 as compared with control). To further evaluate the apoptosis induced by PEE, TUNEL assay was subsequently performed. As shown in Figure 2B, apoptotic index was significantly increased to 33.5%±2.1% in response to PEE (400 μg/mL, P<0.05 as compared with control), and the increase of apoptotic cells was dose-dependent. In addition, similar increased apoptotic index (31.1%±1.8%) was also observed in the presence of AICAR. Collectively, these results showed that PEE treatment remarkably induced apoptosis of 143B cells.
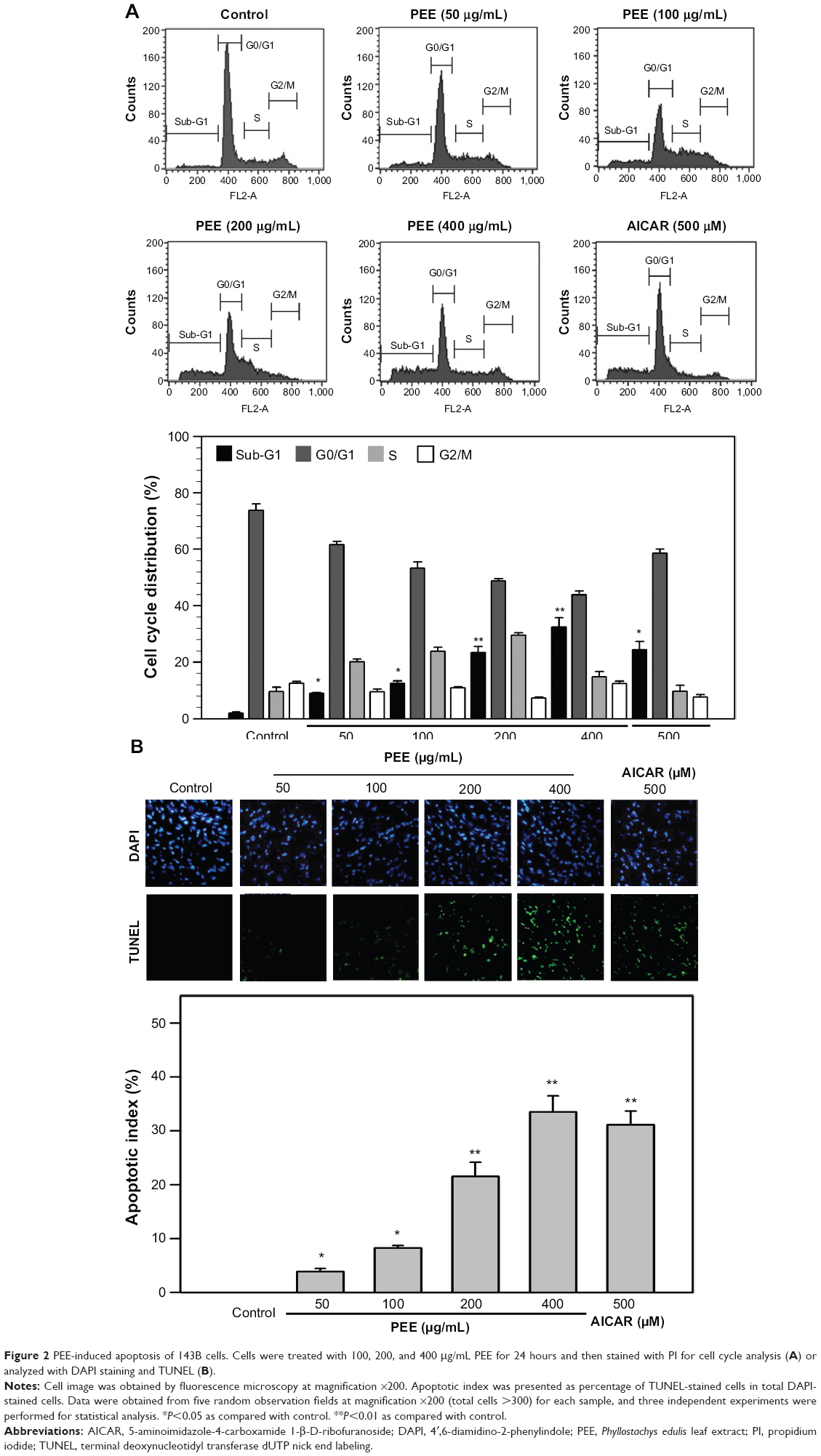 | Figure 2 PEE-induced apoptosis of 143B cells. Cells were treated with 100, 200, and 400 μg/mL PEE for 24 hours and then stained with PI for cell cycle analysis (A) or analyzed with DAPI staining and TUNEL (B). |
PEE activates apoptotic pathway in 143B cells
To further explore apoptosis of 143B cells induced by PEE, the activation of intrinsic and extrinsic caspase cascades was determined. As shown in Figure 3A, the level of procaspase-3, an effecter caspase contributing to DNA fragmentation, was reduced in response to PEE treatments in a dose-dependent fashion. In addition to the reduced procaspase-3, active cleaved caspase-3 was increased in 143B cells with exposure to PEE. We next investigated whether caspase-8 and caspase-9, two upstream activators of caspase-3 in extrinsic and intrinsic pathway, were activated by PEE treatment, and the results revealed that PEE treatments decreased the level of inactive procaspase-8 and procaspase-9 but increased the level of active cleaved caspase-8 (43 kDa) and caspase-9 (35 kDa) in 143B cells.
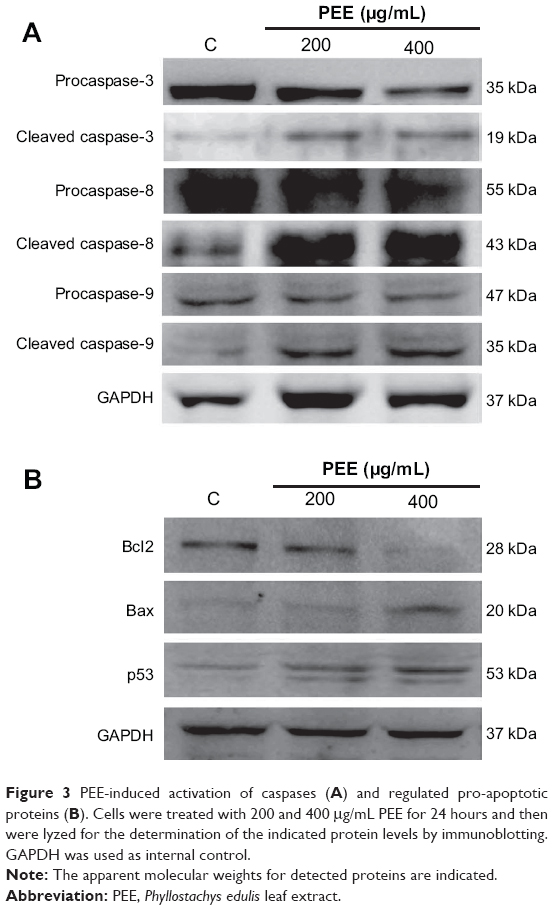 | Figure 3 PEE-induced activation of caspases (A) and regulated pro-apoptotic proteins (B). Cells were treated with 200 and 400 μg/mL PEE for 24 hours and then were lyzed for the determination of the indicated protein levels by immunoblotting. GAPDH was used as internal control. |
For PEE, treatment significantly induced cell apoptosis and activation of caspase cascades, expression of the anti-apoptotic protein Bcl2 and the pro-apoptotic proteins Bax and p53 was determined.23 As shown in Figure 3B, PEE decreased Bcl2 levels but induced increases in Bax and p53 levels. Together, these findings indicate that PEE triggers activation of caspase cascades, which may attribute to suppression of anti-apoptotic Bcl2 and enhancement of pro-apoptotic Bax and p53.
PEE treatment activates AMPK signaling in human osteosarcoma 143B cells
A previous study demonstrated that bamboo leaf extract could induce AMPK signaling and the consequent biological activity.24 Accordingly, effects of PEE on AMPK signaling were examined. After 24 hours treatment, PEE enhanced AMPK phosphorylation at threonine 172 (Figure 4). AMPK activation was associated with a decrease in mammalian target of rapamycin (mTOR) and 70 kDa ribosomal protein S6 kinase (P70S6K) phosphorylation (Figure 4). Using a well-known AMPK activator, AICAR, similar AMPK phosphorylation at threonine 172 and decreased mTOR and P70S6K was observed in 143B cells (Figure 4). The findings indicate that AMPK was activated in the presence of PEE in 143B cells.
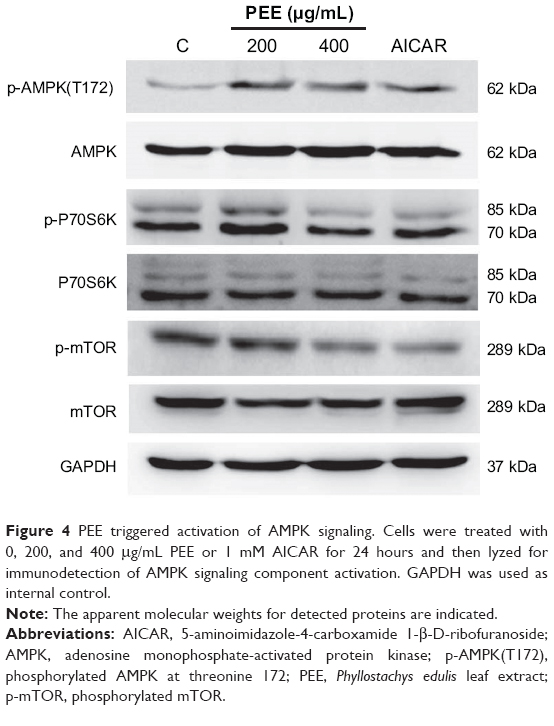 | Figure 4 PEE triggered activation of AMPK signaling. Cells were treated with 0, 200, and 400 μg/mL PEE or 1 mM AICAR for 24 hours and then lyzed for immunodetection of AMPK signaling component activation. GAPDH was used as internal control. |
PEE induced apoptosis and lowered cell viability of 143B cells via AMPK activation
To examine whether AMPK phosphorylation was responsible for PEE-induced cell death, cells were treated with PEE (400 μg/mL) for 24 hours, and apoptotic proteins were analyzed by immunoblotting. The selective AMPK inhibitor compound C diminished PEE-induced caspase-3 and PARP cleavage (Figure 5A) and cell growth inhibition (P<0.01) (Figure 5B). These results suggest that PEE-induced apoptosis and viability suppression are mediated by activating AMPK.
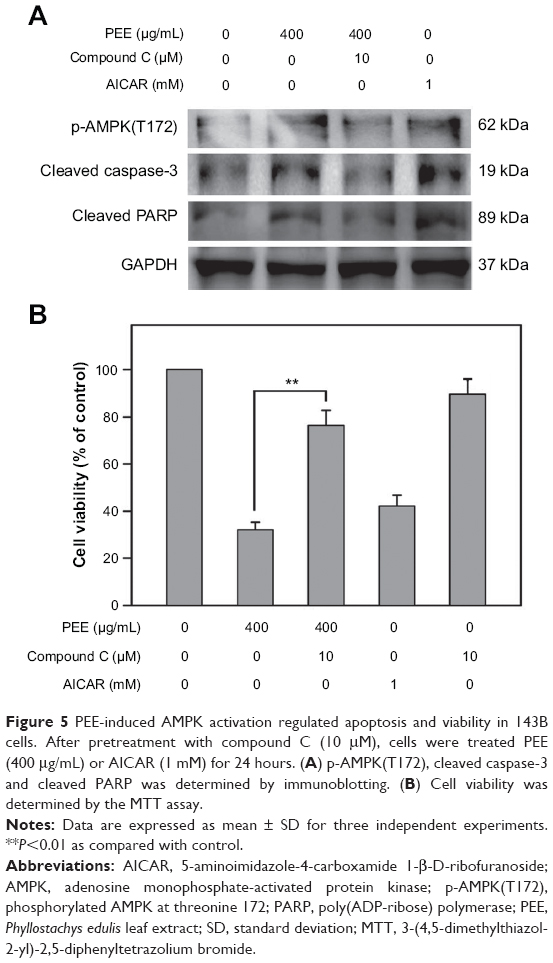 | Figure 5 PEE-induced AMPK activation regulated apoptosis and viability in 143B cells. After pretreatment with compound C (10 μM), cells were treated PEE (400 μg/mL) or AICAR (1 mM) for 24 hours. (A) p-AMPK(T172), cleaved caspase-3 and cleaved PARP was determined by immunoblotting. (B) Cell viability was determined by the MTT assay. |
Discussion
In the past few years, several lines of evidence indicate that Phyllostachys spp. extracts or derived products such as bamboo salt possess cancer-preventive or anticancer effects.8–10 However, the mechanism that bamboo extracts induces apoptosis or autophagy of tumor cells remains sketchy. In the present study, we first demonstrated that a bamboo leaf extract PEE induced apoptosis and growth inhibition of human osteosarcoma 143B cells via both intrinsic and extrinsic pathways attributing to AMPK activation.
AMPK signaling is an important target with potent antitumor activity; thus, identifying new AMPK activators is pivotal for developing anticancer agents.18,25 Recently, several studies have demonstrated that AMPK activators including AICAR and metformin inhibit different types of tumors via AMPK-dependent pathway.26–28 The present study revealed that PEE significantly induced AMPK activation and inhibited the downstream P70S6K and mTOR, which may contribute to decreased Bcl2 level and increased Bax level. In addition, our results show that PEE reduced more cell viability of 143B cells as compared with AICAR alone. Taken together, these findings indicate that AMPK activation plays a pivotal role in PEE-induced apoptosis of osteosarcoma 143B cells, but PEE may also suppress viability of 143B cells via AMPK-independent pathways.
Abnormal cells such as neoplastic cells are eliminated and removed by apoptosis, a programmed cell death.29 The apoptotic cell death pathway is mediated by Bcl2 protein family, a group of anti-apoptotic proteins that regulate the passage of cytochrome c, which binds to apaf-1 to form active apoptosome and then activates the following caspase cascades.30,31 Activated caspase-3 exerts as the key executioner of apoptosis to induce the cleavage and inactivation of key cellular protein such as PARP.32,33 Our findings revealed that PEE treatment increased Bax level and reduced Bcl2 level, as well as elevated the cleaved caspase-3, caspase-8, and caspase-9 levels, suggesting that PEE could alter Bcl2 proteins and activate apoptotic pathway in osteosarcoma 143B cells. In addition, PEE increased p53 level, which may contribute to G1-phase cell cycle arrest as shown in Figure 2C. Taken together, these findings indicate that PEE possesses potent antitumoral activity on osteosarcoma cells, attributing to induction of apoptosis and cell cycle arrest.
In conclusion, we presented the evidence that PEE treatment significantly suppresses viability of osteosarcoma 143B cells through activation of AMPK and a synergistic effort of apoptotic signaling induction and suppressed anti-apoptotic signaling. By regulating both arms of apoptotic and anti-apoptotic signaling, PEE constitutes a pharmacological property that makes it a promising anticancer agent against human osteosarcoma.
Disclosure
The authors declare that there are no conflicts of interest.
References
Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;21(Suppl 7): vii320–vii325. | ||
Steliarova-Foucher E, Stiller C, Lacour B, Kaatsch P. International Classification of Childhood Cancer, third edition. Cancer. 2005;103: 1457–1467. | ||
Marina N, Gebhardt M, Teot L, Gorlick R. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist. 2004;9:422–441. | ||
Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3–13. | ||
Geller DS, Gorlick R. Osteosarcoma: a review of diagnosis, management, and treatment strategies. Clin Adv Hematol Oncol. 2010;8:705–718. | ||
Van Hoyweghen L, De Bosscher K, Haegeman G, Deforce D, Heyerick A. In vitro inhibition of the transcription factor NF-κB and cyclooxygenase by bamboo extracts. Phytother Res. 2014;28(2):224–230. | ||
Zhang Y, Bao B, Lu B, Ren Y, Tie X, et al. Determination of flavone C-glucosides in antioxidant of bamboo leaves (AOB) fortified foods by reversed-phase high-performance liquid chromatography with ultraviolet diode array detection. J Chromatogr A. 2005; 1065:177–185. | ||
Seki T, Maeda H. Cancer preventive effect of Kumaizasa bamboo leaf extracts administered prior to carcinogenesis or cancer inoculation. Anticancer Res. 2010;30:111–118. | ||
Zhao X, Deng X, Park KY, Qiu L, Pang L. Purple bamboo salt has anticancer activity in TCA8113 cells in vitro and preventive effects on buccal mucosa cancer in mice in vivo. Exp Ther Med. 2013;5:549–554. | ||
Zhao X, Kim SY, Park KY. Bamboo salt has in vitro anticancer activity in HCT-116 cells and exerts anti-metastatic effects in vivo. J Med Food. 2013;16:9–19. | ||
Kim A, Im M, Yim NH, Jung YP, Ma JY. Aqueous extract of Bambusae Caulis in Taeniam inhibits PMA-induced tumor cell invasion and pulmonary metastasis: suppression of NF-κB activation through ROS signaling. PLoS One. 2013;8:e78061. | ||
Carotenuto M, Esposito M. Nutraceuticals safety and efficacy in migraine without aura in a population of children affected by neurofibromatosis type I. Neurol Sci. 2013;34:1905–1909. | ||
Esposito M, Carotenuto M. Ginkgolide B complex efficacy for brief prophylaxis of migraine in school-aged children: an open-label study. Neurol Sci. 2011;32:79–81. | ||
Esposito M, Ruberto M, Pascotto A, Carotenuto M. Nutraceutical preparations in childhood migraine prophylaxis: effects on headache outcomes including disability and behaviour. Neurol Sci. 2012;33:1365–1368. | ||
Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. | ||
Hoyer-Hansen M, Jaattela M. AMP-activated protein kinase: a universal regulator of autophagy? Autophagy. 2007;3:381–383. | ||
Luo Z, Saha AK, Xiang X, Ruderman NB. AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci. 2005;26:69–76. | ||
Cho SY, Lee HJ, Jung DB, Kim H, Sohn EJ, et al. Activation of AMP-activated protein kinase α and extracelluar signal-regulated kinase mediates CB-PIC-induced apoptosis in hypoxic SW620 colorectal cancer cells. Evid Based Complement Alternat Med. 2013;2013:974313. | ||
Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5´-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. | ||
Kim CJ, Cho YG, Park JY, et al. Genetic analysis of the LKB1/STK11 gene in hepatocellular carcinomas. Eur J Cancer. 2004;40:136–141. | ||
Lee CW, Wong LL, Tse EY, et al. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012;72:4394–4404. | ||
Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–277. | ||
Ulivieri C. Cell death: insights into the ultrastructure of mitochondria. Tissue Cell. 2010;42:339–347. | ||
Nam JS, Chung HJ, Jang MK, et al. Sasa borealis extract exerts an antidiabetic effect via activation of the AMP-activated protein kinase. Nutr Res Pract. 2013;7:15–21. | ||
Youn SH, Lee JS, Lee MS, et al. Anticancer properties of pomolic acid-induced AMP-activated protein kinase activation in MCF7 human breast cancer cells. Biol Pharm Bull. 2012;35:105–110. | ||
Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer. 2013;13:235. | ||
Sauer H, Engel S, Milosevic N, Sharifpanah F, Wartenberg M. Activation of AMP-kinase by AICAR induces apoptosis of DU-145 prostate cancer cells through generation of reactive oxygen species and activation of c-Jun N-terminal kinase. Int J Oncol. 2012;40:501–508. | ||
Zheng QY, Jin FS, Yao C, et al. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem Biophys Res Commun. 2012;419:741–747. | ||
Hickman JA. Apoptosis induced by anticancer drugs. Cancer Metastasis Rev. 1992;11:121–139. | ||
Slee EA, Harte MT, Kluck RM, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144: 281–292. | ||
Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem. 1999;274:20049–20052. | ||
Charalambous C, Pitta CA, Constantinou AI. Equol enhances tamoxifen’s anti-tumor activity by induction of caspase-mediated apoptosis in MCF-7 breast cancer cells. BMC Cancer. 2013;13:238. | ||
Thornberry NA. Caspases: key mediators of apoptosis. Chem Biol. 1998;5:R97–R103. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.